Organically modified mixed-oxide sol–gel films with complex compositions and pore structures
Marina
Lomoschitz
a,
Herwig
Peterlik
b,
Gernot
Friedbacher
c and
Ulrich
Schubert
*a
aInstitute of Materials Chemistry, Vienna University of Technology, Getreidemarkt 9, A-1060, Wien, Austria. E-mail: Ulrich.Schubert@tuwien.ac.at
bFaculty of Physics, University of Vienna, Boltzmanngasse 5, A-1090, Wien, Austria
cInstitute of Chemical Technologies and Analytics, Vienna University of Technology, Getreidemarkt 9, A-1060, Wien, Austria
First published on 6th November 2008
Abstract
Films with polymodal porosity (macro-, meso- and micropores) as well as complex materials compositions (different metals, different organic groups) were prepared. First, self-assembled polystyrene spheres on glass slides were coated with a sol prepared from tetraethoxysilane, phenyl- or methyltriethoxysilane and ethyl acetoacetate-stabilized zirconium propoxide or titanium iso-propoxide in the presence of a non-ionic surfactant. Macropores were created by dissolving the polystyrene porogen. Subsequent calcination at different temperatures resulted in the sequential formation of meso- and micropores. Post-calcination treatment with phosphonic acids allowed the introduction of a second type of organic group.
Introduction
Surfactant templated mesoporous sol–gel materials have potential applications in catalysis, separation and sensing because of their high surface area, controllable pore size and narrow pore size distribution. Adsorption applications may require extraordinary materials properties such as combination of different adsorption sites. Different metal sites and different organic functionalities may increase the interactions of adsorbate mixtures in one material. The generation of thin films with such properties is a special challenge, because several synthesis and/or post-synthesis treatment methods are not compatible with film formation and film properties (e.g. adhesion). Angelome and Soler-Illia prepared hybrid thin films with ordered mesoporosity, organic bifunctionality and mixed oxide framework from organically modified alkoxysilanes (with aminopropyl, mercaptopropyl and phenyl groups) and zirconium or titanium chloride precursors, followed by post-synthesis grafting of a second organic functionality.1Another very important criterion for adsorption is high mass transport. This can be achieved by macropores. Macroporous metal oxides, including binary mixed oxides,2 can be prepared by colloidal crystal templating (CCT), i.e. infiltration of a pre-formed colloidal crystal with precursor material and subsequent removal of the template. This method also allows the creation of macroporous thin films.3,4 Materials with interconnected macropores and mesopores were obtained by combining self-assembly of surfactants or ionic liquids with CCT by polystyrene (PS) spheres.5,6 Such materials have enhanced properties compared to conventional MCM-type mesoporous materials because of increased mass transport in combination with high surface areas. Even materials with micro-, meso- and macroporosity are known; silicafilms with trimodal porosity were prepared by combining zeolite nanoparticles and templating techniques.7
Functional or non-functional organic groups can be attached to the pore walls of porous materials by several methods: (i) post-synthesis modification of the surface of inorganic materials by grafting of organic groups, (ii) co-condensation of organically substituted and unsubstituted precursors or (iii) incorporation of organic groups as bridging components directly and specifically into the pore walls.
The goal of the current work was to combine approaches for creating films with polymodal (macro-, meso- and micropores) porosity as well as complex materials compositions (different metals, different organic groups) in one material. Thus, a combination of (i) several porogens, (ii) co-condensation of different metal alkoxide precursors and (iii) post-synthesis modification was used to obtain highly porous mixed oxide films with multiple organic functionality and different pore-size regimes. The general approach is shown in Fig. 1. Highly porous mixed oxide films with two different organic groups were prepared by coating a film of self-assembled PS spheres with a sol from a mixture of silicon and metal (Ti, Zr) alkoxides, including an organotrialkoxysilane, and a surfactant. After thermal treatment, micro-, meso- and macroporous organically substituted mixed oxide films were obtained that also allowed the introduction of another organic group by post-synthesis functionalization of the metal sites.
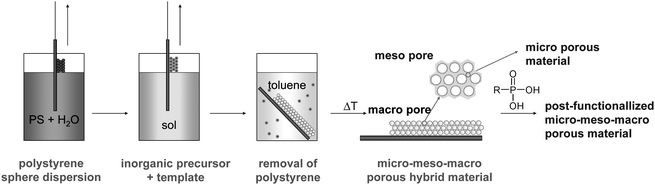 | ||
| Fig. 1 Schematic film preparation procedure. | ||
The reported work has proof-of-principle character using simple organic groups and glass as the substrate. For the subsequent development of coatings as molecular contaminant traps, specific functional organic groups and metal alkoxide mixtures will be used.
Results and discussion
In the first step, commercially available PS spheres of 100 nm diameter were deposited onto cleaned glass slides from a dispersion in water by dip coating. This is a simple and convenient method to obtain densely packed layers of polymer spheres by self-assembly.5 After drying the film of packed PS spheres for 24 h at ambient temperature, the slides, without further treatment, were immersed in a sol of hydrolyzed alkoxide precursors. During this step the PS spheres are coated with an organically modified mixed oxide gel network.The employed sols were prepared from mixtures of three different precursors: (i) 2 molar equivalents of Si(OEt)4 (TEOS), (ii) 2 molar equivalents of either phenyltriethoxysilane (PTES) or methyltriethoxysilane (MTES) as organically modified alkoxysilanes, and (iii) 1 molar equivalent of either Zr(OPr)4 or Ti(OiPr)4. Because the hydrolysis and condensation rates of silicon alkoxides are different to those of zirconium or titanium alkoxides, the mixture of silicon precursors, TEOS and PTES or MTES was pre-hydrolyzed in the presence of the non-ionic surfactant Brij 56 in an acidic water–ethanol mixture, whereas the metal alkoxide was reacted with ethyl acetoacetate to decelerate its reactivity. Three combinations were investigated: TEOS/MTES/Zr(OPr)4 (MeSi-Zr), TEOS/MTES/Ti(OiPr)4 (MeSi-Ti), and TEOS/PTES/Ti(OiPr)4 (PhSi-Ti).
The deposited sol–gel films were thermally treated at 100 °C for further network condensation. To dissolve the PS porogen, the aged films were then placed in toluene for a few hours and then thoroughly washed with fresh toluene. Removal of surfactant was carried out by calcination at 400 °C. This temperature treatment allowed removal of residual PS, chelating agent and surfactant without removing the organic groups covalently bonded to the silicon atoms of the gel network. All steps of film manufacturing are schematically shown in Fig. 1.
The temperature stability of the methyl and phenyl groups was monitored by IR spectroscopy of sol–gel films deposited on a KBr disk but otherwise treated the same as the films of coated PS spheres (Fig. 2). Retention of the bands at 1270 cm−1 and 772 cm−1, corresponding to CH deformation and rocking vibrations, proved the presence of Si–CH3group of MeSi-Zr after calcination at 400 °C. On the other hand, decrease of the bands at 2920 and 2850 cm−1, corresponding to C–H stretching vibrations of the surfactant, was observed. This clearly shows that Si–CH3groups were still present after calcination at 400 °C, whereas the surfactant was removed at this temperature. The band at 1020 cm−1 corresponds to overlapping stretching vibrations of Zr–O–Si and Si–O–Si, thus indicating the formation of a mixed-oxide network during sol–gel processing and after thermal treatment.
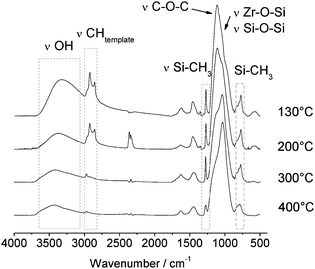 | ||
| Fig. 2 IR spectra of a MeSi-Zr film, deposited onto a KBr disk, during temperature treatment. | ||
Transmission electron microscopy (TEM, Fig. 3) showed that the films had uniform ellipsoidal macropores after calcination at 400 °C, with long axes of about 70 nm. The anisotropic shrinkage during calcination, from originally 100 nm to 70 nm in the present case, is a common effect during calcination of porous films, where a higher tendency of film pore shrinking perpendicular to surface was noticed.7,8
 | ||
| Fig. 3 TEM image of PhSi-Ti film: macroporosity. | ||
Fig. 4 shows a TEM image of a calcined MeSi-Zr film, scratched off with a razor blade. The pore walls of the macropores, consisting of an organically modified mixed oxide material, clearly exhibited (non-ordered) mesopores of 14–16 nm diameter.
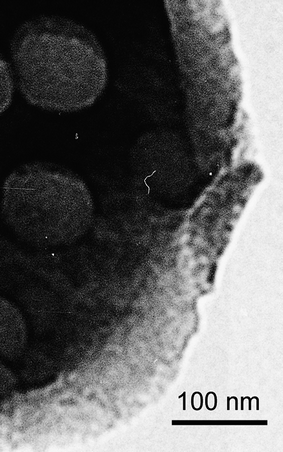 | ||
| Fig. 4 TEM image of MeSi-Zr: macro- and mesopores. | ||
For further investigation of the mesoporosity and the film surface properties, the MeSi-Zr sol was also deposited onto a Si wafer, without prior deposition of a PS film, calcined at 400 °C and investigated by atomic force microscopy (AFM). The topological image of the AFM measurement (Fig. 5a) exhibited a smooth surface with height differences of ∼2 nm. This very small height difference can be explained by the size of AFM tip, which limits the penetration depth of the tip into the pores (the radius of curvature of the tip is typically less than 10 nm, the apex angle is 70 deg). Analyzing the phase image (Fig. 5b), mesopores with size of 15 nm diameter, opened towards the surface, were detected. The phase image also showed that nanoparticles of 20 to 25 nm size are formed. The nanoparticles are condensed with each other, and the voids between these particles apparently correspond to the observed mesoporosity.9 The role of the surfactant thus is to stabilize uniform nanoparticles during sol–gel processing rather than acting as a supramolecular template as in the case of MCM-type materials. This notion was also confirmed by SAXS experiments (see below).
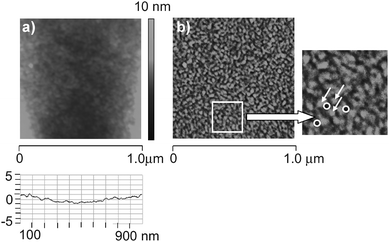 | ||
| Fig. 5 AFM images of a MeSi-Zr film after calcination: a) topological image; b) phase image (white arrow: particles, circles: pores). | ||
There was also strong evidence for particle formation by SAXS measurements (Fig. 6). The approach was used for the numerical description of the scattering curves that was proposed by Beaucage10 for materials containing two structural levels together with an interference function, which takes a short range order of these structures into account.11 A constant background at large q-values arising from residual scattering and a power-law dependent background at low q-values from large objects was also added. The obtained information is the typical size (diameter 2r) and distance (d) of the particles (the diameter is calculated from the respective radius of gyration for spherical particles), the fractal exponent p, which allows one to conclude whether the structure is an open network or consists of particles with a sharp interface, and the packing density k of the particles. The exponent p2 of the basic structural units was set to 4 (i.e. sharp smooth interface) to reduce the number of fit parameters.11,12 The obtained numerical values are summarized in Table 1.
| 2r1/nm | 2r2/nm | p 1 | k 1 | d 1 | p 2 | k 2 | d 2 | |
|---|---|---|---|---|---|---|---|---|
| MeSi-Zr | 1.9 | 0.8 | 2.0 | 1.4 | 2.6 | 4 | 4.9 | 0.9 |
| MeSi-Ti | 3.7 | 1.0 | 3.4 | 3.1 | 3.9 | 4 | 3.2 | 1.0 |
| PhSi-Ti | 7.6 | 1.1 | 4 | 4 | 2.3 | 1.3 |
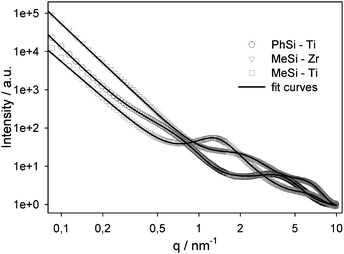 | ||
| Fig. 6 SAXS intensity dependence on the scattering vector q together with fit curves (samples prepared in the absence of PS spheres—see text). | ||
The calculations showed that the material is built of small basic structural units (indicated by the subscript 2) with a size of approximately 1 nm. It is very probable that the microporosity is due to the voids between these structural units, as the size of the micropores is 0.5 to 0.7 nm (see below). This is also consistent with the trend that the basic structural units as well as the micropores are slightly smaller in MeSi-Zr, together with a higher packing factor k2 (i.e., a denser structure at this level) and a lower total porosity compared to MeSi-Ti. These basic structural units agglomerate to larger particles (denoted by the subscript 1). In the case of MeSi-Zr, these particles have a typical diameter of 1.9 nm and a distance of 2.6 nm. They are arranged in a more disordered fractal open network (exponent p1 = 2.0), whereas for MeSi-Ti and PhSi-Ti the particles are larger and exhibit a sharp smooth surface (exponent p1 close to 4). There is some influence of the methyl and phenyl groups on the material formation, because the particles are considerably larger for the latter. Furthermore, they do not exhibit any short range order, but are randomly arranged in PhSi-Ti. A possible interpretation is that the influence of the phenyl group enables titanium-controlled hydrolysis–condensation processes to build larger particles with a smooth surface.
The scattering intensity towards low q values increases strongly, which arises from objects larger than the measuring range of the SAXS equipment used. A third structural level with a size of 20–40 nm cannot be unambiguously identified, because the strong background scattering masks this signal. However, it is probable that the particles agglomerate to larger particles with a size in this range, because these structures are clearly visible in the AFM and TEM images.
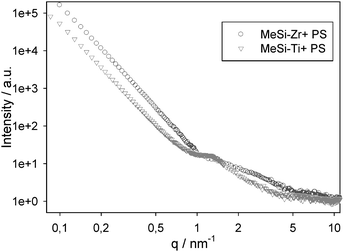
The SAXS measurements discussed above and shown in Fig. 6 were also done with materials prepared in the absence of PS spheres. To exclude the possibility that the structure/porosity of the solid network is influenced by the presence of the PS spheres, we also investigated two samples by SAXS prepared in the presence of PS spheres (Fig. 7). The scattering curves of MeSi-Zr and MeSi-Ti are quite similar independent of the presence of PS during synthesis. In particular, in MeSi-Ti the stronger short range order peak is clearly visible. The broad shoulder in MeSi-Zr is shifted to slightly higher q-values (shorter distances in real space) when the sample was prepared with PS spheres. The lower intensity of all peaks (at smaller as well as at larger q-values) of samples prepared with PS spheres is attributed to the much larger background, possibly arising from residual amorphous moieties after the calcination process.
To quantify the surface area of the porous films, meso- and microporosity was investigated by N2 sorption measurements at 77K. To this end, films were manufactured as mentioned before, but in the absence of PS spheres (i.e. same chemical composition of the coating sol, including Brij 56). The films were deposited on glass sheets, dried at ambient atmosphere and scratched off with a razor blade. The obtained powder was subsequently calcined at 400 °C. The isotherm of MeSi-Zr is shown in Fig. 8 and is representative for the other samples. The sorption isotherms are of type I according to IUPAC classification,13 characteristic for microporous materials. The BET method was applied to calculate the total surface area, while the t-plot was used to determine microporosity (Table 2). Both t-plot and BET methods showed a higher surface area of the titania-containing films. The t-plot resulted in roughly similar surface areas for MeSi-Ti and PhSi-Ti films. Comparing the BET results, MeSi-Ti exhibits the highest and MeSi-Zr the smallest surface area (SBET), which can be attributed to the different condensation properties between Ti and Zr precursors. The average mesopore sizes were calculated according to the BJH model.14 In all samples, most of the pores were <2 nm, but an additional maximum in the region 14–16 nm was noticed. This pore size is in very good agreement with the mesopore size obtained by AFM and TEM measurements discussed before. Mesopores in the range of 3 nm might be also present, but they are not clearly visible in the BJH and nonlocal density functional theory (NLDFT) evaluation, as the large number of micropores masks the signal in this range. NLDFT was applied on adsorption isotherms to evaluate the size of the micropores. The micropores have a wide size distribution and a maximum at very low diameters (∼0.5 nm).
| Sample | SBET (m2/g) | St-plot (m2/g) | Vt (m3/g) | Vmeso (m3/g) | Vmicro (m3/g) | DBJH (nm) |
|---|---|---|---|---|---|---|
| a Vt: single point adsorption total pore volume, Vmeso: BJH desorption cumulative volume, Vmicro: t-plot micropore volume, DBJH: BJH desorption (adsorption) pore diameter. | ||||||
| MeSi-Zr | 385 | 330 | 0.19 | 0.02 | 0.15 | 3.7 (3.2) |
| MeSi-Ti | 470 | 375 | 0.24 | 0.03 | 0.16 | 3.6 (3.0) |
| PhSi-Ti | 410 | 365 | 0.20 | 0.02 | 0.17 | 3.6 (3.2) |
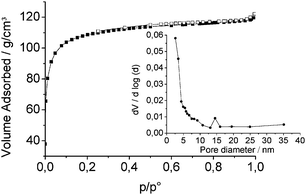 | ||
| Fig. 8 N2 adsorption–desorption isotherms and pore size distribution determined by the BJH method using the desorption branch for MeSi-Zr. | ||
The micropores could originate from two effects: (i) ethyl acetoacetate ligands, used to control the hydrolysis and condensation kinetics of the metal alkoxide precursor, are degraded during calcination, (ii) small nanoparticles as basic unit are formed, as demonstrated by SAXS experiments, and their condensation could also produces micropores.
The use of organo(trialkoxysilanes) as one of the precursors allowed the introduction of one kind of organic group into the material. In this work, we were just using methyl and phenyl groups for proof-of-principle. In a later stage of the project, these simple groups could be replaced by functional organic groups or by mixtures of different organo(trialkoxy)silanes. Introducing a second type of organic group by post-synthesis functionalization is also feasible. To this end, phosphonic acids were grafted to MeSi-Ti or MeSi-Zr after template removal by calcination. Phosphonic acids are known to coordinate tightly to titanium and zirconium; selective modification of the metal sites in MO2 (M = Ti, Zr)/SiO2 composites can be accomplished in aqueous solution.15 The post-functionalization was performed by impregnating the samples with phenyl or decyl phosphonic acid, followed by thorough washing and drying at 80 °C. The presence of organic groups was verified by IR spectroscopy and thermogravimetric analysis (TGA), comparing samples before and after the grafting step. Fig. 9 shows IR spectra of MeSi-Zr after calcination and after post-functionalization with decyl or phenylphosphonic acid. Due to the overlapping of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Si–O–Si bands, the characterization was accomplished by identification of organic groups. After post-functionalization, CH (2960 and 2856 cm−1) and P-phenyl (1143 cm−1) bands were clearly detected, corresponding to decyl- or phenylphosphonate units.
O and Si–O–Si bands, the characterization was accomplished by identification of organic groups. After post-functionalization, CH (2960 and 2856 cm−1) and P-phenyl (1143 cm−1) bands were clearly detected, corresponding to decyl- or phenylphosphonate units.
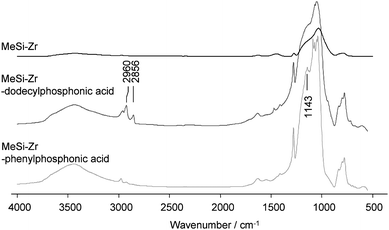 | ||
| Fig. 9 IR measurements of MeSi-Zr: after calcination (top), after post-functionalization with decylphosphonic acid (center) and after post-functionalization with phenylphosphonic acid (bottom). | ||
The efficiency of the post-synthesis functionalization was also monitored by TGA measurements. We observed higher mass loss at corresponding temperatures after post-functionalization. To eliminate mass loss errors of volatile compounds, the data for T = 200–700 °C were analyzed. The achieved mass loss percentages, corresponding to alkyl chains and phenyl groups respectively, were converted into molar percentages and so the amount of post-functionalized metal sites could be determined. The calculations showed that MeSi-Zr exhibited a lower tendency to post-functionalization with phenylphosphonic acid (11% of metal sites post-functionalized) than MeSi-Ti (33% of metal sites post-functionalized). These results can be explained by higher surface area of MeSi-Ti. Post-functionalization of MeSi-Zr with decylphosphonic acid was more efficient (29% of metal sites post-functionalized) than post-functionalization with phenylphosphonic acid (11% of metal sites post-functionalized). Free decylphosphonic acid was also detected by 31P MAS NMR in addition to bound decylphosphonic acid (data not shown). This can be explained by strong intermolecular interaction between the alkyl chains of decylphosphonic acid, which hinders complete removal of unreacted acid by the washing procedure.
The post-synthesis modified MeSi-Zr samples were also investigated by N2 sorption at 77 K. The measurement of the sample modified by phenylphosphonic acid exhibited no significant change in specific surface area, whereas the surface area of the sample modified by decylphosphonic acid was drastically reduced. These results are in very good agreement with TGA data and can also be explained by strong hydrophobic interaction of the alkyl chains of decylphosphonic acid.16
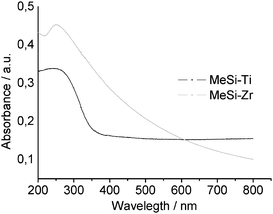
For UV-vis measurements, the sol was deposited onto quartz glass and calcined in the same manner as the previous samples. The UV-vis absorption spectra showed a broad maximum at 240 nm for MeSi-Ti and 250 nm for MeSi-Zr (Fig. 10). The absence of the absorption band at 320 nm in MeSi-Ti implies tetrahedral coordination of Ti and incorporation into the silica network,17 and allows the conclusion that no phase separation during the synthesis and calcination processes took place in MeSi-Ti. The UV-vis spectrum of MeSi-Zr, which also exhibits only one absorption maximum, allows no conclusions to be drawn about the distribution of the silica and zirconia structures.
Conclusions
We have described a simple way to create hybrid mixed oxide films with three different pore size regimes and two different ways of introducing organic groups. Creation of macro-, meso- and microporous films was achieved by coating of a film of self-assembled PS spheres with a sol obtained by hydrolytic polycondensation of a surfactant-stabilized precursor mixture. Macroporosity was achieved by extraction of the PS spheres with toluene. In a second step the formation of mesoporosity was accomplished by calcination of the surfactant. The microporosity was a consequence of the removal of complexing agent used for decelerating the reaction rates of the metal alkoxide precursor.One of the advantages of using mixed oxide networks is the possibility of post-functionalization of metal sites by grafting with organic ligands, such as phosphonates18 or carboxylates.1 We therefore introduced organic groups by two methods, i.e. by using organically substituted precursors and by post-synthesis grafting of organic groups to the pore walls.
Due to the high porosity and the possibility of selective modification with a large variety of organic functionalities, the described protocol qualifies for a wide field of applications.
Experimental
Synthesis
Si(OEt)4 (TEOS, Fluka ≥98%), phenyltriethoxysilane (PTES, ABCR 97%), methyltriethoxysilane (MTES, Fluka ≥98%), Zr(OPr)4 (70% in propanol, ABCR), Brij 56 (Fluka), Ti(OiPr) (Aldrich 97%), phenylphosphonic acid (Aldrich 98%), decylphosphonic acid (synthesized according to ref. 19) and polystyrene particles, average diameter 100 nm, 10 wt% dispersion in water (Aldrich), were used as received.Before film deposition, glass slides were cleaned by storage in concentrated sulfuric acid for 24 h and thorough washing with water. Silicon wafers were cleaned by ultrasonic treatment in acetone and drying in nitrogen flow.
The aqueous PS particle dispersion was diluted to 2 wt% and deposited onto cleaned glass slides by dip coating (withdrawal speed 0.4 cm/s). The PS film was dried at ambient temperature for 24 h.
A sol, prepared by adding diluted HCl to an ethanolic solution of Brij 56, TEOS and MTES or PTES, was stirred for 60 min at ambient temperature. The metal alkoxide was added to ethyl acetoacetate in a 1:2 molar ratio and stirred for 30 min at ambient temperature in a separate vessel. This solution was then added to the alkoxysilane sol. The final sol composition was TEOS : MTES (PTES) : M(OR)4 = 2 : 2 : 1, and sum of inorganic precursors : ethanol : H2O : HCl : Brij 56 = 1 : 40 : 16 : 2.2 : 0.05 (molar ratios). After further 60 min of stirring the sol was deposited onto the PS particle-coated glass substrate by dip coating (withdrawal speed 0.4 cm/s). The films were aged at ambient temperature for 24 h followed by calcination at 400 °C (10 °C/min, 2 h) in air.
Post-functionalization was performed using a 0.01 molar aqueous solution of phenyl or decyl phosphonic acid. After 3 h stirring at ambient temperature, the samples were thoroughly washed three times with water and dried for at least 24 h at 80 °C in vacuo.
Materials characterization
For N2 sorption measurements, TEM, SAXS and post-functionalization experiments the sol was deposited onto large glass sheets and scratched off with a razor blade. The obtained powder was treated in the same way as the thin films. AFM, BET measurements and part of the SAXS measurements (see text) were performed on materials prepared in the absence of PS spheres but otherwise with the same synthesis protocol.Nitrogen sorption measurements were performed on an ASAP 2020 (Micromeritics) instrument. The samples were degassed under vacuum at 60 °C for at least 5 h prior to measurement. The surface area was calculated according to Brunauer, Emmett and Teller (BET) and t-plot methods, and the pore size distribution according to Barrett, Joyner and Halenda (BJH) and nonlocal density functional theory (NLDFT) (using DFT plus (R) V3.00 program).
Transmission electron microscopy (TEM) images were recorded on a JEOL JEM-100CX analytical transmission electron microscope. The samples were attached to Formvar copper grids by coating the grid with the scratched powder.
Atomic force microscopy (AFM) was performed on a NanoScope III multimode SPM from Digital Instruments (Veeco Metrology Inc.). Measurements were conducted in tapping mode under ambient air using single-crystal silicon cantilevers (Arrow NC cantilevers, NanoWorld, spring constant 42 N/m, resonance frequency ∼285 kHz), and an E-scanner (maximum scan range 10 µm × 10 µm), operating at a scanning rate of 4 Hz. The data were evaluated with the NanoScope 6.10b20 software (Digital Instruments, Veeco Metrology Inc.). The phase images (i.e., the phase lag between the piezo-oscillation driving the AFM cantilever and its response) were recorded simultaneously with the topographic images using the second data channel of the instrument.
Infrared spectroscopy (IR) measurements were performed on Bruker Tensor 27 Spectrometer under ambient air (32 scans at resolution of 4 cm−1) using KBr disks.
Thermogravimetric analysis (TGA) was performed on a Netzsch Iris TG 209 C in a platinum crucible with a heating rate of 10 °C/min under synthetic air.
Structural characterization by SAXS was performed with Cu Kα radiation from a rotating anode generator (Nanostar from BRUKER AXS) equipped with a pinhole camera and an area detector (VANTEC 2000). The SAXS intensity patterns were corrected for background scattering and then radially averaged to obtain the function I(q), where q = (4π/λ)sinθ is the scattering vector, 2θ the angle between the incident and the diffracted beam, and λ = 0.1542 nm the X-ray wavelength.
Acknowledgements
This work was supported by the European Space Agency (ESA) and the Austrian Forschungsförderungsgesellschaft (FFG) in the framework of the Austrian Space Application Programme (ASAP).References
- P. C. Angelome and G. J. d. A. A. Soler-Illia, J. Mater. Chem., 2005, 15, 3903–3912 RSC.
- S. M. Hant, G. S. Attard, R. Riddle and K. M. Ryan, Chem. Mater., 2005, 17, 1434–1440 CrossRef CAS.
- A. Stein, F. Li and N. R. Denny, Chem. Mater., 2008, 20, 649–666 CrossRef CAS.
- Y. Li, W. Cai and G. Duan, Chem. Mater., 2008, 20, 615–624 CrossRef CAS.
- M. Sakurai, A. Shimojima, M. Heishi and K. Kuroda, Langmuir, 2007, 23, 10788–10792 CrossRef CAS.
- Y. Kim, C. Kim and J. Yi, Mater. Res. Bull., 2004, 39, 2103–2112 CrossRef CAS.
- R. A. Farrell, N. Petkov, H. Amenitsch, J. D. Holmes and M. A. Morris, J. Mater. Chem., 2008, 18, 2213–2220 RSC.
- G. J. A. A. Soler-Illia, E. L. Crepaldi, D. Grosso and C. Sanchez, J. Mater. Chem., 2004, 14, 1879–1886 RSC.
- J. Prochazka, L. Kavan, V. Shklover, M. Zukalova, O. Frank, M. Kalbac, A. Zukal, H. Pelouchova, P. Janda, K. Mocek, M. Klementova and D. Carbone, Chem. Mater., 2008, 20, 2985–2993 CrossRef CAS.
- G. Beaucage, Journal of Applied Crystallography, 1996, 29, 134–146 Search PubMed.
- H. Peterlik and P. Fratzl, Monatshefte fuer Chemie, 2006, 137, 529–543 CrossRef CAS.
- S. Trabelsi, A. Janke, R. Haessler, N. E. Zafeiropoulos, G. Fornasieri, S. Bocchini, L. Rozes, M. Stamm, J.-F. Gerard and C. Sanchez, Macromolecules, 2005, 38, 6068–6078 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. Perez-Ramirez, Microporous and Mesoporous Materials, 2003, 60, 1–17 CrossRef CAS.
- P. H. Mutin, V. Lafond, A. F. Popa, M. Granier, L. Markey and A. Dereux, Chem. Mater., 2004, 16, 5670–5675 CrossRef CAS.
- J. T. Woodward, A. Ulman and D. K. Schwartz, Langmuir, 1996, 12, 3626–3629 CrossRef CAS.
- N. Huesing, B. Launay, G. Kickelbick and F. Hofer, J. Sol-Gel Sci. Technol., 2003, 26, 615–619 CrossRef CAS.
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC.
- T. Vallant, H. Brunner, U. Mayer and H. Hoffmann, Langmuir, 1998, 14, 5826–5833 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |