A comparative XPS surface study of Li2FeSiO4/C cycled with LiTFSI- and LiPF6-based electrolytes
David
Ensling
,
Mårten
Stjerndahl
,
Anton
Nytén
,
Torbjörn
Gustafsson
and
John O.
Thomas
*
Ångström Advanced Battery Centre, Department of Materials Chemistry, Uppsala University, Box 538, SE-751 21, Uppsala, Sweden. E-mail: josh.thomas@mkem.uu.se
First published on 28th October 2008
Abstract
X-Ray photoelectron spectroscopy (XPS) has been used to characterise the surfaces of carbon-coated Li2FeSiO4 cathodes extracted from Li-ion batteries in both a charged and discharged state. 1 M lithium bis(trifluoromethylsulfonyl)imide (LiTFSI) and lithium hexafluorophosphate (LiPF6) based electrolytes were used with ethylene carbonate (EC) and diethyl carbonate (DEC) as organic solvents. The LiTFSI-based electrolyte exhibited high salt stability and no significant formation of LiF. However, solvent reaction products from EC were found together with lithium carbonate. A LiPF6-based electrolyte, on the other hand, showed inferior salt stability with LixPFy, LixPOyFz and LiF species formed on the surface. Solvent reaction products together with lithium carbonate were also found. There are also indications that Li2FeSiO4 is degraded by the HF formed in the electrolyte by the hydrolysis of LiPF6. A better understanding of the surface chemistry of carbon-coated Li2FeSiO4 after the first cycles in a Li-ion battery has thus been achieved, thereby facilitating the optimisation of Li-ion batteries based on this potentially cheap and electrochemically most promising cathode material giving excellent capacity retention: <3% drop over 120 cycles.
Introduction
Li2FeSiO4 is a recent addition to the group of interesting and potentially cheap cathode materials for large-scale lithium-ion battery applications: it shows great promise, especially in terms of its electrochemical stability. Li2FeSiO4 has a theoretical capacity of 166 mAh g−1 based on the reaction:| Li2FeSiO4 → LiFeSiO4 + Li+ + e− | (1) |
In practice, a capacity of 120 mAh g−1 has been achieved with only a ∼3% loss in capacity over more than 120 cycles.1 Furthermore, the presence of strong Si–O bonds should promote the same lattice stabilization effect exploited in LiFePO4.2 The shift observed in the potential plateau from 3.10 to 2.85 V between the first and second cycles has been suggested to be related to structural rearrangement to a more stable structure.3 While details of this process are still not totally understood, it has been seen that occupation of the Li- and Fe-sites becomes more randomised between the first and subsequent cycles.
Surface-film formation at both the anode and cathode electrode–electrolyte interfaces is an undisputed source for irreversible capacity loss in the Li-ion battery. Extensive studies of graphite anodes in a Li-ion battery have shown that, during the first cycle, the so-called solid electrolyte interphase (SEI) is formed as a film completely covering the graphite particles. This film contains different quantities of polymeric degradation products, as well as inorganic species like LiF and Li2CO3. These inorganic species are normally incorporated into the polymeric matrix of the surface film. Ultimately, the composition of the SEI depends on the specific choice of lithium salt and organic solvent used in the electrolyte.4–8
Surface-film formation on cathodes and its influence on battery performance has also been studied systematically. In particular, the transition-metal oxides have been shown to participate actively in the film formation,9–13 resulting in films comprising a mixture of polycarbonates and various electrolyte-salt derived compounds. When using the LiPF6 salt, these compounds are typically: LiF, LixPFy and LixPOyFz. This surface-film formation is not only electrochemically driven, it can also have a chemical origin as a result of storage in the electrolyte.10–12 There is no significant difference in the elemental composition of the surface-film after cycling and storage at elevated temperature compared to that at room temperature. However, the thickness and degree of coverage of the film are found to increase.11 Indeed, the observed increase in ac-impedance for LiNi0.8Co0.2O2 electrodes has been suggested to originate in the thicker surface layer formed at higher temperatures.14,15
From a study of electrochemically cycled LiFePO4 electrodes,16 it was concluded that no solvent-based reaction products were formed on the surface, indicating that the oxygen in the phosphate group does not take part in any such reactions, unlike the oxygen in the Co and Mn oxides. In LiFePO4, the surface film consisted only of different salt-based products. Depth profiling could show that this film did not completely cover the electrodes or, at least, was not thicker than the detection limit of a few nanometres.
The surface layer formed at 55 °C on fresh uncycled Li2FeSiO4 electrodes compared with that formed on electrodes cycled with a 1 M LiTFSI EC:PC (1 : 1) electrolyte has previously been studied by photoelectron spectroscopy using both synchrotron and monochromatized AlKα radiation.1 At ambient temperature, fresh electrodes exposed to air showed larger amounts of carbonate-based compounds, such as Li2CO3 and LiHCO3, on their surfaces than electrodes stored under an inert atmosphere. This indicates that lithium is withdrawn from the original structure on exposure to air. After electrochemical cycling, the main component of the surface film is LiTFSI salt in its original form, together with small amounts of solvent reaction products, possibly lithium carboxylates. No LiF or carbonate-based compounds are found on the surface.
Here, the Li2FeSiO4 electrode surface layer is studied in detail by photoelectron spectroscopy in the light of earlier results for LiTFSI- and LiPF6-salt based electrolyte systems.
Experimental
Li2FeSiO4 was prepared by the solid-state reaction of Li2SiO3 with FeC2O4·2H2O. These starting materials were dispersed in acetone, mixed thoroughly and then ground together with 10 wt% polyethylene glycol. After evaporating the acetone, the mixture was heated to 700 °C for 20 h in a flow of CO–CO2 (50 : 50) gas to suppress the oxidation of Fe2+. The crystal structure was readily confirmed by X-ray powder diffraction (not shown here). The carbon content of the synthesised material was determined to be 4.68(5) wt% using the carbon combustion method for elemental analysis.The material was cycled electrochemically using “coffee-bag”-type cells. Electrodes were prepared by spreading a mixture of 90 wt% active material, 5 wt% carbon black and 5 wt% EPDM binder dissolved in cyclohexane onto an aluminium foil; the thickness of the active layer was ca. 25 µm. Circular electrodes (3.14 cm2) with a typical loading of 3–5 mg of active material were dried under vacuum at 120 °C, prior to assembly of the electrochemical cells. Half-cells of configuration <Li2FeSiO4|glass-wool separator soaked in electrolyte|Li foil> were assembled in an Ar-filled glove-box (<5 ppm H2O and O2). Two different electrolytes were used for the study: 1 M LiTFSI (lithium bis(trifluoromethylsulfonyl)imide) and 1 M LiPF6 (lithium hexafluorophosphate). Each lithium salt was dissolved in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) in a 2 : 1 volumetric ratio. The LiTFSI salt was dried under vacuum at 120 °C prior to electrolyte mixing; the LiPF6 salt was dried at 80 °C. The solvents were used as received.
Galvanostatic cycling was performed at 55 °C using a Digatron BTS 600 testing system. Cells were precycled three times at a C/20 rate (current density: 4.7 mA g−1) between 2.0 and 3.7 V. Cycling was stopped either in the discharged or the charged state. The cells were then dismantled in the glove-box and small pieces of the electrodes cut out, mounted on a sample-holder, and transported to the XPS equipment, using a specially designed transport chamber to avoid contamination by air or moisture. All measurements were performed on unwashed electrodes to preserve any surface species formed during the cycling. XPS measurements using monochromatized Al Kα at 1486.6 eV were performed on a PHI 5500 system. The spectrometer energy scale was calibrated using Cu2p3/2 (932.7 eV), Ag3d5/2 (386.2 eV) and Au4f7/2 (84.0 eV) emissions of 3keV sputter-cleaned metal foils.
Results and discussion
Electrochemical characterisation
Fig. 1 shows the first charge and discharge cycle for the 1 M lithium bis(trifluoromethylsulfonyl)imide (LiTFSI) EC:DEC (2 : 1) cell (a) and the 1 M hexafluorophosphate (LiPF6) EC:DEC (2 : 1) cell (b). Both cells were cycled at 55 °C and at a C/20 rate, and both exhibited two distinct plateaus: one at 3.10 V during charge and one at 2.76 V during discharge. The following charge (not shown) had a plateau at 2.85 V; this shift in potential from the first to subsequent charge-cycles has been discussed in a previous paper and will not be elaborated upon further here.3 The initial capacity for the LiTFSI-based cell is 100 mAh g−1, while it is a little lower for the LiPF6-based cell: 93 mAh g−1. As reported earlier, Li2FeSiO4 exhibits a stable capacity of 120 mAh g−1 with an electrolyte based on the LiTFSI-salt together with EC and propylene carbonate (PC) in a volumetric ratio of 1 : 1.3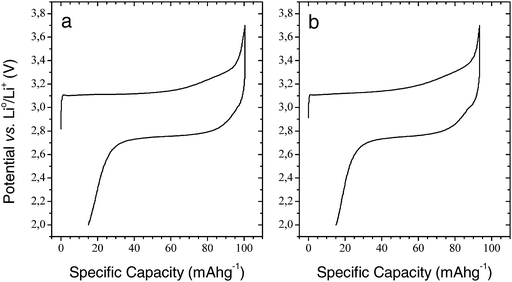 | ||
| Fig. 1 First-cycle voltammograms for carbon-coated Li2FeSiO4 electrodes at 55 °C and C/20 cycle rate for an electrode with 1 M LiTFSI EC:DEC (2 : 1) (a) and 1 M LiPF6 EC:DEC (2 : 1) (b). | ||
XPS characterisation
Surface characterisation by XPS has been conducted for both the 1 M LiTFSI EC:DEC (2 : 1) system and the 1 M LiPF6 EC:DEC (2 : 1) systems. The XPS spectra for electrodes in an intercalated state are compared with those for deintercalated electrodes for both LiTFSI- and LiPF6-based electrolytes in Fig. 2. The respective high-resolution spectra are compared in Fig. 3. The data has been background-corrected by subtracting Tougaard-type background functions. The spectra have been curve-fitted using a combination of Gaussian and Lorenztian curves (Voigt-type). These curves, together with the summation curve, are drawn as thin lines while the raw data appears as points. An overview of peak assignments and binding energies is given in Table 1.| Assignments | Measured binding energy/eV | |||||||
|---|---|---|---|---|---|---|---|---|
| C1s | F1s | Li1s | O1s | P2p3/2 | Si2p3/2 | S2p3/2 | N1s | |
| C/Li2FeSiO4 | 284.5 | — | 55.2 | 531 | — | 101.8 | — | — |
| Carbon black | 284.5 | — | — | — | — | — | — | — |
| Hydrocarbon | 285.3 | — | — | — | — | — | — | — |
| LiN(SO2CF3)2 | 293.3 | 689.1 | 56–57 | 532.5 | — | — | 169.4 | 399.7 |
| LiF | — | 685.5 | 56–57 | — | — | — | — | — |
| Li2CO3 | 290.2 | — | 56–57 | 532 | — | — | — | — |
| R(CH2CH2O)n–R | 286.7 | — | — | 533 | — | — | — | — |
| LiPF6 | — | 688 | 56–57 | — | 137.8 | — | — | — |
| LixPFy | — | 687–688 | 56–57 | — | 136.5 | — | — | — |
| LixPOyFz | — | — | 56–57 | 534 | 134.5–135 | — | — | — |
| –Si–F | — | — | — | — | — | 102.7 | — | — |
| –SOx | — | — | — | — | — | — | 167.5 | — |
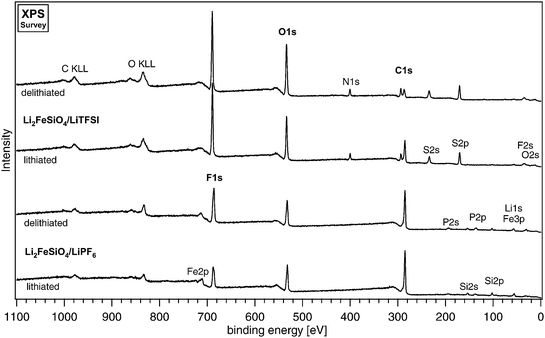 | ||
| Fig. 2 A survey of the XPS AlKα spectra for lithiated and delithiated carbon-coated Li2FeSiO4 electrodes cycled with 1M LiTFSI EC:DEC (2 : 1) and 1M LiPF6 EC:DEC (2 : 1). | ||
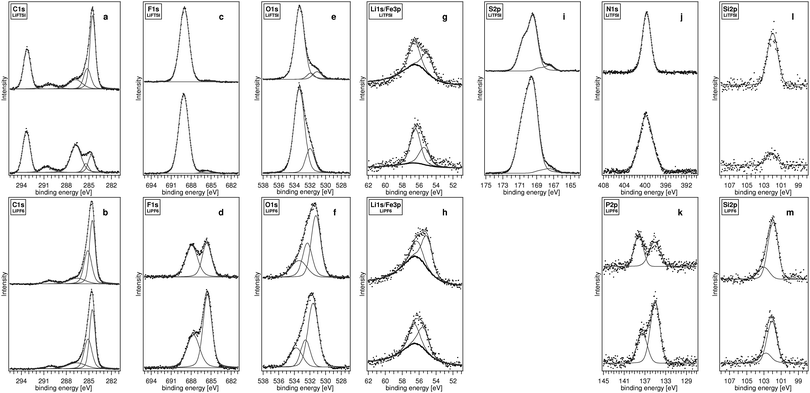 | ||
| Fig. 3 XPS spectra for lithiated (top) and delithiated (bottom) carbon-coated Li2FeSiO4 electrodes cycled with 1M LiTFSI EC:DEC (2 : 1) and 1M LiPF6 EC:DEC (2 : 1). The C1s (a, b), F1s (c, d), O1s (e, f), Li1s/(Fe3p) (g, h), S2p (i), N1s (j), P2p (k) and Si2p (l, m) core-level emissions are presented. In the case of the Li1s/Fe3p emissions (g, h), the Fe3p peak for Fe(III)O is superimposed (thick solid line) to facilitate the separation of the overlapping Li1s and Fe3p levels. | ||
LiTFSI
The C1s spectrum for the intercalated electrode with LiTFSI (Fig. 3a) displays a sharp dominant peak at 284.5 eV (FWHM: 0.8 eV) due to the presence of different (aliphatic) carbon species, i.e., the carbon black, the carbon coating on the Li2FeSiO4 particles and the EPDM binder. A smaller peak is observed at 285.3 eV within the main emission peak, which can be attributed to various hydrocarbons, including the EPDM binder. The –CF3 groups of the LiTFSI salt can be found at a fairly high binding energy of 293.3 eV. Since the line width of this peak is quite narrow (FWHM: 1.05 eV) with a symmetric peak shape, the LiTFSI salt seems to be present on the surface and has not reacted significantly or become degraded. This is confirmed for the F1s spectra (Fig. 3c), where the large peak at 689.0 eV is assigned to the –CF3 group in LiTFSI, and the much smaller peak at 685.5 eV is attributed to a minimal amount of LiF. A similar situation has been observed for the previously reported LiFeSiO4/LiTFSI/EC:PC (1 : 1) electrode system in the intercalated state.1 However, the peak at 290.3 eV corresponding to lithium carbonate and that at 286.8 eV were not reported in the previous study. The latter peak originates most probably from an oxygen-containing polymeric compound of polyethylene oxide (PEO) type, formed by a salt-anion initiated polymerisation of EC on the electrode.6,17–20 These two findings are confirmed in the O1s spectrum (Fig. 3e), where the large peak at 533.4 eV contains both the LiTFSI oxygen and PEO-type polymer contributions. The smaller peaks at 531.9 eV and 531.1 eV can probably be attributed to Li2CO3 and the silicate oxygen in Li2FeSiO4, respectively. The Li1s/Fe3p emissions are shown in Fig. 3g. Due to the overlap of those regions, the Fe3p emission of a Fe(III)O (99%, Merck) powder sample is superimposed as a reference. The intensity has been normalized to that found for the Si2p peak of Li2FeSiO4 (discussed later), and an empirical correction constant has been applied for all measurements to fit the slope of the Li1s/Fe3p signal of the electrodes. To a first approximation the lithium contributions have thus been separated using this procedure. The emission found at 55.2 eV represents the Li+ in Li2FeSiO4, whereas that centred at 56.6 eV can be attributed to a number of components in the SEI (see Table 1).The S2p emission of the –SO2– group of the LiTFSI salt is shown in Fig. 3i. In the following, the binding energies of spin–orbit split core-level emissions are always referred to the main emission. The doublet separation (DS) of the S2p3/2 and S2p1/2 components is 1.185 eV. The peak is composed of a main emission at 169.4 eV and a weak shoulder component at 167.5 eV. This group must therefore be slightly reduced, leading to a shift to lower binding energies. The N1s spectrum in Fig. 3j is composed of only one component located at 399.7 eV. The peak reveals a FWHM of 1.7 eV, corresponding to the imide group of LiTFSI. The Fe2p core-level, however, cannot be addressed, since it overlaps with strong satellite peaks of the intense F1s emission.
The deintercalated electrode (Fig. 3a) shows more or less the same binding energies for the features in the C1s emission structure as the intercalated electrode, but with grossly differing relative intensities. The largest peak is no longer the carbon-coating and carbon-black/EPDM binder peak (at 284.6 eV), but the –CF3 emission of the LiTFSI salt at 293.3 eV, while the intensity of hydrocarbon peak at 285.4 eV is slightly decreased. The decrease of the emissions at lower binding energies is accompanied by a strongly enhanced component (at 286.7 eV) corresponding to the PEO-type polymer. The products seem to contain mainly carboxylic groups, but also carbonates, since the Li2CO3 peak in the 290.6 eV region has increased slightly. This observation is supported by the normalized peak intensities given in Table 2. This is also reflected in the O1s spectra (Fig. 3e), which shows an intense LiTFSI and PEO peak at 533.3 eV and a Li2CO3 peak at 532.0 eV. This latter peak only indicates the presence of Li2CO3, contrary to the intercalated electrode, where an additional peak was observed attributed to Li2FeSiO4. The F1s spectrum of the deintercalated electrode (Fig. 3c) remains more or less the same compared to that for the intercalated electrode. The feature at 685.3 eV reveals a slightly increased though still minimal amount of LiF. As expected for a charged electrode, the Li1s signal at 55.2 eV (Fig. 3g) corresponding to Li+ in the Li2−xFeSiO4 structure is notably reduced compared to that for the intercalated electrode. On the other hand, the Li emission at 56.7 eV is increased as a result of the thicker surface layer.
| Core-level | Normalized intensity (cps/ASF) | ||||
|---|---|---|---|---|---|
| C1s | P2p | S2p | C1s | Si2p3/2 | |
| Binding energy of components/eV | 286.7 | all | all | 284.5 | 101.8 |
| Assignments | PEO | LiPF6 | LiTFSI | C-coating | Li2FeSiO4 |
| ASF | 0.314 | 0.525 | 0.717 | 0.314 | 0.368 |
| Sample | |||||
| LiTFSI lithiated | 0.16 | — | 0.25 | 0.55 | 0.03 |
| LiTFSI delithiated | 0.39 | — | 0.47 | 0.14 | 0.01 |
| LiPF6 lithiated | 0.18 | 0.03 | — | 0.74 | 0.05 |
| LiPF6 delithiated | 0.22 | 0.06 | — | 0.69 | 0.04 |
The S2p and N1s features of the electrolyte (shown in Fig. 3i and 3j) are broadened and the intensities of the main lines are increased. The increased FWHM for both N1s and S2p suggests an inhomogeneous LiTFSI salt distribution, possible degradation products or a slight partial charging of the surface species.
These results clearly indicate an increased thickness in the surface layer, containing reaction products as well as unreacted electrolyte species on the deintercalated electrode. The intensities from features in the underlying electrode are therefore reduced. The amount of oxidized/reacted species, mainly decomposition products of the solvents, increases significantly compared to the intercalated electrode (see Table 2). The overall increase in electrolyte and decomposition products, i.e., LiTFSI, PEO compounds and Li2CO3, suggests a dynamic and possibly reversible SEI coverage of the electrode, dependant on the electrode potential.
LiPF6
The situation using an LiPF6-based electrolyte is somewhat different for both intercalated and deintercalated electrodes compared to that for LiTFSI. The C1s spectra of the intercalated and deintercalated electrodes (Fig. 3b) are basically the same, with insignificant changes in relative intensities. The major contribution to these spectra is the peak at 284.6 eV, assigned to the carbon-coating, carbon-black and EPDM binder, together with the hydrocarbon and EPDM binder peak at 285.2–285.3 eV. Similar peaks are found at 286.4–286.6 eV and 290.1–290.2 eV to those found for the LiTFSI electrodes; these originate from PEO-type polymer compounds as well as Li2CO3. However, the amounts of these compounds appear less here than on the LiTFSI electrodes. Meanwhile, the overall carbon intensity is about three times higher than for LiTFSI, indicating a larger carbon content at the surface and hence a lower SEI coverage. No increase is observed on deintercalation (Table 2).The F1s spectra (Fig. 3d) reveal large amounts of LiF at 685.5–685.6 eV both for the intercalated and the deintercalated electrodes. The relative intensity of the LiF peak for the deintercalated electrode is roughly double that for the intercalated. The LiPF6-salt contribution is found at binding energies of 687.7–687.9 eV; it is ∼1.2 eV lower than for the –CF3 groups of LiTFSI. However, the quite broad peak widths (FWHM: 1.8 and 2.0 eV, respectively) are a further indication that the LiPF6 salt is unstable during cycling, and has degraded to LixPFy-type compounds. The same situation has been found in a previous study of LiFePO4 with a 1M LiPF6 EC:DEC (2 : 1) electrolyte.16 It can be noted that the amount of fluorine observed for LiPF6 is 3–4 times lower than for LiTFSI. However, the amount of reacted LiF found for the LiTFSI system is only ∼10% of that observed for LiPF6. This amount may be higher at the interface, but the signal is reduced due to the higher SEI coverage.
The peak contributing most to the O1s spectra both for the intercalated (531.2 eV) and deintercalated (531.6 eV) electrodes is assigned to Li2CO3, with probable contributions also from the silicate oxygen in Li2FeSiO4 (Fig. 3f). The peaks at higher binding energies are generally attributed to PEO-type polymer compounds and, to a lesser extent, possible oxygen-containing decomposition products of LiPF6, e.g., LixPOyFz. The Li1s/Fe3p region (shown in Fig. 3h) has been treated in the same way as the LiTFSI sample. Two lithium emissions are observed (at 55.0 and 56.5 eV), corresponding to the Li2FeSiO4 electrode and surface phases, respectively. On deintercalation, the relative intensity of the peak at lower binding energy is decreased, as seen earlier for the LiTFSI salt.
Two distinct peaks appear in the P2p spectrum (DS: 0.85 eV) at 134.8 and 137.9 eV for the intercalated and at 134.7 and 137.2 eV for the deintercalated electrodes (Fig. 3k). The LiPF6 salt, with a binding energy of 137.9 eV for the intercalated electrode, moves to lower binding energy (137.2 eV) and thus represents not only the LiPF6 salt but also its degradation product LixPFy (at 136.5 eV). It has been shown earlier16 that LiPF6 can decompose in the electrolyte to LiF and LixPFy according to the reactions:
| LiPF6 → LiF + PF5 | (2) |
| PF5 + 2xLi+ + 2xe− → LixPF5−x + xLiF | (3) |
| LiPF6 + H2O → LiF + POF3 + 2HF | (4) |
The oxygenated salt-derived species formed here are best summarized as LixPOyFz, and are responsible for the 134.8 and 134.7 eV peaks in Fig. 3k, respectively.
XPS Si2p
It has been debated as to whether the LiPF6 electrolyte will react and subsequently degrade the Li2FeSiO4 framework. As seen in eqn (3), LiPF6 can hydrolyse to form HF, which is thought to induce degradation of Li2FeSiO4.Fig. 3l and 3m show the Si2p emission (DS: 0.61 eV) for intercalated and deintercalated electrodes from cells with LiTFSI- and LiPF6-based electrolytes. A dominant Si emission is seen at 101.7–101.9 eV for both electrolytes. For LITFSI, only a single emission can be found at 101.8 eV for the intercalated and deintercalated electrode (Fig. 3l). The Si2p emission reveals an asymmetric peak shape for LiPF6, suggesting a second emission at higher binding energies. This weak emission at 102.7 eV (Fig. 3m) indicates the presence of Si–F groups at the electrode surface, as expected when Li2FeSiO4 is degraded on reaction with the HF formed by hydrolysis of LiPF6.
However, the Si2p emission is weakened due to the carbon-coating on the Li2FeSiO4 particles, and is even less visible for LiTFSI, where the SEI coverage is greater (see Table 2). There are some indications of degradation in the Li2FeSiO4/LiPF6 system. Studies continue of the SEI phase on uncoated electrodes to help clarify this picture.
Conclusions
Here, the surface layer of Li2FeSiO4 has been investigated thoroughly for two electrolyte systems, and a better understanding of the overall surface chemistry has emerged. The experimental picture of the SEI layers formed is summarized schematically in Fig. 4.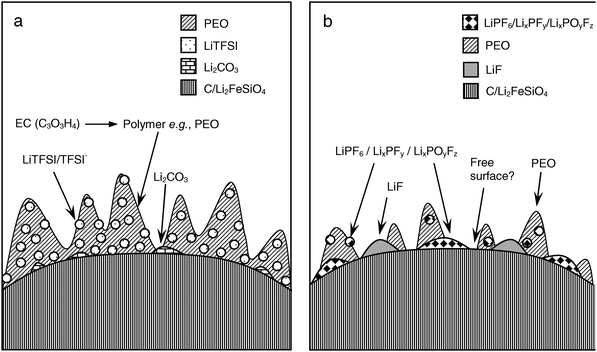 | ||
| Fig. 4 Schematic representation of the SEI formed on Li2−xFeSiO4 cathode surfaces for (a) LiTFSI and (b) LiPF6 salt-containing electrolytes. | ||
For the LiTFSI-based electrolyte, a polymeric surface layer incorporating LiTFSI and TFSI− salt anions is formed from the polymerized solvent species, along with lithium carbonates. The amount of carboxylic/carboxalate bonding is strongly enhanced, especially for the delithiated LiTFSI/LiFeSiO4 system. This has not been observed previously for the EC/PC electrolyte system, and may indicate the formation of a dynamic and even reversible SEI coverage, dependant on the state of charge/potential of the electrode. Further studies are needed to validate this observation.
The LiPF6-based electrolyte forms a thin surface layer with only small amounts of carbon-based surface reaction products, both in the lithiated and delithiated states. The main contribution to the C1s spectra comes from the electrode itself. The commonly observed instability of the LiPF6 salt, resulting in formation of LixPFy, LixPOyFz and LiF species, was further confirmed in this work. Lithium carbonate is found on all electrodes studied, suggesting that the lithium carbonate initially present on the Li2FeSiO4 surface is insoluble in the EC/DEC electrolyte, as in the case of the EC/PC electrolyte. There are also indications that Li2FeSiO4 is degraded by the HF formed in the hydrolysis of LiPF6.
Our study demonstrates well the difference in surface chemistry for two electrolyte systems. The most striking features are the spectral changes in the C1s region between the discharged and charged state for the LiTFSI system, while virtually no changes are observed for the LiPF6-based samples. On the other hand, evidence has been found to suggest electrolyte-induced formation of LiF and the degradation of the Li2FeSiO4 particle surface by HF for the LiPF6-containing electrolyte.
Acknowledgements
This work has been supported by the Global Climate and Energy Project (GCEP) of Stanford University (USA), the Swedish Energy Agency (STEM), the Swedish Science Council (VR) and the Swedish Governmental Agency for Innovation Systems (Vinnova).References
- A. Nytén, M. Stjerndahl, H. Rensmo, H. Siegbahn, M. Armand, T. Gustafsson, K. Edström and J. O. Thomas, J. Mater. Chem., 2006, 16, 3483 RSC.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CAS.
- A. Nytén, S. Kamali, L. Häggström, T. Gustafsson and J. O. Thomas, J. Mater. Chem., 2006, 16, 2266 RSC.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CAS.
- A. M. Andersson, A. Henningsson, H. Siegbahn, U. Jansson and K. Edström, J. Power Sources, 2003, 119–121, 522 CrossRef CAS.
- A. M. Andersson and K. Edström, J. Electrochem. Soc., 2001, 148, A1100 CrossRef CAS.
- A. M. Andersson, M. Herstedt, A. Bishop and K. Edström, Electrochim. Acta, 2002, 47, 1885 CrossRef CAS.
- K. Edström, M. Herstedt and D. P. Abraham, J. Power Sources, 2006, 153, 380 CrossRef.
- D. Aurbach, K. Gamolsky, B. Markovsky, G. Salitra, Y. Gofer, U. Heider, R. Oesten and M. Schmidt, J. Electrochem. Soc., 2000, 147, 1322 CrossRef CAS.
- T. Eriksson, A. M. Andersson, A. G. Bishop, C. Gejke, T. Gustafsson and J. O. Thomas, J. Electrochem. Soc., 2002, 149, A69 CrossRef CAS.
- T. Eriksson, A. M. Andersson, C. Gejke, T. Gustafsson and J. O. Thomas, Langmuir, 2002, 18, 3609 CrossRef CAS.
- G. Pistoia, A. Antonini, R. Rosati and D. Zane, Electrochim. Acta, 1996, 41, 2683 CrossRef CAS.
- A. M. Andersson, D. P. Abraham, R. Haasch, S. MacLaren, J. Liu and K. Amine, J. Electrochem. Soc., 2002, 149, A1358 CrossRef CAS.
- C. H. Chen, J. Liu and K. Amine, J. Power Sources, 2001, 96, 321 CrossRef CAS.
- D. P. Abraham, J. Liu, C. H. Chen, Y. E. Hyung, M. Stoll, N. Elsen, S. MacLaren, R. Twesten, R. Haasch, E. Sammann, I. Petrov, K. Amine and G. Henriksen, J. Power Sources, 2003, 119–121, 511 CrossRef CAS.
- M. Herstedt, M. Stjerndahl, A. Nytén, T. Gustafsson, H. Rensmo, H. Siegbahn, N. Ravet, M. Armand, J. O. Thomas and K. Edström, Electrochem. Solid-State Lett., 2003, 6, A202 CrossRef CAS.
- M. Herstedt, A. M. Andersson, H. Rensmo, H. Siegbahn and K. Edström, Electrochim. Acta, 2004, 49, 4939 CrossRef CAS.
- E. Peled, D. Bar Tow, A. Merson, A. Gladkich, L. Burstein and D. Golodnitsky, J. Power Sources, 2001, 97–98, 52 CrossRef CAS.
- L. Vogdanis and W. Heitz, Macromol. Chem. Rapid Commun., 1986, 7, 543 CrossRef CAS.
- J.-C. Lee and M. Litt, Macromolecules, 2000, 33, 1618 CrossRef CAS.
- M. Herstedt, D. P. Abraham, J. B. Kerr and K. Edström, Electrochim. Acta, 2004, 49, 5097 CrossRef CAS.
- H. Ota, Y. Sakata, X. Wang, J. Sasahara and E. Yasukawa, J. Electrochem. Soc., 2004, 151, A437 CrossRef CAS.
- N. M. Brown, J. A. Hewitt and B. J. Meenan, Surf. Interface Anal., 1992, 18, 187 CAS.
- L. J. Rendek, G. S. Chottiner and D. A. Scherson, J. Electrochem. Soc., 2002, 149, E408 CrossRef CAS.
- J. F. Moulder, W. Stickle, P. E. Sobol and K. D. BombenHandbook of X-Ray Photoelectron Spectra, Perkin-Elmer, 1995 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |