Advances in combinatorial and high-throughput screening of biofunctional polymers for gene delivery, tissue engineering and anti-fouling coatings
J. Carson
Meredith
*
School of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, 30332-0100, USA. E-mail: carson.meredith@chbe.gatech.edu
First published on 23rd October 2008
Abstract
The past ten years have witnessed the emergence of a new set of tools, combinatorial and high-throughput screening, in polymeric biomaterials development. These tools, developed initially in the drug-discovery industry, and later applied to catalysts and inorganic materials, allow orders of magnitude increases in the rate of exploration and characterization of new materials. This feature article covers recent examples of high-throughput and combinatorial studies of biofunctional polymers. Biofunctional polymers exhibit chemistry or physical properties specifically designed to function in a biological context. Examples include polymers with unique binding affinities or surface physical properties for gene and drug delivery, tissue engineering scaffolds, anti-fouling and anti-bacterial coatings, and biocatalytic applications.
 Carson Meredith | Carson Meredith obtained his PhD in Chemical Engineering from U. Texas-Austin, then spent 2 years at the NIST Polymers Division developing combinatorial/high-throughput methods for polymers. He is now an associate professor of Chemical & Biomolecular Engineering at Georgia Tech with research interest in design of biological/materials interfaces, characterization of advanced materials interfaces and development of novel measurement tools. |
Introduction
Research and development of biofunctional polymers, including applications in biomaterials, tissue engineering, diagnostic devices, drug delivery, and gene therapy, are critical to the advancement of health care. Related technologies that also involve materials/biological interfaces include biocatalysis and anti-biofouling coatings. The past ten years have witnessed the emergence of a new set of tools, combinatorial and high-throughput screening (CHTS), in polymeric biomaterials development. These tools, developed initially in the drug-discovery industry, and later applied to catalysts and inorganic materials, allow orders of magnitude increases in the rate of exploration and characterization of new materials. While the terms combinatorial and high-throughput are often used synonymously, they actually refer to different aspects of a highly efficient discovery and development process. Combinatorial sample libraries expand significantly the range of parameters (composition, temperature, etc.) that are examined in a single experiment, by using a relatively small sample footprint, e.g., a titer plate or microscope slide. To achieve significant increases in exploration rate, the libraries must then be fed into high-throughput screening experiments chosen judiciously to detect meaningful properties. Depending on the scale of the problem, data exploration and interpretation can be performed manually or automatically via numerical or logical algorithms, e.g., data mining and informatics. It is desirable that the timescale for synthesis and screening of an entire library of hundreds or more samples be on the same scale as a conventional one-sample synthesis and screening.An important aspect of CHTS is the need to use experimental design (at the front end) and data mining (at the back end) in order to make effective use of throughput. Data mining consists of methods for detecting anomalies (hits), clustering results, and classifying data into predictive models that may be used for subsequent experimental design. The development of new data mining techniques is not covered in depth here, except where applications specific to biofunctional polymers have been developed, for example see the work of Smith et al.1 or Su and Meredith.2 New directions and classical methods are covered well in a number of recent articles.1,3–7 Commercially oriented development of new materials is likely to depend heavily on previous data and computed descriptors of properties, combined with factorial-design algorithms to choose new library conditions. Hypothesis-driven research is more likely to support experimental design via theory. It is in these cases, in particular, where CHTS relies on a good understanding of the physicochemical and biological premises for a particular system. This is a unique challenge for biofunctional materials, as theories for describing biological interactions with synthetic materials are not well developed.
This feature article will cover recent examples of high-throughput and combinatorial studies of biofunctional polymers. Biofunctional polymers exhibit chemistry or physical properties specifically designed to function in a biological context. Examples include polymers with unique binding affinities or surface physical properties for gene and drug delivery, tissue engineering scaffolds, anti-fouling and anti-bacterial coatings, and biocatalytic applications. A number of significant differences exist between development of polymers for non-biological versus biofunctional materials. One challenge is the prominence of surface chemical and physical properties in governing interactions with biomolecules (proteins, nucleic acids, etc.) and live cells. Experimentally, the preparation and maintenance of clean surfaces with specific chemistry and physical properties is challenging, which complicates materials screening. Another challenge is the use of bioassays based upon in vitro culture of cells and organisms and, ultimately, in vivo implantation studies. Considerable complexity is introduced when materials are subjected to cell culture, even in a controlled laboratory environment. This includes interactions with culture media, serum proteins, and adherent cells and their metabolites. Cell response is governed by a complex interaction between soluble and surface-born biomolecules, surface physical properties, as well as cell–cell interactions. In addition, living cells in the same population differ in their growth rate and stage in the cell cycle. Hence, measurement uncertainty is often larger than that encountered in non-biological systems and statistical identification of outliers and trends is more challenging.
Combinatorial and high-throughput methods offer significant benefits to biofunctional polymers research because they provide a mechanism for efficient screening of experimental parameters. This is a significant consideration whether the goal is oriented towards materials discovery, optimization of a design, or knowledge generation. Libraries also offer the ability to expose a large set of samples uniformly to the same environmental conditions. Given the empirical nature of biomaterials research, an exploratory screening experiment can identify the significant regions of parameter space prior to detailed in vitro and in vivo studies. In this way, detailed and costly efforts are focused on the most interesting (as defined by the user) material compositions. An additional attraction of CHTS is the small sample size, since exploratory polymer synthesis often yields limited amounts of material.
Strictly speaking, the term combinatorial refers to a particular way of combining structures, compositions, and other parameters, which evolved with the use of these techniques in pharmaceutical development. The word finds its origin in the branch of mathematics known as combinatorics, which explores how a finite set of elements can lead to infinite varieties of combinations. Examples of true combinatorial synthesis include split-and-pool methods for successive reagent addition and production of antibodies in the immune system. High-throughput screening is a more general term encompassing a wide range of techniques for increasing throughput of synthesis or characterization steps, and may not necessarily involve combinatorial enumeration of possible structural and compositional combinations. For example, high-throughput methods include rapid point-to-point sampling for chromatography, parallel spectroscopic or imaging techniques, and robotic mechanical testing.
