Organosilane-assisted synthesis of ordered mesoporous poly(furfuryl alcohol) composites†
Yunpu
Zhai
,
Bo
Tu
and
Dongyuan
Zhao
*
Department of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Key Laboratory of Molecular Engineering of Polymers of the Chinese Ministry of Education, Laboratory of Advanced Materials, Fudan University, Shanghai, 200433, P. R. China. E-mail: dyzhao@fudan.edu.cn; Fax: +86-21-6564-1740; Tel: +86-21-5566-4194
First published on 12th November 2008
Abstract
In the present work, we report an organosilane-assisted synthesis of ordered mesoporous poly(furfuryl alcohol) (PFA)-silica composites by employing TEOS, 3-(triethoxysilyl)furan and furfuryl alcohol as precursors, and triblock copolymer F127 as a structure-directing agent via an EISA process. After thermal curing of the PFA, the triblock copolymer can be removed by calcination at 350 °C in N2 atmosphere. The PFA/silica nanocomposites have been characterized by SAXS, TEM, N2 sorption, FT-IR, elemental analysis, 13C-NMR, 29Si-NMR and TGA techniques. The results show that the organic–inorganic nanocomposites have ordered 2-D centered-rectangular (c2mm) mesostructure even when the organic content in the samples is as high as 60 wt%, but their surface areas (200–510 m2/g), pore volumes (0.15–0.54 cm3/g) and pore sizes (4.8–5.7 nm) gradually reduce with increasing organic component. Air (600 °C, 5 h) or HF (10 wt%) secondary treatments of the nanocomposite with 60 wt% PFA lead to a collapse of the mesostructure as shown by SAXS and N2 sorption techniques, implying the existence of an interpenetrating PFA/silica framework, in which both silica and organic polymers synergistically support the structure. The adsorption performances of the hybrid materials for toluene were also conducted. Because of the hydrophobic property, i.e. affinity with toluene molecules, the PFA/silica nanocomposite having a similar surface area as ordered mesoporous silica shows an adsorption quantity twice as large as that of ordered mesoporous silica.
1. Introduction
Organic mesoporous materials, particular those constructed with ordered uniform nanopore arrays, have been of longstanding interest in fundamental research and industry due to their large internal surface area, high sorption capacity, and catalytic activity toward large molecules. In general, these porous organic materials can be prepared by phase separation1 and hard templating2 approaches. However, most of the pore structures constructed by the phase separation approach are disordered with wide pore size distributions because of the contraction and swelling from changes in volume. For the hard template approach, the multi-step process makes it elaborate, expensive, and thus industrially unfeasible; moreover structure defects are always formed during template removal. Recently, highly ordered mesoporous polymers synthesized from organic–organic assembly of block copolymer and phenolic resins, have been reported by several research groups.3–6 The successful synthesis is mainly related to the unique characteristics of the thermosetting phenolic resins, including three-dimensional (3-D) cross-linked networks, abundant benzyl and phenolic hydroxyl groups, and controllable polymerization rate.7 Inspired by such one-pot fabrication of ordered mesoporous phenolic polymers from resols, it would be worthwhile to devote much effort to the synthesis of organic polymer frameworks with elaborately designed functional blocks by manipulating polymerization.The incorporation of inorganic materials, for example silica, into organic polymeric networks can assist the organic polymers, especially hydrophobic and soft ones, to organize into ordered mesostructures, since abundant Si–OH groups can provide strong interaction with the surfactants to avoid macro-phase separation, and relatively high mechanical strength can guarantee mesostructure maintenance after the template removal. In addition, the silica–polymer composites take advantage of the properties of both organic polymers, like flexibility, toughness, hydrophobicity, and versatility for further functionalization, and inorganic components, such as good mechanical and thermal stability. These composites are thus considered as innovative advanced materials, and promising applications are expected in many fields, including protective coatings, catalysis, separations, adsorption, etc. The organization degree viz. the nanostructures of these silica-reinforced mesoporous polymers certainly depends on the synergy between the organic and inorganic components and the surfactant templates. Therefore, to form a homogeneous hybrid network, an appropriate organofunctionalized alkoxysilane is usually necessary to bridge the siloxane networks and the organic species in order to make it easy for surfactants to modulate the hybrids into an ordered mesostructure.
Poly(furfuryl alcohol) (PFA) is a common thermosetting resin usually prepared by the cationic condensation of furfuryl alcohol (FA) monomer. In general, FA polymerizes exothermically in the presence of cationically active initiators, e.g. CF3COOH, silica, HCl, or Lewis acids,8 producing black, amorphous, and branched and/or cross-linked polymer structures. In addition to a flexible choice of catalysts, the polymerization process can be carried out at different temperatures and in various solvents. Importantly, PFA is compatible with many organic and inorganic materials, resulting in high carbon yields. Therefore, PFA is not only important for use as adhesives, binders, corrosion-resistant materials, but also widely used to synthesize nanoporous carbons and polymer nanocomposites for a wide range of applications, such as adsorbents, separation membranes, catalysts, electrodes of fuel cells, lithium batteries, electric double-layer capacitors, etc.9 Moreover, FA is derived from renewable sources, viz. the wastes from agricultural crops (maize, cotton, and sugar-cane), which will contribute to the sustainable development in a social, environmental and economic sense.10
Despite the fact that FA has high solubility in water and many common solvents, cross-linked PFA is hydrophobic. PFA chains pack together and the products suspend in the surfactant solution,11 which leads to failure of the attempts to solvothermally synthesize ordered mesoporous PFA polymers.11,12 The incorporation of hydrophilic inorganic precursors such as tetraethyl orthosilicate (TEOS), which can copolymerize with FA to fabricate fiber-reinforced composites, effectively improves the assembly of PFA around the hydrophilic chains of surfactants. However, only wormlike mesostructure was obtained,13 possibly because the C–O–Si bonds are too weak to moderate the different polymerization rates of TEOS and FA, which destroyed their co-assembly and lead to the damage of the mesostructure. Therefore, it is still a challenge to get ordered mesoporous PFA composites using a one-step convenient approach.
Herein, we demonstrate an organosilane-assisted approach to prepare ordered mesoporous hybrid PFA/silica composites via a solvent evaporation-induced self-assembly (EISA) process.14 A rationally selected organosiloxane, 3-(triethoxysilyl)furan (TESF), is used to form strong covalent linkages between TEOS and FA, and triblock copolymer F127 is used as a structure-directing agent. The inorganic reinforced polymers have ordered 2-D centered-rectangular (space group of c2mm) mesostructures, even when the organic content in the samples increases as high as 60 wt%. The silica and organic polymer species interpenetrate homogeneously and support the framework synergistically. The existence of hydrophobic PFA, which has an affinity with toluene molecules, benefits the toluene adsorption.
2. Experimental
2.1 Chemicals
Poly(propylene oxide)-block-poly(ethylene oxide)-block-poly(propylene oxide) triblock copolymer Pluronic F127 (Mw = 12 600, EO106PO70 EO106) was purchased from Acros Corp. Organosiloxane 3-(triethoxysilyl)furan (TESF, 96 wt%) was purchased from Sigma-Aldrich Corp. Tetraethyl orthosilicate (TEOS, 97 wt%), furfuryl alcohol (FA, 90 wt%), hydrochloric acid (HCl, 37 wt%), and ethanol were purchased from Shanghai Chemical Corp. All chemicals were used as received without further purification. Millipore water was used in all experiments.2.2 Synthesis of mesoporous PFA/silica composites
Mesoporous PFA/silica hybrid materials were prepared by co-assembly of FA, TEOS, and copolymer F127 with organosilane-assistant TESF under acidic conditions. In a typical preparation, 1.04 g (5.0 mmol) of TEOS, 1.09 g (10.0 mmol) of FA and 0.23 g (1.0 mmol) of TESF were dissolved in 2.0 g (0.04 mol) of ethanol with 0.27 g (0.05 mmol) of HCl (0.2 M, aqueous solution). The solution (solution A) was kept stirring for 5 h at 40 °C. In solution B, 1.0 g (0.08 mmol) of F127 was dissolved in 5.0 g (0.11 mol) of ethanol. Solutions A and B were mixed together and vigorously stirred for another 0.5 h at 40 °C. The mixture was transferred into several dishes for ethanol evaporation under ambient conditions. After 1–2 h, the dishes were covered with glass slides and heated in an oven for 24 h at 60 °C, followed by heating at 130 °C for a further 24 h. The as-made products (denoted as FS-60-as-made), flaxen membranes, were scraped from the dishes and ground into fine powders. Calcination was carried out under N2 flow in a tubular furnace at 350 °C for 3 h with a heating rate of 1 °C/min to get mesoporous PFA/silica composites, named as FS-60. “FS-x” denotes the mesoporous PFA/silica nanocomposites, wherein x represents the weight percentage of the polymer in the products after calcination at 350 °C. To obtain PFA/silica composites with variable polymer contents, different weight ratios of FA to TEOS were used as summarized in Table 1, with the molar ratio of TEOS/TESF/HCl/EtOH fixed at 1.0:0.2:0.01:30.0.| Sample | TEOS/g | TESF/g | FA/g | F127/g | 0.2M HCl/g | Polymer a (wt%) | SiO2a (wt%) |
|---|---|---|---|---|---|---|---|
| a Polymer% and SiO2% are the mass percentages in the mesoporous polymer/silica nanocomposites, determined from TG results. | |||||||
| FS-33 | 1.04 | 0.23 | 0.54 | 0.7 | 0.27 | 33 | 67 |
| FS-41 | 1.04 | 0.23 | 0.82 | 0.7 | 0.27 | 41 | 59 |
| FS-60 | 1.04 | 0.23 | 1.09 | 1.0 | 0.27 | 60 | 40 |
| MSiO2 | 1.04 | 0 | 0 | 0.5 | 0.50 | 0 | 100 |
2.3 Preparation of mesoporous silica
Sample with pure silica framework (M-SiO2) was synthesized by an EISA method with copolymer F127 as a template according to the previous report,15 except for different reaction temperature. Typically, 0.5 g of F127 was dissolved in 6.0 g of ethanol containing 0.5 g of 0.2 M HCl, and then 1.04 g of TEOS was added followed by stirring for 1.5 h at 40 °C. The homogeneous solution was transferred into several dishes for ethanol evaporation under ambient conditions for 5 h, and then heated at 60 °C for 24 h. The as-made products were calcined at 350 °C for 3 h in air to remove the template.2.4 Characterization and measurements
Small-angle X-ray scattering (SAXS) measurements were taken on a NanoSTAR system (Bruker, Germany) using Cu Kα radiation (40 kV, 35 mA). The d-spacing values were calculated by the formula d = 2π/q. Transmission electron microscopy (TEM) images were taken on a JEOL JEM2011 electron microscope operating at 200 kV. For TEM measurements, the samples were dispersed as a slurry in ethanol and dried on a holey carbon film on a Cu grid. N2 adsorption–desorption isotherms were measured with a Micromeritics Tristar 3000 analyzer at 77 K. Before measurements, the samples were degassed at 150 °C for more than 5 h. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas (SBET) using adsorption data at a relative pressure range from 0.04 to 0.2. The pore size distributions were derived from the adsorption branches of the isotherms based on the Barrett–Joyner–Halanda (BJH) model. The total pore volume, VP, was estimated from the amount adsorbed at a relative pressure (P/P0) of 0.99. Fourier transform infrared (FT-IR) spectra were collected on Nicolet Fourier spectrophotometer, using KBr pellets of the solid samples. Weight changes of the products were monitored using a Mettler Toledo TGA-SDTA851 analyzer (Switzerland) from 25 to 900 °C under nitrogen or air with a heating rate of 5 °C/min. 13C and 29Si solid-state nuclear magnetic resonance (NMR) experiments were performed on a Bruker DSX300 spectrometer (Germany) under conditions of cross polarization (CP) and magic angle sample spinning (MAS). 13C CP-MAS NMR spectra were collected at room temperature with a frequency of 75 MHz (2 s recycle, 2.5 ms contact time) by using adamantane as a reference. 29Si solid-state MAS NMR spectra were collected at room temperature with a frequency of 59.6 MHz, a recycling delay of 600 s, a radiation frequency intensity of 62.5 kHz, and a reference sample of Q8M8 ([(CH3)3SiO]8Si8O12). The C, H, and O contents were measured on a Vario EL III elemental analyzer (Germany). Before the measurements, the silica components were removed from polymer–silica nanocomposites.2.5 Water and toluene adsorption measurements
Water and toluene adsorption isotherms were recorded by using a digital microbalance (Hiden Isochema Instrument, model IGA-002) with a sensitivity of 0.1 µg at 25 °C. Before measurements, the samples were degassed at 180 °C for at least 5 h in a high-vacuum system.3. Results
3.1 Mesostructure of PFA/silica nanocomposites
Mesoporous PFA/silica hybrid composites can be synthesized by organosilane-assisted co-assembly of FA, TESF, TEOS and triblock copolymer F127 via an EISA approach. The as-made samples are brown membranes without obvious macrophase separation. After pyrolysis at 350 °C in N2 atmosphere, the products changed to dark brown color. The SAXS pattern of as-made hybrid polymer composite (FS-33-as-made) with low PFA content of 33 wt% displays two scattering peaks with d-spacings of 11.8 and 10.3 nm (Fig. 1A(a)). After calcination at 350 °C under N2, the SAXS pattern becomes more resolved and four additional scattering peaks can be observed in the q range from 1.0 to 1.6 nm−1 (Fig. 1B(a)), which are attributed to 02, 31, 22 and 40 reflections of 2-D centered rectangular mesostructure (space group of c2mm), similar to previously reported ordered mesoporous materials.16,17 Compared with those of the FS-33-as-made sample, the 11 and 20 scattering peaks shift to higher values, indicating framework shrinkage. The cell parameters are calculated as a = 20.6, b = 14.5 nm, a/b = 1.42 and a = 17.6, b = 11.8 nm, a/b = 1.49 for the as-made and calcined samples respectively (Table 2). Therefore, the mesostructure undergoes a shrinkage of 16%, due to further condensation of the polymer/silica framework at high temperature. With an increase of PFA content, as expressed in FS-41-as-made and calcined FS-41, the SAXS patterns show that the products have ordered c2mm mesostructure (Fig. 1A(b) and 1B(b)), similar to those for FS-33-as-made and FS-33. With further increasing polymer contents, the SAXS patterns of composites such as FS-60-as-made and FS-60 exhibit poorly resolved scattering peaks (Fig. 1A(c) and 1B(c)), implying partial collapse of the mesostructure. The calculated mesostructural shrinkage increases with the increase in PFA content, further suggesting the introduction of more organic PFA polymers into the framework.| Sample | a/nm | b/nm | DBJH/nm | V P /cm3 g−1 | SBET/m2 g−1 | Q t /mmol g−1 | Q w /mmol g−1 |
|---|---|---|---|---|---|---|---|
a
a and b correspond to the lattice parameters of the unit cell (a > b) with rectangular columnar phase of c2mm symmetry, the values were calculated following the formula  . The dhk0 values were obtained from SAXS data using the formula d = 2π/q. DBJH is the pore size diameter. SBET is the BET surface area. VP is the total pore volume. Qt and Qw are the adsorption amounts for toluene and water, respectively.
b The unit cell parameter of the mesoporous silica was calculated by using the formula a0 = 2d100/√3. . The dhk0 values were obtained from SAXS data using the formula d = 2π/q. DBJH is the pore size diameter. SBET is the BET surface area. VP is the total pore volume. Qt and Qw are the adsorption amounts for toluene and water, respectively.
b The unit cell parameter of the mesoporous silica was calculated by using the formula a0 = 2d100/√3.
|
|||||||
| FS-33-as-made | 20.6 | 14.5 | — | — | — | — | — |
| FS-33 | 17.6 | 11.8 | 5.7 | 0.54 | 510 | 4.6 | 27.2 |
| Si- FS-33 | 15.8 | 10.8 | 6.2 | 0.66 | 530 | — | — |
| FS-41-as-made | 20.5 | 14.7 | — | — | — | — | — |
| FS-41 | 18.2 | 12.4 | 5.4 | 0.37 | 410 | 3.5 | 20.3 |
| Si- FS-41 | 15.4 | 10.3 | 6.7 | 0.57 | 380 | — | — |
| FS-60-as-made | 21.0 | 15.1 | — | — | — | — | — |
| FS-60 | 16.8 | 11.3 | 4.5 | 0.15 | 200 | 2.1 | 15.1 |
| Si- FS-60 | 13.7 | 9.5 | 5.9 | 0.37 | 240 | — | — |
| MSiO2-as-made | 15.7b | — | — | — | — | — | — |
| MSiO2 | 13.6b | — | 6.2 | 0.51 | 520 | 2.3 | 21.7 |
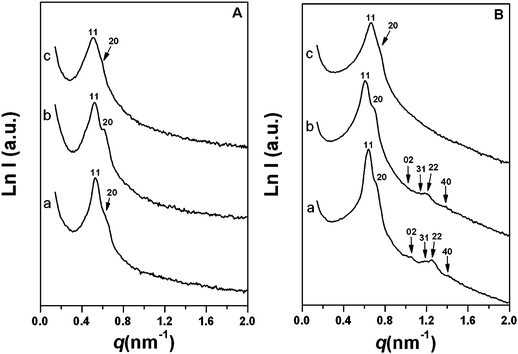 | ||
| Fig. 1 SAXS patterns of mesoporous silica/polymer composites with different polymer contents before (A) and after (B) template removal by calcination at 350 °C under N2 flow. In (A): (a) FS-33-as-made, (b) FS-41-as-made, and (c) FS-60-as-made. In (B): (a) FS-33, (b) FS-41, and (c) FS-60. | ||
TEM images of the mesoporous PFA/silica composite FS-33 show large domains of cylinder arrays along the [11] direction (Fig. 2A) and a distorted hexagonal arrays along the [10] zone plane (Fig. 2B), clearly indicating that the composite has ordered 2-D mesostructure. The corresponding Fourier diffractograms further confirm that the mesostructure is centered rectangular cmm symmetry (Fig. 2A, B, insets). With increase of organic polymer content (in cases of FS-41 and FS-60), well-defined 2-D centered rectangular mesostructure can also be observed (Fig. 2C, D, E, F). A high resolution TEM (HRTEM) image of sample FS-60 viewed along the [10] direction clearly reveals a centered rectangular mesostructure (Fig. 2G). The corresponding FFT diffractogram can be indexed up to the 4th order of a c2mm space group as shown in Fig. 2H, according to the previous reports.17,18 It suggests that the structural integrity of the mesoporous hybrid materials can be retained with up to 60 wt% of PFA moiety. The cell parameters estimated from TEM measurements are consistent with those from SAXS analysis.
![TEM images of mesoporous PFA/silica composites with different polymer contents after calcination at 350 °C in N2 flow, viewed from the [11] (A, C, E) and [10] (B, D, F) directions: (A) and (B) FS-33; (C) and (D) FS-41; (E) and (F) FS-60. The insets are the corresponding FFT diffractograms. HRTEM image (G) of FS-60 along the [10] direction and its indexed FFT pattern (H).](/image/article/2009/JM/b813688b/b813688b-f2.gif) | ||
| Fig. 2 TEM images of mesoporous PFA/silica composites with different polymer contents after calcination at 350 °C in N2 flow, viewed from the [11] (A, C, E) and [10] (B, D, F) directions: (A) and (B) FS-33; (C) and (D) FS-41; (E) and (F) FS-60. The insets are the corresponding FFT diffractograms. HRTEM image (G) of FS-60 along the [10] direction and its indexed FFT pattern (H). | ||
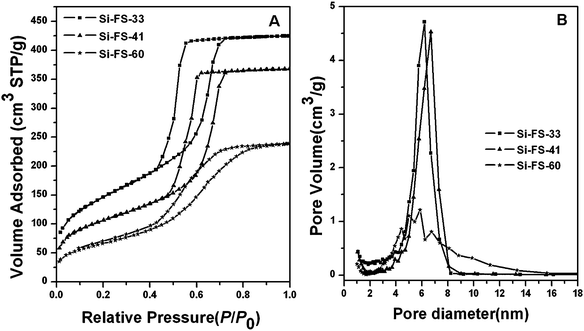
Nitrogen sorption isotherms for the mesoporous hybrid composites with different organic polymer contents after pyrolysis at 350 °C show representative type-IV curves with H2 hysteresis loops (Fig. 3A). Well-defined and steep steps occur approximately at P/P0 = 0.40–0.60. The behavior is associated with nitrogen filling of the mesopores owing to capillary condensation, reflecting a uniformity of mesopore sizes. The shift of capillary condensation steps towards lower relative pressure indicates a decrease of the pore size with increasing PFA content, which is consistent with the trend of framework shrinkage. Notably, different from those for FS-33, the adsorption and desorption isotherms of FS-41 are not close at low relative pressures, indicative of a typical organic polymer framework.7,19 This phenomenon is more obvious for FS-60, implying that more organic polymer is incorporated into the framework. The mean pore sizes gradually decrease from 5.7 to 4.5 nm as the organic content increases from 33 to 60 wt% (Fig. 3B), which is owing to the severe shrinkage of the framework. The BET surface areas and total pore volumes also reduce from 510 m2/g and 0.54 cm3/g to 200 m2/g and 0.15 cm3/g, respectively, with increasing organic content. This is possibly due to the shrinkage and even closure of the micro/mesopores during the pyrolysis, as well as deteriorated mesostructural features.
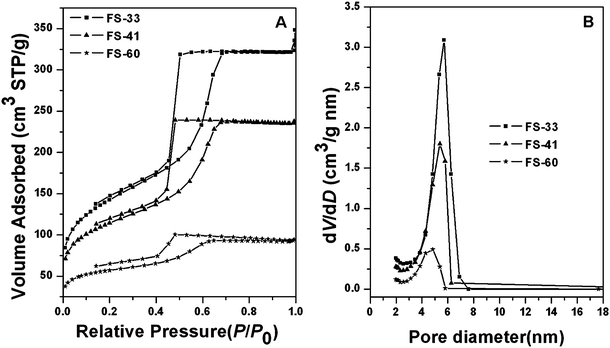 | ||
| Fig. 3 Nitrogen sorption isotherms (A) and pore size distribution curves (B) of mesoporous PFA/silica composites with different polymer contents after calcination at 350 °C under N2 flow. | ||
3.2 Constitution of the framework
The TGA curve of pure copolymer F127 in N2 atmosphere shows an obvious weight loss of ca. 98.3 wt% in the temperature of 300 to 400 °C (Fig. 4A), suggesting that most of the templates can be decomposed at 350 °C. Four detectable weight loss steps can be observed in the TGA curve of FS-60-as-made nanocomposite (Fig. 4A). The large weight loss of 50.2 wt% occurs in the range of 300 to 400 °C, which is mainly attributed to the template degradation. The weight loss observed below 200 °C (4.6 wt%) is caused by the desorption of physisorbed water, and the subsequent weight loss (6.7 wt%) between 200 and 300 °C coincides with the cross-linking of PFA.20 The weight loss (16.7 wt%) which occurred at 400–900 °C corresponds to the dehydrogenation and carbonization of PFA and condensation of silicate species. For the nanocomposites with different organic contents after calcination at 350 °C in nitrogen, the TGA curves recorded in air flow display similar trends, except their distinct weight losses (Fig. 4B). Weight losses of ∼5 wt% below 200 °C result from physisorbed water. The minor loss peaks at 300–450 °C are assigned to the degradation of the PFA. Significant weight losses of 450–600 °C are attributed to the combustion of organic constituents. The contents of organics and silica in the nanocomposites are listed in Table 1. The values may fluctuate a bit because condensation of silicate species may also contribute to the weight loss.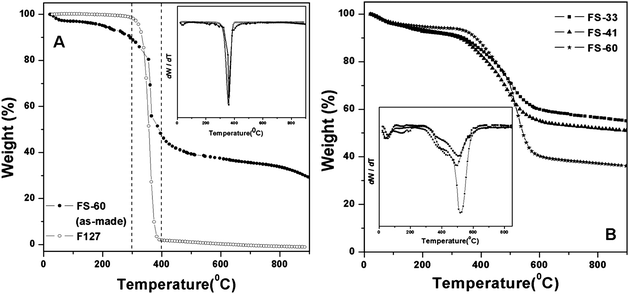 | ||
| Fig. 4 TG and DTG curves (insets) recorded (A) in N2 of triblock copolymer F127 and the as-made mesoporous PFA-silica nanocomposite FS-60-as-made, and (B) in air of mesoporous FS-33, FS-41 and FS-60. | ||
The FT-IR spectrum of nanocomposite FS-60-as-made (Fig. 5a) shows characteristic bands at 1635 and 1365 cm−1 from the ring stretch modes of 2,3-bisubstituted and 2-substituted furan rings.21 The band at 1461 cm−1 is attributed to aliphatic CH2groups in the polymer.12 Due to the opening of some furan rings by acid-catalyzed electrophilic attack, carbonylic groups are formed with a feature band at 1730 cm−1.12,22,23 These results confirm the polymer frameworks from FA. The bands at 948 and 1100 cm−1 are observed, which are contributed to Si–OH and Si–O–Si vibrations.21,22,24–26 The strong but rather broad band at 3435 cm−1 can be assigned to the OH end group stretching in PFA and silanol groups.12,22,23 The band at 2900 cm−1 is ascribed to C–H stretching of copolymer F127. After calcination at 350 °C under nitrogen, the FT-IR curve of FS-60 (Fig. 5b) shows that vibrations arising from PFA and silicates at 1635, 1461, 1730, 1100 and 948 cm−1 are retained, suggesting coexistence of polymer and silicate solids. The band at 1635 cm−1 is strengthened while the 1365 cm−1 band is weakened, implying that most furan rings are 2,3-bisubstituted. It indicates that the cross-linking degree of PFA is improved. The band at 2900 cm−1 almost disappears, giving evidence for template decomposition in FS-60.
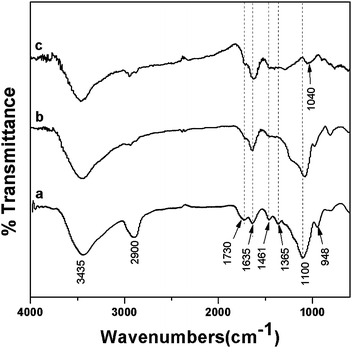 | ||
| Fig. 5 FT-IR spectra of (a) FS-60-as-made, (b) FS-60, and (c) FA-FS-60 derived from (b) by silica dissolution in HF solution. | ||
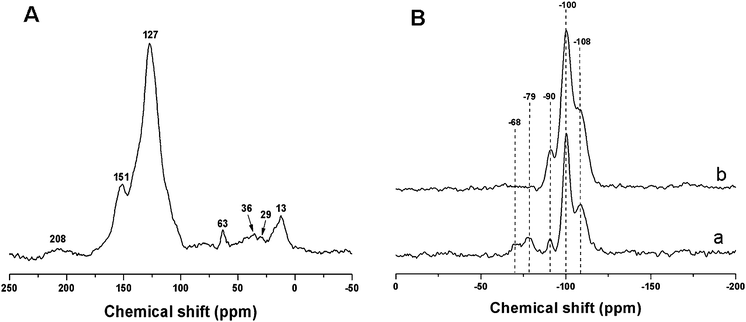
The solid-state 13C CP-MAS NMR spectrum shows that mesoporous nanocomposite FS-60 calcined at 350 °C in N2 is typical of cured PFA resins (Fig. 6A). The peak at 151 ppm corresponds to the 2- and 5-positions of internal furan rings –[–C4H2O–CH2–]– or the 2-position of terminal furan rings in PFA resins.8,22,27 The peak around 29 ppm can be assigned to CH2groups between the furan rings, and a broad peak at 36 ppm due to the cross-linking between oligo-FA and PFA sequences through these methylene groups.8,22,27,28 Occurrence of cross-linking branches through 3- and 4-positions of furan ring is detected by the presence of a peak around 127 ppm.22 A broad peak at 208 ppm can be assigned to the diketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species,22 due to some furan ring opening during polymerization. Furthermore, a 13C signal for methyl groups is observed at δ = 13 ppm.8,27 The signal at 63 ppm indicates Si–O–C bonds in the nanocomposites.8,27,29 These results clearly reveal a polymeric framework.
O species,22 due to some furan ring opening during polymerization. Furthermore, a 13C signal for methyl groups is observed at δ = 13 ppm.8,27 The signal at 63 ppm indicates Si–O–C bonds in the nanocomposites.8,27,29 These results clearly reveal a polymeric framework.
 | ||
| Fig. 6 Solid-state 13C CP/MAS NMR spectrum (A) of mesoporous nanocomposites FS-60 after calcination at 350 °C in N2. Solid-state 29Si MAS NMR spectra (B) of mesoporous nanocomposites FS-60: (a) as-made and (b) calcined at 350 °C in N2. | ||
The solid-state 29Si MAS NMR spectrum of as-made mesoporous nanocomposite, FS-60-as-made (Fig. 6B(a)) shows five resolved signals. The signals at −108, −100 and −90 ppm can be assigned to chemical shifts of Q4 [Si(OSi)4], Q3 [Si(OSi)3(OH)] and Q2 [Si(OSi)2(OH)2] species for amorphous silica, respectively.30,31 The peaks at −79 and −68 ppm are characteristic Tm signals ascribed to Si species covalently bonded with carbon atoms T3 ([C–Si(OSi)3]: δ = −79 ppm), and T2 ([C–Si(OSi)2(OH)]: δ = −68 ppm). After calcination at 350 °C, the peaks at −79 and −68 ppm disappear as shown in Fig. 6B(b), indicating the cleavage of Si–C bonds.
In order to obtain the composition of PFA polymers pyrolyzed at 350 °C, elemental analysis was carried out for the residual materials after removing silica species in the nanocomposites by HF acid. The result reveals that it is composed of C, 78.8 wt%; H, 4.4 wt%; and O, 16.8 wt%, the molar ratio of C:H:O (6.25:4.16:1) close to that in ideal cross-linked PFA resins (C:H:O = 6:4:1).21 It indicates that the polymer networks of PFA polymers are preserved well.
3.3 Burning off PFA or etching silica
Mesoporous silicas are derived from PFA/silica nanocomposites after combustion at 600 °C in air. SAXS patterns of Si-FS-33 and Si-FS-41 show five or six resolved scattering peaks analogous to mother polymer/silica nanocomposites, suggesting that the 2-D centered rectangular (c2mm) mesostructure is retained (Fig. 7). However, for Si-FS-60 with high PFA content after the removal of the PFA polymers, the mesostructure is degraded severely judged by the broadened and weakened scattering peaks. The unit cell parameters of silica mesostructures gradually reduce as polymer content increases (Fig. 7 and Table 2), which is caused by the increased voids of PFA content.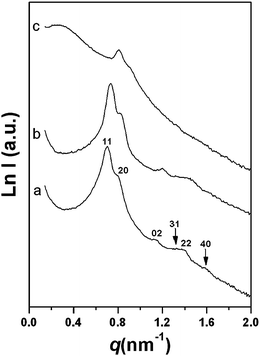 | ||
| Fig. 7 SAXS patterns of the mesoporous silica derived from the PFA-silica composites (a) FS-33, (b) FS-41, and (c) FS-60 by combustion at 600 °C in air. | ||
Type-IV N2 sorption isotherms with capillary condensation steps at a relative pressure of 0.50–0.70 are observed for mesoporous silica Si-FS-33 and Si-FS-41 after burning out the PFA in air (Fig. 8A), suggesting a uniform pore size distribution. On comparison with their mother PFA/silica nanocomposites, the pore sizes calculated from adsorption branches for the mesoporous silica are a little larger (Fig. 8B and Table 2). It may be caused by the combustion of inner-wall polymer PFA. In the case of Si-FS-60, the nitrogen sorption isotherm shows an ill-expressed curve with very broad pore size distribution (Fig. 8), which indicate a disordered mesostructure, in accordance with the SAXS results. These mesoporous silica materials have pore volumes ranging from 0.37 to 0.66 cm3/g. (Table 2), a little larger than their mother PFA/silica composites. It can be explained by the enlargement of their pore sizes after burning off the PFA components.
 | ||
| Fig. 8 Nitrogen sorption isotherms (A) and pore size distribution curves (B) of mesoporous silica Si-FS-33, Si-FS-41 and Si-FS-60 derived from PFA/silica composites FS-33, FS-41, and FS-60 after combustion at 600 °C in air, respectively. | ||
Pure organic materials were obtained by etching silica species in the nanocomposite with HF acid. The disappearance of 1100 and 948 cm−1 bands in the corresponding IR spectrum (Fig. 5c) indicates complete silica removal. The residual bands of 1730, 1635, 1461 and 1040 cm−1 stem from PFA, wherein the weak band at 1040 cm−1 is attributed to C–O–C stretching in furan rings.12,21 It further confirms that the organic framework of PFA is retained after calcination at 350 °C in N2 flow. SAXS patterns of pure organic PFA polymers (not shown) display only one broad and weak scattering peak, and their BET surface areas and pore volumes reduce almost to zero calculated from nitrogen sorption isotherms (not shown), which imply collapse of the mesoscopic regularity.
TEM images show that mesoporous silica Si-FS-33 after the combustion of PFA contents exhibits large domains of typically ordered stripe-like arranged images (Fig. 9A), suggesting that the ordered mesostructure similar to its mother composite FS-33 is well retained. TEM image of Si-FS-41 displays some stripe intermittence in pore walls (Fig. 9B), suggesting mesostructural disfigurement. The evident deformation of strip arrays in some domains for Si-FS-60 implies the severe decrease of mesostructure ordering (Fig. 9C). Almost no ordering can be observed in the TEM image of PFA polymer FA-Si-60 (Fig. 9D), demonstrating the mesostructure collapse, in accordance with the SAXS and N2 sorption results.
 | ||
| Fig. 9 TEM images of silica (A, B, C) and PFA polymer (D) derived from PFA/silica composites after combustion at 600 °C in air and dissolution in HF solution, respectively: (A) Si-FS-33, (B) Si-FS-41, (C) Si-FS-60 and (D) FA-FS-60. | ||
3.4 Adsorption properties
Toluene adsorption isotherms of the nanocomposites FS-33, FS-41 and FS-60 show typical type-IV curves (Fig. 10(A)). Besides increased adsorption at low P/P0 value (< 0.2) due to micropores, well-defined steep steps occur approximately in the P/P0 range of 0.6–0.8, which are associated with toluene filling in the mesopores. For comparison, ordered mesoporous silica (MSiO2) prepared by the EISA method with the pore size, surface area and pore volume of 6.2 nm, 520 m2/g and 0.51 cm3/g (Table 2), respectively, displays a type-I adsorption curve, similar to the previously reported toluene isotherm of MCM-41.32 Steep toluene adsorption in micropores occurs at a low relative pressure, analogical to PFA/silica hybrids; however, capillary condensation in mesopores can not be observed. It has been shown that the capillary condensation step is related to the surface properties of mesoporous materials.32,33 The distinct capillary condensation behavior between mesoporous silica and PFA/silica composites may be owing to their different hydrophilic/hydrophobic properties. As illustrated in Table 2, the amount of adsorbed toluene over nanocomposite FS-33 (4.6 mmol/g) is twice as large as that of mesoporous silica MSiO2 (2.3 mmol/g), despite the fact that they have similar surface areas and pore volumes. With increasing PFA content, the toluene adsorption amount decreases, mainly due to the severe reduction in their pore volumes and surface areas. However, even for nanocomposite FS-60, whose surface area and pore volume are much less than those of mesoporous silica MSiO2, it shows comparable toluene adsorption amount (2.1 mmol/g) with the value of MSiO2. The distinct adsorption performances are possibly associated with their different surface properties. The PFA polymers, which are hydrophobic, exhibit improved affinity to toluene molecules, resulting in the PFA-silica nanocomposites showing enhanced toluene adsorption capacities.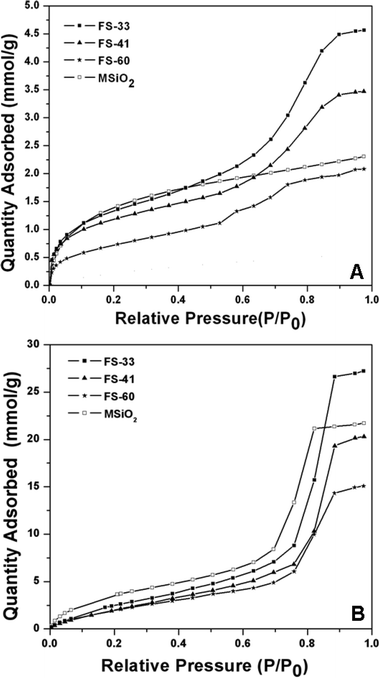 | ||
| Fig. 10 Toluene (A) and water (B) adsorption isotherms of ordered mesoporous silica MSiO2 and the hybrid nanocomposites FS-33, FS-41 and FS-60 with different organic contents at 25 °C. | ||
The hydrophilic/hydrophobic nature of mesoporous PFA/silica composites and silica can further be estimated by water adsorption (Fig. 10(B)). The isotherms are of type-V, indicative of weak adsorbent–adsorbate interaction. At low relative pressures, the adsorption amount is low. The Henry constants calculated from the adsorption data at low pressures (P/P0 < 0.1) are 18.3, 16.8, 15.9 and 34.2 for FS-33, FS-41, FS-60 and MSiO2, respectively, which reflect the adsorption affinities. The Henry constants decrease in the order MSiO2 > FS-33 > FS-41 > FS-60, indicating that pure silica MSiO2 has a higher water affinity than PFA/silica composites. The hydrophobic property is enhanced by increasing PFA loading. A steep increase in adsorption occurs at medium relative pressures of 0.70–0.85, suggesting capillary condensation. However, mesoporous PFA/silica hybrids have capillary condensation at higher P/P0 values than MSiO2. This value is extremely large for FS-60, which has an indistinct capillary condensation step at a P/P0 value of 0.76. The shift could be attributed to the increased hydrophobicity.34
4. Discussion
On the basis of the above observations, we propose that the formation of mesoporous nanocomposites undergoes an organosilane-assisted co-assembly process of organic polymer FA precursors, silicate oligomers hydrolyzed from TEOS and amphiphilic triblock copolymer F127. The final nanocomposites have an interpenetrating framework with “reinforced-concrete” structure, in which both “reinforcing-steel-bar” silicate and “concrete” PFA polymers synergistically support the ordered mesostructure (Scheme 1).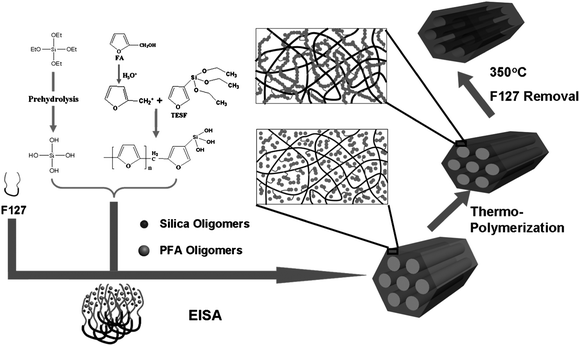 | ||
| Scheme 1 The process for silica-assisted organization of ordered mesoporous PFA polymers. | ||
It is well known that FA can polymerize in the presence of HCl as a cationic active initiator through the interaction of terminal CH2OH groups.8,23 It is different from the system of phenolic resin and TEOS,25 wherein both the resol and silicate precursors have abundant hydroxyl groups to interact with PEO groups of triblock copolymer template viahydrogen bonds and form the framework individually. The polymer/SiO2 mass ratios of the constituents in the phenolic resin/silicate nanocomposites can thus be ranged from zero to infinity, even though only weak Si–O–C bonding occurs between the silicate surface and phenolic resin species. In contrast, the consumption of terminal hydroxyl groups during the FA polymerization makes PFA polymers become hydrophobic and therefore lack the interaction with both PEO segments of triblock copolymer F127 and the silicate species. It is difficult to form an ordered mesostructure without the assistance of the silicates. So, the introduction of organosilane TESF, which can link the PFA and silicates with covalent C–C and Si–O–Si bonds, becomes essential to acquire the ordered mesostructure by the surfactant-assembly approach. In the case of low acidity and medium temperature, the polymerization of FA proceeds at a moderate rate, and it can also react with furan groups of TESF to form stable C–C bonds. With the continuation of stirring at 40 °C for 5 h, the solution gradually turned dark green from primrose, suggesting the cross-linking of FA and TESF. Meanwhile, the ethoxy groups of TESF and TEOS hydrolyze into hydroxyl groups, they can cross-link together to form silicate oligomers. The silicate oligomers together with the connection of the organic PFA precursors can co-assemble with amphiphilic triblock copolymer F127 by hydrogen bonds to form ordered mesostructures of the nanocomposites.
29Si MAS NMR measurement reveals the cleavage of Si–C bonds during the process of triblock copolymer F127 removal, which is inconsistent with the previous results that Si–C bonds are stable at 350 °C.35 It gives a hint that during the thermal treatment, PFA polymers and silicate species undergo further cross-linking and segmental motions of chains, resulting in microphase separation, because organic polymer–inorganic component systems, as a rule, are thermodynamically incompatible.36,37 The Si–C bonds, as the linkage of organic and inorganic components, therefore could suffer intensive strain force, and become easier to break down. Furthermore, the PFA, different from the phenolic resin, cross-link into linear macromolecules during the initial phase. The bond angle needed for the linear cross-linkage may result in an insufficient polymerization of PFA in the nanocomposites. After HF etching the silicate, the separate and soft PFA polymers could not sustain the framework, leading to a collapse of the mesostructure. For the mesoporous silicates, obtained by burning out the organic constituent in the composites with low organic contents (< 41 wt%), the mesostructure can be retained well due to the rigid framework. However, with increasing organic content, such as FA-Si-60, many voids from organic PFA cause collapse of the mesostructure. It implies a homogeneous interpenetrating framework, in which both silicate and furan resins synergistically support the ordered mesostructure.
4. Conclusions
Ordered mesoporous PFA-silica nanocomposites have been successfully prepared with an organosilane-assisted synthesis approach by employing TEOS, 3-(triethoxysilyl)furan and FA as precursors and F127 as a structure-directing agent via an EISA process. The resultant organic–inorganic nanocomposites show ordered 2-D centered-rectangular c2mm mesostructures with uniform pore size (4.8–5.7 nm). The mesoporous PFA-silica nanocomposites have suitable surface areas (200–510 m2/g) and high pore volumes (0.15–0. 54 cm3/g). The organosilane TESF, which can link the PFA polymers and silicates to make a 3-D network, can assist the PFA precursor to involve the self-assembly of silicate species and amphiphilic triblock copolymer template, yielding an ordered PFA mesostructure. The organosilane-assisted approach enables the mesoporous nanocomposites to be produced with high organic content of ∼60 wt%. However, with increasing PFA content, their BET surface area, pore volume and size reduce. After burning out the PFA in the nanocomposites, resultant mesoporous silicas have similar ordered mesostructures with a slightly larger pore size. The mesoporous PFA-silica nanocomposites have homogeneous interpenetrating frameworks, in which both silicate and PFA resins synergistically support the ordered mesostructure. The water adsorption isotherms show the hydrophobic property of PFA-silica nanocomposites, which have an affinity with toluene molecules. The amount of adsorbed toluene over the nanocomposite (4.6 mmol/g) is twice as large as that of mesoporous silica (2.3 mmol/g), despite the fact that they have similar surface areas and pore volumes.Acknowledgements
This work was supported by NSF of China (20721063, 20521140450), State Key Basic Research Program of PRC (2006CB202502) and Shanghai Leading Academic Discipline Project (B108).References
- S. A. Jenekhe and X. L. Chen, Science, 1999, 283, 372 CrossRef CAS.
- S. A. Johnson, P. J. Ollivier and T. E. Mallouk, Science, 1999, 283, 963 CrossRef CAS.
- C. D. Liang, K. L. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem. Int. Ed., 2004, 43, 5785 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem. Int. Ed., 2005, 44, 7053 CrossRef CAS.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Comm., 2005, 2125 RSC.
- F. Q. Zhang, Y. Meng, D. Gu, Y. Yan, C. Z. Yu, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2005, 127, 13508 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, L. Cheng, D. Feng, Z. X. Wu, Z. X. Chen, Y. Wan, A. Stein and D. Y. Zhao, Chem. Mater., 2006, 18, 4447 CrossRef CAS.
- H. Muller, P. Rehak, C. Jager, J. Hartmann, N. Meyer and S. Spange, Adv. Mater., 2000, 12, 1671 CrossRef CAS.
- H. T. Wang and J. F. Yao, Ind. Eng. Chem. Res., 2006, 45, 6393 CrossRef CAS.
- A. V. Voronkov, V. M. Kopylov and I. W. Parsons, Euro. Poly. Jour., 1997, 33, 979 Search PubMed.
- J. Liu, H. T. Wang and L. X. Zhang, Chem. Mater., 2004, 16, 4205 CrossRef CAS.
- Y. Yan, H. F. Yang, F. Q. Zhang, B. Tu and D. Y. Zhao, Small, 2006, 2, 517 CrossRef CAS.
- J. F. Yao, H. T. Wang, K. Y. Chan, L. X. Zhang and N. P. Xu, Micro. Meso. Mater., 2005, 82, 183 CrossRef CAS.
- C. J. Brinker, Y. F. Lu, A. Sellinger and H. Y. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS.
- D. Zhao, P. Yang, N. Melosh, J. Feng, B. F. Chmelka and G. D. Stucky, Adv. Mater., 1998, 10, 1380 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, J. M. Kim, Y. J. Han and G. D. Stucky, Chem. Mater., 1999, 11, 2668 CrossRef CAS.
- U. Breiner, U. Krappe, E. L. Thomas and R. Stadler, Macromolecules, 1998, 31, 135 CrossRef CAS.
- M. Klotz, P. A. Albouy, A. Ayral, C. Menager, D. Grosso, A. Lee, V. Cabuil, F. Babonneau and C. Guizard, Chem. Mater., 2000, 12, 1721 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem. Euro. J., 2005, 11, 2610 CrossRef CAS.
- S. M. Manocha, D. Y. Vashistha and L. M. Manocha, J. Mater. Sci. Lett., 1997, 16, 705 CrossRef CAS.
- Z. Wang, Z. Lu, X. Huang, R. Xue and L. Chen, Carbon, 1998, 36, 51 CrossRef CAS.
- A. J. G. Zarbin, R. Bertholdo and M. Oliveira, Carbon, 2002, 40, 2413 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839 CrossRef CAS.
- C. M. Yang, B. Zibrowius, W. Schmidt and F. Schuth, Chem. Mater., 2003, 15, 3739 CrossRef CAS.
- R. L. Liu, Y. F. Shi, Y. Wan, Y. Meng, F. Q. Zhang, D. Gu, Z. X. Chen, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2006, 128, 11652 CrossRef CAS.
- D. H. Sun, R. Zhang, Z. M. Liu, Y. Huang, Y. Wang, J. He, B. X. Han and G. Y. Yang, Macromolecules, 2005, 38, 5617 CrossRef CAS.
- S. Spange, H. Muller, C. Jager and C. Bellmann, Macromolecular Symposia, 2002, 177, 111 CrossRef CAS.
- S. Spange and H. S. R. Martinez, Makromol. Chem., 1993, 194, 1537 CrossRef CAS.
- D. M. Hoebbel and D. M. N. H. Schmidt, J. Sol-Gel Sci. Techn., 1998, 12, 169 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- Y. Wan, D. Q. Zhang, Y. P. Zhai, C. M. Feng, J. Chen and H. X. Li, Chem. Asian J., 2007, 2, 875 CrossRef CAS.
- M. P. Kapoor, N. Setoyama, Q. H. Yang, M. Ohashi and S. Inagaki., Langmuir, 2005, 21, 443 CrossRef CAS.
- S. Inagaki and Y. Fukushima, Micropor. Mesopor. Mater., 1998, 21, 667 CrossRef.
- J. I. Jung, J. Y. Bae and B. S. Bae, J. Mater. Chem., 2004, 14, 1988 RSC.
- A. D. Pomogailo, Colloid J., 2005, 67, 658 CrossRef CAS.
- G. Kickelbick, Prog. Poly. Sci., 2003, 28, 83 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SAXS pattern , nitrogen adsorption–desorption isotherms and pore size distribution of mesoporous siliceous MSiO2. See DOI: 10.1039/b813688b |
| This journal is © The Royal Society of Chemistry 2009 |