Since a number of excellent reviews of earlier work (starting from the year 1997) in combinatorial polymer development have been published,8–15 this paper focuses on the most recent work since 2003. In addition, numerous recent reviews deal with the broader subject of combinatorial development of polymeric materials in general.16–19 This feature article focuses solely on synthetic polymers, and omits discussion of the engineering of natural or synthetic biomolecules. Recent progress in oligonucleotide-based functional polymers that have molecular recognition and catalytic ability was reviewed by Yoshihiro.20
High-throughput cell culture methods
Without methodologies specifically adapted to the preparation and high-throughput screening of combinatorial libraries of biofunctional polymer candidates, the field would not have advanced to its current state. Hence, it is fitting to describe the methodologies that have developed. The first published high-throughput screening of cell responses to combinatorial libraries of polymers was reported by Brocchini et al. in 1997.21 The discrete 96-well titer plate format was selected for the synthesis of biodegradable poly(arylate) copolymers, followed by culture of fibroblasts directly in the titer plate.22 The discrete library format is convenient and compatible with a variety of commercially available liquid handling robots and has been adopted in many studies reviewed herein. Discrete methods such as these may also be implemented in either a parallel or a serial fashion, giving them flexibility in coupling with either parallel or discrete analytical equipment.In contrast to discrete sample preparation, continuous-gradient libraries are inherently parallel since a single sample actually contains a range of conditions. Gradient-based libraries can, however, be sectioned for characterization with serial or point-to-point analytical measurements. The use of samples containing continuous gradients in polymer properties, a method complimentary to discrete library synthesis was developed by Meredith et al.12,23,24 Libraries containing the equivalent of several hundred distinct chemistries, microstructures, and roughnesses were prepared on samples the size of a microscope slide by using composition spread and temperature gradient techniques, as indicated in Fig. 1a.24 These libraries were used to investigate the response of an osteoblast cell line to microstructures of blends of poly(D,L-lactide) (PDLA) and poly(ε-caprolactone) (PCL). The phenomenon of heat-induced phase separation in these polymer mixtures was used to generate the diverse surface features on the libraries shown in Fig. 1a. Surface features at a narrow range of composition and process temperature were discovered to enhance dramatically alkaline phosphatase expression, not previously observed for osteoblasts on polymer blends. It is likely that this small region of composition and temperatures would have been missed using conventional approaches that focus on detailed characterization of a few sample compositions. The gradient-based method was extended to investigate other cell functions on demixed polymer blends including proliferation, spreading, and viability for osteoblasts2,25 and for primary mouse aortic smooth muscle cells.26 The method has also been extended to include synthesis of polyurethane copolymers with variations in block composition,27,28 such as shown in Fig. 1b. In this example, PCL was blended with a segmented polyurethane composed of poly(ethylene oxide) and PCL crosslinked with MDI, butanediol and propylene triol. Fig. 1b shows a cross-polarized optical image of regions from the library indicating the sensitivity of microstructure to annealing temperature and the ratio of triol to PEG.
 | ||
| Fig. 1 Temperature–composition gradient libraries of (a) a physical blend of PLGA and PCL (white regions are crystalline PCL from polarized optical microscopy) and (b) a blend of PCL with a copolymerized poly(ethylene oxide)-based urethane. (c) Fabrication of a universal gradient substrate for biofunctionalization, and subsequent RGD peptide immobilization by click chemistry. From ref. 29. | ||
Fig. 1c shows a schematic of a the recent development of “universal” functional gradient substrates to which a variety of species can be attached by “click” chemistry, e.g., azide-functionalized biomolecules.29 The NIST-based (U.S. National Institute of Standards and Technology) researchers described how a self-assembled monolayer (SAM) of n-octyldimethylchlorosilane was exposed to a gradient in UV energy, as in Fig. 1c, resulting in ozone oxidation to form a linear gradient in carboxylated end groups. The resulting oxidized SAM showed water contact angles varying from 15° to 103° on a single slide. This gradient was derivatized into a gradient in alkyne functionalities appropriate for surface conjugation of biomolecules. The technology was demonstrated by preparing an arginine–glycine–aspartate (RGD) peptide density gradient and followed by screening the RGD-density-dependent control of CRL-1476 smooth muscle cell adhesion and morphology.
“Metabolomics” is a rapidly emerging field that finds tremendous utility in understanding normal and diseased cellular processes, toxicity, and discovery of biomarkers. However, metabolomics depends on high-throughput analytical tools to profile biofluid metabolites. Recently, a high-throughput HPLC (high-performance liquid chromatography) method was developed for this purpose,30 by using a short monolithic column, rapid gradient generation and high flow-rates.
Over the past decade, cell-based assays have become an important tool in drug development and testing, as opposed to purely molecular bioactivity assays. This represents a shift towards early identification of side effects, toxicity, and other interactions that make drug candidates non-viable, preferably before clinical or animal trials. Hence, high-throughput assays that mimic the human tissue environment are needed. For example, Kunz-Schughart et al. used a 3-D multicellular spheroid cell culture system in developing a HTS system for novel antitumor drugs.31
Nanoparticles and microparticles have many potential biomedical applications ranging from imaging to drug delivery. Researchers at MIT reported a microfluidic high-throughput system (Fig. 2a) for analysis of the interaction of particles with cells.32 The binding of particles to cells can be studied quickly and reliably as a function of the fluid chemistry, flow characteristics, and particle size and chemistry. Farokhzad et al. evaluated the binding affinity of polymeric nanoparticles and microparticles conjugated to aptamers for prostate specific membrane antigen (PSMA).32 Cells were seeded into the device microchannels (see Fig. 2a) and the binding of particles to PSMA-negative or -positive cells was evaluated with respect to changes in fluid shear stress and particle size. These results demonstrate that the interaction of particles with cells can be studied under both high-throughput and controlled conditions, which may aid in the engineering of desired particle characteristics.
 | ||
| Fig. 2 (a) Schematic diagram of the nanoparticle–aptamer bioconjugate and the microfluidic device development. (A) Pegylated PLA particles were modified with RNA aptamers for the PSMA protein. (B) Several model cell lines, which differ in the pattern of PSMA expression, were patterned on glass substrates. A microfluidic mold was aligned over the cell pattern to develop microchannels. Adapted with permission from reference 32. (b) Microfluidic cell culture array fabricated on a 2 × 2 cm device. Adapted with permission from reference 36. (c) Single microfluidic culture unit. Multiple perfusion channels surround the main culture chamber, 40 µm in height with a diameter of 1 mm. Each culture unit had four fluidic access paths. Adapted with permission from reference 36. | ||
Khademhosseini et al. recently reviewed the utilization of 3D microfabrication fabricating devices for high-throughput in vitro assay of cells under controlled microenvironments.33 By combining microfluidics and soft lithography, surface patterning and patterned cocultures can be used to regulate various aspects of cellular microenvironments to direct cell fate and discover underlying biology. For example a multiphenotype cell array was constructed by capturing cells within an array of reversibly sealed elastomeric polydimethylsiloxane (PDMS) microfluidic channels.34 Reversible sealing of channels is used to deliver culture fluids or cells sequentially onto specific locations on a substrate. Capture and immobilization of cells in low-shear regions was accomplished by manipulating shear stress via flow rate. By using an array of channels it was possible to sequentially deposit hepatocytes, fibroblasts, and embryonic stem cells. The PDMS is subsequently removed to reveal a multiphenotype array of cells. In addition, the orthogonal alignment and subsequent attachment of a secondary array of channels on the patterned substrates could be used to deliver fluids to the patterned cells. The ability to position different cell types with spatial control could potentially lead to improved high-throughput methods in drug screening and tissue engineering.
In a similar development, Figallo et al. have developed a micro-bioreactor array with twelve independent micro-bioreactors perfused with culture medium.35 The array enables cultivation of cells attached to substrates or encapsulated in hydrogels at variable levels of hydrodynamic shear. This system was used to investigate factors that regulate differentiation in primary rat cardiac myocytes, human embryonic stem cells and the C2C12 cell line.
Hung and coworkers recently presented a novel microfluidic cell culture array for assay of 100 different cell-based experiments in parallel (Fig. 2b).36 The 10 × 10 discrete array device was designed to integrate the processes used in typical cell culture experiments on a single self-contained microfluidic system, including cell growth and passage cycles, reagent introduction, and in situ optical analysis. As Fig. 2c shows, each circular chamber in the array has four ports connecting to narrow perfusion channels for fluidic access. Human carcinoma (HeLa) cells were cultured inside the device with continuous perfusion of medium at 37 °C. Cell assay was demonstrated by monitoring the fluorescence localization of calcein AM from 1 min to 10 days after reagent introduction.
Many approaches for screening biomaterials utilize a two-dimensional film or surface for cell culture. However, cells cultured in vitro display more natural in vivo characteristics when cultured in a three-dimensional environment. Recently, researchers at NIST designed a novel 3-D combinatorial polymer library.37 Their method is based upon gradient libraries in the form of three-dimensional, porous, salt-leached, polymer scaffolds, shown in Fig. 3a. Two syringe pumps are used to pump polymer solutions into a static mixer and then deposit them into a three-dimensional mold pre-filled with NaCl particles. The method was demonstrated for poly(D,L-lactic acid) (PDLLA) containing a gradient in Sudan IV (a red dye), although it is equally applicable to a blends of polymers with other gradient additives, e.g., proteins, drugs, other polymers, etc.
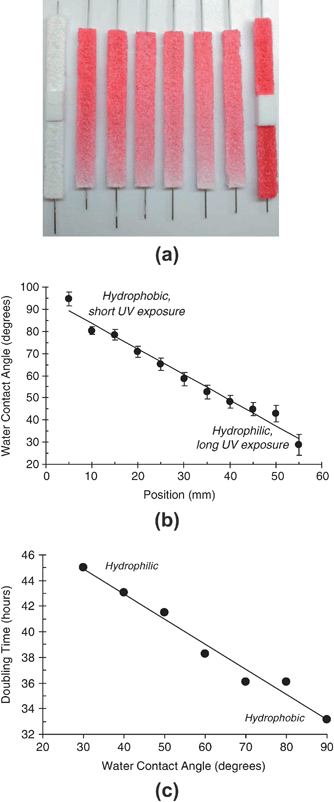 | ||
| Fig. 3 (a) Three-dimensional, porous PDLLA polymer scaffolds containing gradients in red dye Sudan IV illustrate the ability to form libraries with open interconnected porous structures. Adapted with permission from reference 37. (b) Contact angle versus position on UV-gradient treated SAMs. (c) Proliferation rate of MC3T3-E1 osteoblast cells cultured on linear-gradient surface energy substrates. Adapted with permission from reference 38. | ||
Surface energy is a fundamental material property that influences protein adsorption and cell attachment. A combinatorial approach for investigating the effect of surface energy on cell response has been developed, also at NIST.38 Gradients in surface energy were created by using an automated stage to decelerate a glass slide coated with an n-octyldimethylchlorosilane SAM beneath a UV lamp such that the SAM is exposed to the UV light in a graded fashion (as in Fig. 1c). This approach yielded substrates having a linear gradient in water contact angle from 25° to 95° shown in Fig. 3b. Surface energy gradients were coated with fibronectin and cells were seeded on the fibronectin-coated gradients and adhesion, spreading and proliferation were assessed with automated fluorescence microscopy. While surface energy did not affect initial cell adhesion, the rate of proliferation was linearly dependent on surface energy and increased with increasing hydrophobicity, indicated in Fig. 3c.
A simple yet versatile method was developed to prepare a low-density polymerization initiator gradient, which was combined with surface-initiated atom transfer radical polymerization (ATRP) to produce a well-defined poly(2-hydroxyethyl methacrylate) (HEMA) gradient substrate (Fig. 4).39 A smooth variation in film thickness was measured across the gradient, ranging from 20 Å to over 80 Å, corresponding to variation in the HEMA grafting density. The density of fibronectin adsorbed to the substrate was controlled by the HEMA grafting density. This method was used to investigate osteoblast response to fibronectin density, which is modulated by the HEMA gradient (discussed below with Fig. 7).
 | ||
| Fig. 4 Schematic illustrating substrates with well-defined gradients in HEMA grafting density produced using a low-density polymerization initiator gradient in an OTS self assembled monolayer. The HEMA gradient affects the amount of fibronectin (FN) that can be subsequently absorbed (see Fig. 7 later). The schematic representations of both FN and poly(HEMA) in the figure are not to scale. Adapted with permission from reference 45. | ||
Polymer–cell interactions and tissue engineering
Designing materials that regulate cell function in a desired manner is a major goal of biomaterials engineering. Challenges include the vast material property space to be explored, the complexity of cell–surface interactions, and the empirical nature of research in this field. To address these challenges, combinatorial methods have been developed in recent years for screening adherent cell responses to material surfaces. Smith and coworkers demonstrated the utility of combining combinatorial and computational approaches for the rational design of polymers for biomedical applications.1,6,40,41 A library of 112 biodegradable polyarylates, discussed above,21 was used to develop efficient screening techniques to determine biorelevant polymer properties (fibrinogen adsorption, gene expression in macrophages, growth of fetal rat lung fibroblasts (RLFs)). A surrogate (semiempirical) model was developed (i) to determine molecular-scale polymer properties that correlate to various biological responses, and (ii) to predict fibrinogen adsorption and RLF growth on polymeric surfaces, shown in Fig. 5a. For 38 out of 45 polymers, the model predicted the amount of fibrinogen adsorbed correctly within the error of the experimental measurements. The growth of rat lung fibroblasts was correctly predicted by the model for 41 out of 48 polymers. Continued work with the semiempirical surrogate models included prediction of the effect of polymer surface chemistry on fibrinogen adsorption using molecular dynamics approaches.42 The significance of the new model is that it allows high accuracy prediction of fibrinogen adsorption without the need for experimentally derived descriptors and it has better predictive quality than the original 2D surrogate model due to utilization of realistic polymer representations. There is a strong potential for the use of predictive strategies in conjunction with HTS of biofunctional polymers. Reynolds described a generalized approach for designing diverse and focused combinatorial libraries of synthetic polymers for biomedical materials.7 | ||
| Fig. 5 (a) The validation set for the five input ANN surrogate model: prediction vs. mean experimental data, taken from reference 40. (b) Cell number versus roughness of PLLA surfaces for MC3T3-E1 osteoblasts cultured on gradient crystallinity libraries. Adapted with permission from reference 43. (c) One day proliferation and (d) one day viability for primary rat aortic smooth muscle cells cultured on composition–temperature gradient libraries of PLGA and PCL. Parts (c) and (d) adapted with permission from reference 26. | ||
Washburn et al.43 used gradient-based libraries to investigate osteoblast response to polymer crystallinity. Gradients of polymer crystallinity were fabricated on films of poly(L-lactic acid) (PLLA) using a gradient in annealing temperature. This resulted in roughness values ranging from 0.5 to 13 nm on a single sample. The rate of proliferation of MC3T3-E1 osteoblastic cells on the smooth regions of the films was much greater than on the rough regions. As Fig. 5b shows, a monotonic variation in proliferation rate was observed as a function of roughness. Results from enzyme-linked immunofluorescence assays indicated no change in the accessibility of adhesion proteins, suggesting the cells were directly responding to substrate topography. The use of the gradient library approach yielded the functional dependence of cell proliferation on nanometer-scale roughness and gave a sensitive estimate of the critical roughness change (approximately 1.1 nm) associated with observable decreases in proliferation. Simon et al.44 used a similar composition-gradient between amorphous and crystalline forms of PLA, PDLLA and PLLA, to produce variations in surface roughness. Specifically, roughness increased with the composition of the crystalline component PLLA. MC3T3-E1 osteoblast adhesion was similar on all regions of the gradients while proliferation was faster on the smooth, PDLLA-rich, end of the gradients than on the rough, PLLA-rich, end of the gradients. This finding was in agreement with the above-mentioned study of roughness effects of PLLA, although the two studies used different methods for producing gradients in crystallinity and roughness (temperature gradients of PLLA versus composition gradients of PLLA + PDLLA).
Previous work using composition-temperature 2-D gradient libraries of biodegradable polymers PCL and PDLA showed qualitatively that alkaline phosphatase activity of MC3T3-E1 osteoblasts was dramatically enhanced at specific blend compositions and temperatures. Zapata et al. recently expanded the gradient combinatorial screening to include quantitative measurements of early events in the osteoblast life cycle: attachment, spreading, and proliferation.25 Cell spreading was strongly influenced by phase-separated microstructures on the polymer surfaces. Regions of enhanced cell proliferation shifted from one microstructural region to others as the culture progressed from 3 to 8 days.
Sung and collaborators also used temperature–composition libraries to screen quantitatively materials for tissue engineered vascular grafts that regulate cell adhesion and growth.26 The effects blending poly(D,L-lactic-co-glycolic acid) (PLGA) and PCL on primary murine aortic smooth muscle cells (SMC) were investigated. SMCs were exposed to ∼1000 distinguishable surfaces in a single experiment, allowing the discovery of optimal polymer compositions and processing conditions. SMC adhesion, aggregation, proliferation, and protein production were highest in regions with medium to high PCL concentrations and high annealing temperatures, illustrated in Fig. 5c and d. These regions exhibited increased surface roughness, increased microscale PLGA-rich matrix stiffness, and significant change of bulk PCL-rich crystallinity relative to other library regions. This study revealed a previously unknown processing temperature and blending composition for two well-known polymers that optimized SMC interactions.
The screening experiments on two-dimensional gradient libraries mentioned above resulted in quantitative measures of the range of preparatory conditions and microstructures associated with cell responses. This information could be used to design future confirmatory studies that examine the fundamental mechanisms of cell response to these heterogeneous patterned surfaces. However, it is desirable to obtain more detailed fundamental information directly from the library screening, especially the quantitative relationships between cell assays and measures of polymer surface microstructure and topography. For this reason, Su and Meredith2 have recently developed an informatics approach to extracting relationships between cell function and surface microstructures, while also filtering the large amount of noise that exists in combinatorial experiments. This method, termed local cell-feature metrics (LCFA), utilizes all of the individual cell and microstructure positions to construct histograms of cell properties as a function of distance from surface features, as illustrated in Fig. 6a. The distances from each proliferated cell (P) to other cells and other microstructures are measured and stored in a database: PP (proliferating-to-proliferating cells), PC (proliferating cell-to-PCL island), and PR (proliferating-to-resting cells). The same information is measured for resting cells: RP, RR, and RC. Fig. 6b shows a contour plot of the relative probability of proliferation for cells positioned a certain distance from various sizes of PCL islands. This plot shows four major regions (in dashed boxes) of enhanced proliferation that are associated with four distinct microstructural sizes. In addition, this type of screening illustrates the non-linear, rugged ‘landscape’ of cell responses and the sensitivity to distance from nearby microstructures. LCFA allows for filtering of microstructural information beyond a ‘cutoff’ distance from each cell under consideration, which is a considerable source of noise in library experiments. By using the conventional approach of averaging the microstructure size and cell responses in each microscope image from the library, this noise masks the cell-microstructural signals.
 | ||
| Fig. 6 (a) Schematic illustrating concept of cell-based local feature metrics on a composite microscope image from osteoblasts cultured on a gradient library of PLGA and PCL. PCL islands are shown in blue, F-actin is labeled in red, and proliferated cell nuclei are labeled green. Labels are P (proliferated cells), R (cells at rest), C (PCL islands). Solid arrows represent measured distances between proliferated cells and other objects: PP (proliferated-to-proliferated cells), PR (proliferated-to-resting cells) and PP (proliferated cell-to-PCL island). Dotted arrows represent measurements of distances RP, RR, and RC for resting cells. (b) Screening of enhancement of cell proliferation (ΔPC|AC) as a function of cell-to-PCL lateral distance and PCL diameter. Adapted with permission from reference 2. | ||
Mei and coworkers developed a versatile method to prepare a low-density polymerization initiator gradient, which was combined with surface-initiated ATRP to produce well-defined HEMA gradient substrates (see Fig. 4).39,45 The gradient libraries were characterized with X-ray reflectivity, which indicated a gradual change in polymer chain structure from a “mushroom” to a “brush” regime. Fibroblasts were seeded on gradients precoated with fibronectin to test cellular responses to this novel substrate, shown in Fig. 7a. The gradient was increasingly adhesive to fibroblasts as the grafting density decreased (into the mushroom regime), indicating that cell adhesion could be tuned by controlling the density of the polymer grafts. Cell adhesion did not follow the expected trend; instead, saturated cell adhesion and spreading was found at the low grafting density region, as indicated in Fig. 7b. This finding suggests that protein adsorbs in accessible state for cell adhesion preferentially at lower HEMA grafting densities.
 | ||
| Fig. 7 (a) Fibronectin density and (b) NIH3T3 osteoblast cell area versus grafting density of PHEMA on gradient-based substrates. Adapted with permission from reference 45. | ||
Drug and gene delivery
Anderson et al.46 utilized combinatorial synthesis to generate a library of more than 500 degradable poly(β-amino esters) (PBAEs) for screening as nonviral DNA vectors. This library of novel polymers was screened in vitro for transfection efficiency and cytotoxicity at six different polymer : DNA ratios, by using plasmid DNA to encode the firefly luciferase reporter gene. In all, more than 12![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 transfection experiments (data obtained in quadruplicate) were done in a 96-well plate format. A number of the best polymers from the in vitro assays were then screened for transfection in a mouse-human prostate cancer xenograft model. The best in vitro screened polymer, called C32, transfected the luciferase gene 4-fold better than one of the best commercially available reagents, jetPEI™ (polyethyleneimine). In addition, C32 performed 26-fold better than the naked luciferase gene. C32 delivery of diphtheria toxin DNA (a suicide gene) to prostate cancer xenografts caused 40% of tumors to regress in size. Because C32 transfects tumors locally at high levels, transfects healthy muscle poorly, and displays no toxicity, it may provide a vehicle for the local treatment of cancer.
000 transfection experiments (data obtained in quadruplicate) were done in a 96-well plate format. A number of the best polymers from the in vitro assays were then screened for transfection in a mouse-human prostate cancer xenograft model. The best in vitro screened polymer, called C32, transfected the luciferase gene 4-fold better than one of the best commercially available reagents, jetPEI™ (polyethyleneimine). In addition, C32 performed 26-fold better than the naked luciferase gene. C32 delivery of diphtheria toxin DNA (a suicide gene) to prostate cancer xenografts caused 40% of tumors to regress in size. Because C32 transfects tumors locally at high levels, transfects healthy muscle poorly, and displays no toxicity, it may provide a vehicle for the local treatment of cancer.
This approach was extended to synthesize acrylate-terminated PBAEs in parallel via a condensation reaction that combines primary or secondary amines with diacrylates.47 This library of macromers was then photopolymerized to form networks with a wide range of degradation rates (from less than 1 day to minimal mass loss after three months) and mechanical properties (Young's modulus from 4 to 350 MPa), indicated in Fig. 8. Because of the diversity in properties, these polymers could be developed and screened for numerous applications ranging from microdevices to biomaterials. Recently, the same library was optimized for DNA delivery to human vascular endothelial cells (HUVECs).48,49 Endothelial cells are an important cell type to both cardiovascular disease and cancer, as they play critical roles in vascular function and angiogenesis. However, effective and safe gene delivery to primary endothelial cells in the presence of serum proteins is known to be particularly challenging. Vector parameters including polymer type, polymer weight, and DNA loading were varied, and biophysical properties including particle size, zeta potential, and particle stability over time were studied. The best PBAE vectors were found to transfect HUVECs in the presence of serum significantly higher (47 ± 9% positive, n = 10) than the commercial transfection reagents jetPEI™ (p < 0.001) and Lipofectamine 2000™ (p < 0.01).
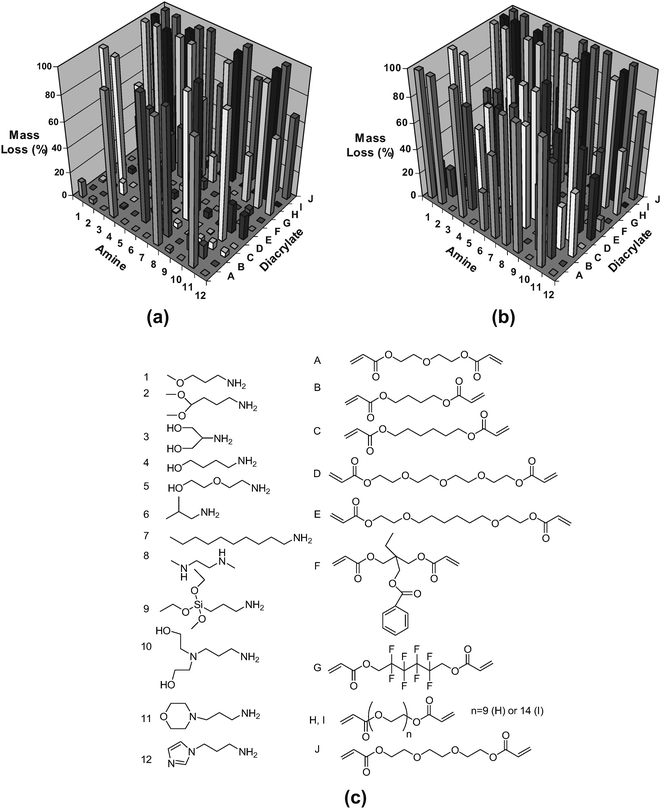 | ||
| Fig. 8 Degradation behavior (a, b) of polymers fabricated from the macromer library (c). The mass loss after (a) 1 and (b) 57 days for the polymers formed from the macromer array synthesized with 12 amines (1–12) and 10 diacrylates. Adapted with permission from reference 47. | ||
Zugates et al. synthesized novel PBAEs using a new monomer, 2-(pyridyldithio)ethylamine, and showed that these respond to intracellular conditions, behavior that may enable DNA unpacking inside the cell.50 The pyridyldithio functionalities react with high specificity toward the thiol peptide RGDC, a ligand that binds with high affinity to certain integrin receptors. The DNA binding affinity was reduced significantly in response to intracellular glutathione concentrations, which may aid in DNA unpacking inside the cell. Zugates et al. also presented a generalized method to modify PBAEs at the chain ends to improve delivery performance.51 End-chain coupling reactions were developed to allow diverse end-chain structures to be evaluated without an intervening purification step. End-modification of the PBAE C32, mentioned above, significantly enhanced its in vitro transfection efficiency. In vivo, intraperitoneal gene delivery using this polymer resulted in expression levels ten times higher than unmodified C32 and jet-PEI™.
The effect of chemical transport enhancers was investigated for the transdermal delivery of the anesthetic lidocaine across pig and human skin in vitro.52 The lipid disrupting agents oleic acid, oleyl alcohol, butenediol, and decanoic acid showed no significant flux enhancement, either by themselves or in combination with isopropyl myristate (IPM). However, a combination of IPM and n-methyl pyrrolidone (IPM/NMP) improved drug transport 25-fold over IPM alone at 2% lidocaine dose. These findings allow a more rational approach for designing oil-based formulations for the transdermal delivery of lidocaine free base and similar drugs.
Low adhesion and anti-fouling coatings
Combinatorial and high-throughput capabilities have been established to aid in the rapid development of new antifouling marine coatings.53–55 A biological screening process involving marine bacteria was developed that allows for rapid and effective quantification of bacterial biofilm growth and retention on large numbers of coating surfaces in parallel.56,57 The screening process involves (1) multiwell plate modifications for coating deposition, (2) deposition of combinatorial coating libraries via an automated liquid dispensing robot, (3) coating thickness measurements of cured coatings, (4) preconditioning of coatings via immersion in deionized water, (5) bacterial incubation, (6) plate processing, and (7) data analysis for identification of promising candidates. Majumdar and coworkers presented a detailed optimization of the high throughput coating deposition, which uses an automated doctor-blade unit.58 In order to facilitate a semi-high throughput approach to the evaluation of novel fouling-release coatings, a unique ‘spinjet’ apparatus was constructed.59,60 The apparatus, pictured in Fig. 9a, delivers a jet of water of controlled, variable pressure into the wells of 24-well plates in order to facilitate measurement of the strength of adhesion of algae growing on the base of the wells. Fig. 9b shows evaluation of H. pacifica removal from polyurethane and Silastic T2® coated plates. | ||
| Fig. 9 (a) Automated water jet simplified process diagram. Pressure for the jet is supplied via an electronic pressure regulator feeding into the dispensing tank. A solenoid valve provides on and off control while a gear motor rotates the nozzle mount. Up to 39 multiwell plates can be presented to the water jet for cleaning by the five axis robotic arm while selecting plates from three plate stacking hotels. A total of 936 individual coating samples can be challenged by the rotating, pressurized jet in a single experiment. All movements, process parameters, and database activities are controlled by the computer. (b) Evaluation of H. pacifica removal from polyurethane and Silastic T2®. Plate images are shown after crystal violet staining. Adapted with permission from reference 60. | ||
This integrated screening system has been applied to study a variety of crosslinked siloxane-containing polyurethanes for fouling-release coatings. Specific high-throughput studies include an investigation of the effects of variations in acrylic polyol composition,61 and characterization of microtopographical surfaces in thermosetting siloxane-urethane coatings.62 Ekin et al. investigated the synthesis, formulation, and characterization of these coatings for underwater marine applications.63 Starting from hydroxyalkyl carbamate-terminated poly(dimethylsiloxane) (PDMS) oligomers and their carbamate-linked block copolymers with poly(ε-caprolactone) (PCL), a series of 288 polyurethane coatings were synthesized. Based upon performance in algal, bacterial, and barnacle laboratory screening two coatings were identified as potential candidates for ocean testing.
This system was investigated further by varying the molecular weight of PDMS, amount and length of PCL blocks, and siloxane polymer level in the coating formulation.64 In addition, the coating formulation included a trifunctional isocyanate crosslinker, trifunctional PCL-polyol, 2,4-pentanedione (a pot-life extender), dibutyltin diacetate (a catalyst), and a blend of solvents. From high-throughput screening of pseudo-barnacle adhesion and water and methylene–iodide contact angles, relationships between composition, structure, surface energy and anti-fouling behavior were developed.
Dental materials
The increased usage of composite dental restorations underscores the need for continued improvements in material properties. Lin et al. fabricated polymer libraries that systematically varied the comonomer ratio of 2,2-bis[4-(2-hydroxy-3-methacryloxypropoxy)phenyl]propane and triethylene glycol dimethacrylate and irradiation time.65,66 Incomplete conversion, an ongoing challenge facing photopolymerized methacrylate-based polymers, leaves potentially toxic leachable compounds. Lin et al. studied, using an in vitro screening methodology, cells cultured directly on cross-linked films with a gradient in methacrylate double bond conversion ranging from 43.0% to 61.2%. RAW 264.7 macrophage-like cells were cultured on aged substrates to evaluate the cell response to conversion, with possible contributions from the polymer network and local leached components. Conversions of 45% and 50% decreased viability (via calcein/ethidium staining) and increased apoptosis (via annexin-V staining) (Fig. 10a and b).65,66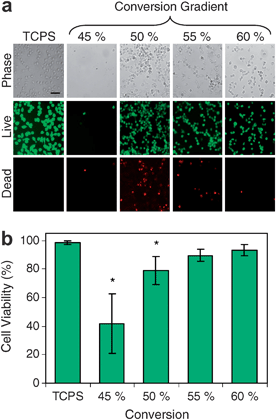 | ||
| Fig. 10 Viability of RAW 264.7 cells cultured for 24 h on conversion gradient substrates. (a) Representative images shown include phase contrast, live cells (calcein AM) and dead cells (ethidium homodimer-1). Scale bar = 50 μm. (b) Quantification of cell viability on TCPS controls and at various locations on whole gradient substrates. Adapted with permission from reference 65. | ||
Conclusions
The past ten years have witnessed the emergence of combinatorial and high-throughput screening as a new set of tools in polymeric biomaterials development. These tools, developed initially in the drug-discovery industry, and later applied to catalysts and inorganic materials, allow orders of magnitude increases in the rate of exploration and characterization of new materials. This feature article covers recent examples of high-throughput and combinatorial studies of biofunctional polymers. Examples include polymers with unique binding affinities or surface physical properties for gene and drug delivery, tissue engineering scaffolds, anti-fouling and anti-bacterial coatings, and biocatalytic applications.CHTS promise markedly improved throughput, with modest sacrifices in accuracy, and it appears from the body of work reviewed here that this is achieved in most cases. When speed and efficiency are the only concerns, it is clear that CHTS methods are advantageous over conventional throughput methods. However, another central tenet of CHTS is that enhanced throughput will enable discovery of new materials or new knowledge in significantly shorter times than by using conventional methods alone. The ultimate ‘payoff’ for investing in CHTS is the development of new, commercial products or unforeseen advances in knowledge. There are numerous examples, such as the discovery of polymers with unusually high transfection efficiencies for specific genes,46 where the likelihood of making a similar discovery with conventional synthesis in the same amount of time is exceedingly small. However, the hypothesis that CHTS will speed discovery in general remains difficult to test by looking at published work alone. For example, it is not often that researchers carry out the exact same experiments both combinatorially and conventionally under controlled conditions. In addition, screening relies on large numbers of ‘failures’, or negative responses, in order to identify a few winners. Failures are less likely to be published than successes. Finally, commercial successes, and the methods used to discover them, may be delayed in publication due to intellectual property and trade secret concerns. It is hoped, however, that this feature article will inspire such comparisons and evaluations of the CHTS field.
References
- J. R. Smith, V. Kholodovych, D. Knight, J. Kohn and W. J. Welsh, Polymer, 2005, 46, 4296–4306 CrossRef CAS.
- J. Su and J. C. Meredith, Comb. Chem. High Throughput Screening, 2008 Search PubMed , in press.
- W. Zhang, M. Fasolka, A. Karim and E. Amis, Meas. Sci. Technol., 2005, 1, 261–269 CrossRef.
- S. D. Abramson, G. Alexe, P. L. Hammer and J. Kohn, J. Biomed. Mater. Res., A, 2005, 73, 116–124 CrossRef.
- V. Kholodovych, J. R. Smith, D. Knight, S. Abramson, J. Kohn and W. J. Welsh, Polymer, 2004, 45, 7367–7379 CrossRef CAS.
- J. R. Smith, A. Seyda, N. Weber, D. Knight, S. Abramson and J. Kohn, Macromol. Rapid Commun., 2004, 25, 127–140 CrossRef CAS.
- C. H. Reynolds, J. Comb. Chem., 1999, 1, 297 CrossRef CAS.
- E. J. Amis, Nat. Mater., 2004, 3, 83–85 CrossRef CAS.
- J. Kohn, Nat. Mater., 2004, 3, 745–747 CrossRef CAS.
- J. Kohn, W. J. Welsh and D. Knight, Biomaterials, 2007, 28, 4171–4177 CrossRef CAS.
- M. A. R. Meier, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 21–33 CrossRef CAS.
- J. C. Meredith, A. Karim and E. J. Amis, MRS Bull., 2002, 27, 330–335 CAS.
- H. Q. Zhang, R. Hoogenboom, M. A. R. Meier and U. S. Schubert, Meas. Sci. Technol., 2005, 16, 203–211 CrossRef CAS.
- M. A. R. Meier and U. S. Schubert, J. Mater. Chem., 2004, 14, 3289–3299 RSC.
- P. G. Schultz and X. D. Xiang, Curr. Opin. Solid State Mater. Sci., 1998, 3, 153–158 CrossRef CAS.
- J. Genzer and R. R. Bhat, Langmuir, 2008, 24, 2294–2317 CrossRef CAS.
- W. F. Maier, K. Stowe and S. Sieg, Angew. Chem., Int. Ed., 2007, 46, 6016–6067 CrossRef CAS.
- D. C. Webster, Macromol. Chem. Phys., 2008, 209, 237–246 CrossRef CAS.
- D. C. Webster, B. J. Chisholm and S. J. Stafslien, Biofouling, 2007, 23, 179–192 CrossRef CAS.
- I. Yoshihiro, Polym. Adv. Technol., 2004, 15, 3–14 CrossRef.
- S. Brocchini, K. Kames, V. Tangpasuthadol and J. Kohn, J. Am. Chem. Soc., 1997, 119, 4553–4554 CrossRef CAS.
- S. Brocchini, K. James, V. Tangpasuthadol and J. Kohn, J. Biomed. Mater. Res., 1998, 42, 66–75 CrossRef CAS.
- J. C. Meredith, A. Karim and E. J. Amis, Macromolecules, 2000, 33, 5760–5762 CrossRef CAS.
- J. C. Meredith, J. L. Sormana, B. G. Keselowsky, A. J. Garcia, A. Tona, A. Karim and E. J. Amis, J. Biomed. Mater. Res., A, 2003, 66, 483–490.
- P. Zapata, J. Su, A. J. Garcia and J. C. Meredith, Biomacromolecules, 2007, 8, 1907–1917 CrossRef CAS.
- F. J. Sung, J. Su, J. D. Berglund, B. V. Russ, J. C. Meredith and Z. S. Galis, Biomaterials, 2005, 26, 4557–4567 CrossRef.
- J. L. Sormana and J. C. Meredith, Macromolecules, 2004, 37, 2186–2189 CrossRef CAS.
- G. Wingkono and J. C. Meredith, in Polymers for Biomedical Applications, ed. A. Mahapatro and A. S. Kulshrestha, American Chemical Society, Washington DC, 2007, ACS Symposium Series vol. 977, pp. 299–309 Search PubMed.
- N. D. Gallant, K. A. Lavery, E. J. Amis and M. L. Becker, Adv. Mater., 2007, 19, 965–969 CrossRef CAS.
- H. Pham-Tuan, L. Kaskavelis, C. A. Daykin and H.-G. Janssen, J. Chromatogr., B, 2003, 789, 283–301 CrossRef CAS.
- L. A. Kunz-Schughart, J. P. Freyer, F. Hofstaedter and R. Ebner, J. Biomol. Screening, 2004, 9, 273–285 CrossRef CAS.
- O. C. Farokhzad, A. Khademhosseini, S. Y. Yon, A. Hermann, J. J. Cheng, C. Chin, A. Kiselyuk, B. Teply, G. Eng and R. Langer, Anal. Chem., 2005, 77, 5453–5459 CrossRef CAS.
- A. Khademhosseini, R. Langer, J. Borenstein and J. P. Vacanti, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2480–2487 CrossRef CAS.
- A. Khademhosseini, J. Yeh, G. Eng, J. Karp, H. Kaji, J. Borenstein, O. C. Farokhzad and R. Langer, Lab Chip, 2005, 5, 1380–1386 RSC.
- E. Figallo, C. Cannizzaro, S. Gerecht, J. A. Burdick, R. Langer, N. Elvassore and G. Vunjak-Novakovic, Lab Chip, 2007, 7, 710–719 RSC.
- P. J. Hung, P. J. Lee, P. Sabounchi, R. Lin and L. P. Lee, Biotechnol. Bioeng., 2005, 89, 1–8 CrossRef CAS.
- J. C. G. Simon, J. S. Stephens, S. M. Dorsey and M. L. Becker, Rev. Sci. Instrum., 2007, 78, 072207 CrossRef.
- S. B. Kennedy, N. R. Washburn, C. G. Simon and E. J. Amis, Biomaterials, 2006, 27, 3817–3824 CrossRef CAS.
- Y. Mei, T. Wu, C. Xu, K. J. Langenbach, J. T. Elliott, B. D. Vogt, K. L. Beers, E. J. Amis and N. R. Washburn, Langmuir, 2005, 21, 12309–12314 CrossRef CAS.
- J. R. Smith, V. Kholodovych, D. Knight, W. J. Welsh and J. Kohn, QSAR Comb. Sci., 2005, 24, 99–113 Search PubMed.
- J. R. Smith, D. Knight, J. Kohn, K. Rasheed, N. Weber, V. Kholodovych and W. J. Welsh, J. Chem. Inf. Comput. Sci., 2004, 44, 1088–1097 CrossRef CAS.
- A. V. Gubskaya, V. Kholodovych, D. Knight, J. Kohn and W. J. Welsh, Polymer, 2007, 48, 5788–5801 CrossRef CAS.
- N. R. Washburn, K. M. Yamada, C. G. Simon, S. B. Kennedy and E. J. Amis, Biomaterials, 2004, 25, 1215–1224 CrossRef CAS.
- C. G. Simon, N. Eidelman, S. B. Kennedy, A. Sehgal, C. A. Khatri and N. R. Washburn, Biomaterials, 2005, 26, 6906–6915 CrossRef CAS.
- Y. Mei, J. T. Elliott, J. R. Smith, K. J. Langenbach, T. Wu, C. Xu, K. L. Beers, E. J. Amis and L. Henderson, J. Biomed. Mater. Res., A, 2006, 79, 974–988 CrossRef.
- D. G. Anderson, W. Peng, A. Akinc, N. Hossain, A. Kohn, R. Padera, R. Langer and J. A. Sawicki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16028–16033 CrossRef CAS.
- D. G. Anderson, C. A. Tweedie, N. Hossain, S. M. Navarro, D. M. Brey, K. J. Van Vliet, R. Langer and J. A. Burdick, Adv. Mater., 2006, 18, 2614–2618 CrossRef CAS.
- J. J. Green, E. Chiu, E. S. Leshchiner, J. Shi, R. Langer and D. G. Anderson, Nano Lett., 2007, 7, 874–879 CrossRef CAS.
- J. J. Green, J. Shi, E. Chiu, E. S. Leshchiner, R. Langer and D. G. Anderson, Bioconjugate Chem., 2006, 17, 1162–1169 CrossRef CAS.
- G. T. Zugates, D. G. Anderson, S. R. Little, I. E. B. Lawhorn and R. Langer, J. Am. Chem. Soc., 2006, 128, 12726–12734 CrossRef CAS.
- G. T. Zugates, W. D. Peng, A. Zumbuehl, S. Jhunjhunwala, Y. H. Huang, R. Langer, J. A. Sawicki and D. G. Anderson, Mol. Ther., 2007, 15, 1306–1312 CrossRef CAS.
- P. J. Lee, N. Ahmad, R. Langer, S. Mitragotri and V. P. Shastri, Int. J. Pharm., 2006, 308, 33–39 CrossRef CAS.
- S. J. Stafslien, J. A. Bahr, J. M. Feser, J. C. Weisz, B. J. Chisholm, T. E. Ready and P. Boudjouk, J. Comb. Chem., 2006, 8, 156–162 CrossRef CAS.
- B. J. Chisholm and D. C. Webster, J. Coat. Technol. Res., 2007, 4, 1–12 Search PubMed.
- D. C. Webster, J. Bennett, S. Kuebler, M. B. Kossuth and S. Jonasdottir, J. Coat. Technol., 2004, 1, 34–39 Search PubMed.
- B. J. Chisholm, D. A. Christianson and D. C. Webster, Prog. Org. Coat., 2006, 57, 115–122 CrossRef CAS.
- B. J. Chisholm, S. J. Stafslien, D. A. Christianson, C. Gallagher-Lein, J. W. Daniels, C. Rafferty, L. V. Wal and D. C. Webster, Appl. Surf. Sci., 2007, 254, 692–698 CrossRef CAS.
- P. Majumdar, D. A. Christianson, R. R. Roesler and D. C. Webster, Prog. Org. Coat., 2006, 56, 169–177 CrossRef CAS.
- F. Casse, S. J. Stafslien, J. A. Bahr, J. Daniels, J. A. Finlay, J. A. Callow and M. E. Callow, Biofouling, 2007, 23, 121–130 CrossRef CAS.
- S. J. Stafslien, J. A. Bahr, J. W. Daniels, L. V. Wal, J. Nevins, J. Smith, K. Schiele and B. Chisholm, Rev. Sci. Instrum., 2007, 78, 072204 CrossRef.
- R. J. Pieper, A. Ekin, D. C. Webster, F. Casse, J. A. Callow and M. E. Callow, J. Coat. Technol. Res., 2007, 4, 453–461 Search PubMed.
- P. Majumdar, S. Stafslien, J. Daniels and D. C. Webster, J. Coat. Technol. Res., 2007, 4, 131–138 Search PubMed.
- A. Ekin, D. C. Webster, J. W. Daniels, S. J. Stafslien, F. Casse, J. A. Callow and M. E. Callow, J. Coat. Technol. Res., 2007, 4, 435–451 Search PubMed.
- A. Ekin and D. C. Webster, J. Comb. Chem., 2007, 9, 178–188 CrossRef CAS.
- N. J. Lin, L. O. Bailey, M. L. Becker, N. R. Washburn and L. A. Henderson, Acta Biomater., 2007, 3, 163–173 CrossRef CAS.
- N. J. Lin, P. L. Drzal and S. Lin-Gibson, Dent. Mater., 2007, 23, 1211–1220 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |