Synthesis of remarkably stabilized metal nanostructures using polyoxometalates
Bineta
Keita
a,
Tianbo
Liu
b and
Louis
Nadjo
*a
aLaboratoire de Chimie Physique, UMR 8000, CNRS, Electrochimie et Photoélectrochimie, Université Paris-Sud 11, Bâtiment 350, 91405, Orsay, Cedex, France. E-mail: louis.nadjo@lcp.u-psud.fr
bDepartment of Chemistry, Lehigh University, 6 E. Packer Ave., Bethlehem, Pennsylvania 18015, USA
First published on 18th November 2008
Abstract
This report reviews the state of the art as concerns the synthesis of metal nanostructures synthesized with polyoxometalates acting as both reducing and capping agents. Two main strategies appear in the literature: the reduced polyoxometalates, necessary for carrying out the process, can be obtained by photochemical reduction or be synthesized directly with reducing capabilities. This last strategy is recent and is shown to feature a “green chemistry-type” synthesis of metal nanostructures.
Dr Bineta Keita is Directeur de Recherche in the French CNRS, (a position equivalent to full professor) working in the Laboratoire de Chimie Physique at Orsay (Université Paris-Sud 11). Her current research interests concern electrochemistry (fundamental aspects and applications) with a particular focus on polyoxometalate solution and solid state electrochemistry and applications in electrocatalysis, photoelectrocatalysis and bioelectrochemistry. |
Tianbo Liu was born in Beijing, China. He got his BS degree in Chemistry from Peking University, China in 1994, and his Ph.D. degree in Physical Chemistry from Stony Brook University in 1999 (advisor: Benjamin Chu). After spending two more years as a postdoctoral associate at Stony Brook, he joined Brookhaven National Laboratories as a physicist. Since January 2005 he has been an assistant professor at Lehigh University. His recent research interest is in the physical chemistry of complex solution systems, especially the solution behavior and self-assembly of hydrophilic macroions. |
Louis Nadjo is Professor of Physical Chemistry at Paris-Sud University. His current research interests focus on polyoxometalate chemistry, from fundamental mechanistic aspects to applications. His work includes their interaction with proteins, their electrochemical and photoelectrochemical properties in conjunction with their electronic structure, the electrocatalytic and photoelectrocatalytic approaches of their behavior in solution and in the solid state. |
Introduction
The nanometer length scale is currently recognized to impart unique chemical and physical properties on metals, semiconductors and metal oxides.1–9 Realized and putative applications are very numerous with an ensemble of requirements on shape, size and geometry. A non-exhaustive list includes quantum dots10 or quantum computers,11 chemical sensors,12 optical, electronic or magnetic nanodevices,13 chemical and, correlatively, electrochemical catalysis.14 Such diversity of possibilities and needs has generated a plethora of experimental recipes for the synthesis of these nanostructures in search of the control of their shape, size and geometry along with a suitable control of their environment.15 Throughout the literature, it has always been difficult to clearly separate the various parameters that govern the different encountered issues. Specifically, the abundance can be illustrated in the case of transition-metal nanoclusters. While their synthesis from the appropriate metallic salts, per se, is triggered by the redox potential of the selected reductant, the subsequent events, including stabilization, are controlled through a series of recipes which seem to defy a general unified rationale, despite a considerable colloidal literature16 and the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory describing the interparticle interaction and setting down the general stabilization mechanisms of colloidal materials.17 Recently, a careful and detailed study of the formation and stabilization of Ir(0) nanoparticles has permitted the establishment of the first “anion series” of relative nanocluster formation and stabilizing abilities, at least for the Ir(0) nanocluster: [(P2W15Nb3O61)2O]16− (a Brönsted-basic polyoxoanion) > [C6H5O7]3− (citrate trianion) > [–CH2CH(CO2)-]nn− (polyacrylate) > Cl−, with emphasis on a lattice size-matching model.18Generally, two main concerns appear in the literature, regarding either the recovery of precious and/or toxic metals or the preparation of zero valent state metal nanoparticles for both fundamental and application purposes. The presence of organic contaminants, the ineffectiveness of most methods at low concentrations of the organics, the use of drastic conditions including high temperature, the difficult search for environmentally benign conditions, are among the recognized disadvantages encountered in the recovery of metals and the synthesis of metal nanostructures. In the following, these two aspects will be considered, with most emphasis on stabilized metal colloidal nanostructures in solution.
In this context, this report highlights the results of a recent metal nanostructure synthesis strategy that uses polyoxometalates19 as both reducing and capping agents. As a matter of fact, it turns out that selected polyoxometalates can act simultaneously for this dual purpose.
Polyoxometalates (POMs for convenience) constitute a unique class of molecular metal–oxygen clusters, remarkable in several respects: the multitude of their properties based on their sizes, shapes, charge densities and reversible redox potentials; their enormous diversity of structures. Even though there are metal oxide clusters that are neutral or cationic, most POMs are anionic structures constituted of early transition-metal elements in their highest oxidation state, hence their usual designation as early transition-metal–oxygen-anion clusters. These atoms in positive oxidation state constitute the so-called “addenda” atoms. Condensation of MO6 octahedra, mostly with Mz+ = W6+, Mo6+ and V5+, gives isopolyanions or isopoly compounds. Heteropolyanions are obtained when a large number of MO6 octahedra is condensed with a much smaller proportion of another atom in positive oxidation state (the so-called X “heteroatom”). The Keggin structure and the Wells–Dawson structure from which the vast majority of POMs studied to date are primarily derived are sketched in Scheme 1. POMs of these groups are characterized by the M/X ratio where M designates the addenda atom and X the heteroatom. The Keggin structure belongs to the 12/1 series and the Dawson structure to the 18/2 series. POMs are a versatile family of molecular metal-oxide clusters. Of central interest for the present purpose are their redox behaviours. Their oxidized forms may only accept electrons; in contrast, their reduced forms, owing to their electron and proton transfer and/or storage abilities, may behave as donors or acceptors of several electrons without structural change. Such reversible charge transfer ability makes POMs ideal candidates for homogeneous-phase electron exchange reactions. Taking into account the ability of unsubstituted POMs to generate lacunary species and, then, metal cation-substituted derivatives, it appears that an enormous family of compounds might result. As a consequence, their redox behaviours may be very flexible and finely tuned on purpose, by changing smoothly their composition, with a tremendously diverse variety of structures. Application of these properties to the synthesis and stabilization of colloidal nanostructures is reviewed in the following.
 | ||
| Scheme 1 | ||
This report is restricted exclusively to metal nanostructures synthesized with POMs acting as both reducing and capping agents. As a consequence, the nanostructures fabricated with other reductants or directly purchased and, then, capped with a POM and related oxothiometalates will not be described.18a,20 However, among the relevant papers, it is worth emphasizing the important series of syntheses carried out by Maksimov et al.20f, g who used several reducing agents, including H2, CO, NaBH4, NaOOCH, N2H4·H2SO4, for preparing noble metal colloidal solutions and a variety of POMs as stabilizing agents.
Synthesis and encapsulation of metallic nanostructures by POMs
Basic features
In a simplified view, the nanoparticle synthesis step, per se, can be sketched by the following overall equation in the case of a one-electron exchange process resulting in metallic silver:| (reduced POM) + Ag+ → (oxidized POM) + Ag0 | (1) |
Then, in the stabilization process, adsorption of a layer of POM polyanions on the nanoparticle surface provides both charge stabilization and steric stabilization. In addition to the kinetic stabilization through coulombic repulsion between negatively charged particles, the steric stabilization helps also in slowing the rate of particle agglomeration.
Obviously, even such an oversimplified scheme requires that some conditions be fulfilled. A first point of concern is the knowledge of the pH domain where the selected POM is stable in its oxidized and its reduced forms. As a matter of fact, such domains were determined for classical and even commercial POMs including α-[SiW12O40]4−, α-[PW12O40]3−, α-[P2W18O62]6− and α-[H2W12O40]6−. In short, the domains were found to be shifted to more alkaline pH values as the number of added electrons increases.19i,21 Another condition is the adequate match between the redox potentials of the POMs on the one hand and the metal salts on the other hand, in order that the electron exchange can be driven in the appropriate direction. Table 1 permits the comparison of redox potentials of some representative POMs and those of commonly studied metal ions. Even a superficial inspection of this table indicates the thermodynamic possibility to reduce most classical metal ions to the zero state. Stabilization issues will be considered elsewhere.
| Polyoxometalates | Redox potential/V vs SCE | Metal ions | Redox potential/V vs SCE |
|---|---|---|---|
| [H2W12O40]6−/8− | −0.380 (0.5 M H2SO4) | Ni2+/Ni | −0.493 |
| [SiW12O40]5−/6− | −0.393 (1M HClO4) | Cu2+/Cu+ | −0.089 |
| [PW12O40]4−/5− | −0.250 (1M HClO4) | Cu2+/Cu | +0.095 |
| [SiW12O40]4−/5− | −0.193 (1M HClO4) | [PtCl4]2−/Pt | +0.513 |
| [P2W18O62]7−/8− | −0.090 (1M HClO4) | Ag+/Ag | +0.557 |
| [PW12O40]3−/4− | +0.005 (1M HClO4) | Hg2+/Hg | +0.608 |
| [P2W18O62]6−/7− | +0.053 (1M HClO4) | Pd2+/Pd | +0.709 |
| α-[P2Mo18O62]6−/8− | +0.460 (0.5 M HCl) | [AuCl4]−/Au | +0.760 |
In short, it is necessary to generate or to have at one's disposal an appropriate reduced POM to realize the electron transfer step in reaction (1). POMs can be reduced by a large family of reducing chemical reagents or by a variety of techniques, including electrochemistry19i photolysis22 and radiolysis.23 However, for clarity, the POM-based syntheses of metal nanostructures can roughly be considered to follow two main strategies in the literature: the necessary reduced POMs can be obtained by photochemical reduction or be synthesized directly with reducing capabilities.
As will appear in the following, reduction of metallic salts and encapsulation of the resulting nano-objects by POMs not only can give the average size distributions obtained with other reducing agents, but also, in appropriate experimental conditions, can generate various nanostructures, including monodisperse spherical nanoparticles, nanowires, nanoribbons, etc., thus providing a fair generality to this chemistry.
POMs as UV-switchable photocatalysts
The state of the art
Photoreduction of POMs with the possibility of subsequent transfer of the excess electron to other species in solution has been known for several decades.24,25 Specifically, the maximum absorption for the classical POM H3PW12O40 peaks at 266 nm in the UV spectral regime. Even so, it is worth noting that sunlight could be used to carry out its one-electron photoreduction, albeit at a slow rate25 followed by the quantitative reduction and precipitation of silver ion to the metallic silver state by the reduced POM:| [PW12O4O]4− + Ag+ → [PW12O4O]3− + Ag0 |
| S + M+ (oxidized POM + hν) → Sox + M0 (oxidized POM) |
This overall scheme can be split into its two constitutive steps which highlight both the photocatalyst role of the POM and the electron donor ability of S (S is sometimes designated as a sacrificial electron donor):
| hν + (oxidized POM) + S → (reduced POM) + Sox |
| (reduced POM) + M+ → (oxidized POM) + M0 |
In short, this achievement laid the foundations for further developments. Revival and recent expansion along these lines are due to Papaconstantinou et al.26 in the framework of recent depollution, metal recovery and/or material science concerns. They have reviewed their work26a and recently completed it by a patent.26b Therefore, only a short survey of this work is given in the following. It provides a comprehensive study of the interactions of several UV-switched POMs, including [SiW12O40]4−, [PW12O40]3−, [H2W12O40]6−, [P2W18O62]6− and [P2Mo18O62]6− with various metal ions like Ag+, Pd2+, Au3+, Cu2+, Ni2+ or Hg2+. UV through near visible light from a high-pressure Hg or Xe arc lamp was used. Propan-2-ol is the favourite sacrificial electron donor in most studies, even though a large variety of other organics could be used, including phenols, hydride, chlorophenols, cresols, benzene derivatives, organohalogenated compounds, aliphatics or triazine pesticides.22a,26b Both one-pot and two-pot systems proved efficient. Deoxygenated solutions are used throughout. Typically, in a one-pot system, the reagents are premixed in a POM/propan-2-ol/Ag+ solution. Photolysis of this solution results in the in-situ reduction of the POM which, in turn, converts the already present silver cation to metallic silver. It must be noted that the POM acts in a catalytic way. In this case, the question might arise whether any in-situ reduction of the POM actually occurs. Even though spectroscopic evidence for the reduction of the POM was not the focus of these authors, the two-step procedure (vide infra) and the absence of formation of metal nanoparticles in the absence of POM strongly support the proposed stepwise mechanism.26g Alternatively, the POM can be used in a stoichiometric manner in a two-pot system. In that case, irradiation by UV-near-visible light of the POM/propan-2-ol solution results in the formation of the reduced POM characterized by its intense blue color and the corresponding UV-visible spectrum.26g Then mixing of this solution with the Ag+ solution gives the desired Ag0 colloidal nanoparticles. Even though these two procedures could be used whatever the POM, it is clear that the two-pot system is easily handled only when the reduced POM is not sensitive to re-oxidation by dioxygen. In addition, spectrophotometric monitoring of the reduced POM is straightforward in a two-pot system and facilitates the kinetic study of the thermal reaction between the metal cation and the reduced POM. Whichever the technique, the final result is the synthesis of metallic nanoparticles, but differences might appear in the kinetic measurements. In two-pot systems, it was noted that the measured kinetics strictly comply with the order of redox potentials: as a specific illustration, the rate of electron transfer to palladium ions, and for several metal ions,26d,e,f follows thermodynamics, i.e. it depends on the relative reduction potentials of the species involved. Thermodynamics also constitutes the basis for the selective recovery of palladium by [P2Mo18O62]6− in the presence of Ni2+ ions. In contrast, in a one-pot system where both photoreduction and homogeneous electron transfer reactions take place, the efficiency of the photocatalytic recovery of palladium was found to follow the order [PW12O40]3− > [SiW12O40]4− > [H2W12O40]6−. In this case, a kinetic phenomenon was observed, in which although [SiW12O40]5− is a better reductant than [PW12O40]4− [E0([SiW12O40]4−/5−) = −0.193 V; E0 ([PW12O40]3−/4−) = 0.005 V versus SCE] the rate of photoformation of POM (e−) prevails over the rate of delivering the electrons to Pd2+. Thus, POM catalysts that are more efficient in the photodegradation of organic compounds are also more effective in recovery of palladium in the one-pot system. Interesting observations are made in the case of Cu2+ which shows stepwise reductions through a stable or stabilizable intermediate Cu+ to the metallic state [E0 (Cu2+/Cu0) = +0.095 V, E0 (Cu2+/Cu+) = −0.089 V, E0 (Cu+/Cu0) = +0.279 V vs. SCE]. The observed induction period before the start of copper deposition is attributed to the formation of the stable Cu+ intermediate. As a matter of fact, the extent of copper reduction depends on kinetic parameters, that is the competition of the further reduction of the obtained Cu+ to Cu0 with its reoxidation to Cu2+.
Most of the early papers of Papaconstantinou's group are oriented toward the depollution/precipitation/metal recovery issue. The presence of dioxygen is usually found to retard metal ion precipitation and is therefore avoided. However, most metal cations are more efficient electron scavengers than dioxygen in the competition for capturing electrons from reduced POMs. As a consequence, the slight detrimental role of dioxygen is mainly attributed to the re-oxidation of the intermediates obtained upon the reduction of metal ions, for example Cu+ or colloidal Ag0. Increase of the concentration of either POM or organic or metal ions leads to an increase of the rate of metal recovery, however up to a saturation value. The metal recovery process is effective in a wide range of metal concentration, varying from ca. 3 to 1300 ppm, thus accounting for the environmentally useful assessment of the process. Last, but not least, the composition of the solution in terms of anions present and ionic strength constitutes an extremely important operational parameter. Anions with complexing and/or stabilizing abilities toward the metal cations will modify importantly the reduction potentials of ions and affect strongly the efficiency of their electron transfer with reduced POMs. The ionic strength will have a determining influence by affecting both the rate and also the nature of the metal product obtained, thus favoring precipitation or the formation of colloidal metal nanoparticles. At high ionic strength values (> 3 × 10−2 M) elemental metals precipitate. However, at low values (< 10−2 M) a slower reduction of metal ions takes place, resulting in the formation of colloidal metal particles.26a It thus appears that a clever choice of operational parameters drives the overall process toward the precipitation/recovery of metals or, alternatively, the synthesis of colloidal metal nanoparticles. In suitable ionic strength conditions, the size of the synthesized nanoparticles was controlled by manipulating the following parameters: i) changing the reaction rate through variation of the initial concentration of reduced POM, the kind of POM or the extent of reduction of the same POM; ii) varying the amount of metal cation. These guidelines proved successful and metal nanoparticles with average diameter less than ca. 15 nm and small size distribution (< 25%) have been formed from Ag+, Pd2+, Au3+ and Pt4+.26b,j The authors provide four indirect lines of evidence in support of the idea that the restriction of the size of the metal particles to the nanoscale is due to the stabilizing role of the POM:26j a) the colloidal solution contains no other potential stabilizing reagent but POM, b) the volume of 12-tungstosilicate (considered to be spherical, with a diameter of about 12 Å) and its (4−) negative charge could account for the prevention of agglomeration of the metal particles, as for other POM; c) the stabilizing role of POM in organic systems (acetone or acetonitrile solutions) has been reported in a process in which H2 was the reducing agent for Ir or Rh complexes, while the POM {[Bu4N]9[P2W15Nb3O62]} functioned solely as a stabilizer. In addition, TEM, ion-exchange, and electrophoresis experiments revealed that POMs attach to the surface of Ir0 or Rh0 nanoclusters,18a,20a and d) the ability of [SiW12O40]4− not only to attach but also to self-assemble onto Ag(111) or Au(111) surfaces has been demonstrated.20b,27 Addition of NaClO4 0.1M into the colloidal solutions leads to immediate precipitation in all cases.26j
Sastry and coworkers caught hold of the same basic procedures to generate nanostructures with increased complexity28,29 and solvent adaptable nanostructures.30 A particularly elegant multi-step synthesis of Au core–Ag shell nanoparticles was achieved.28 The first step consisted of irradiating [PW12O40]3− in the presence of propan-2-ol in a pH = 2.5 solution, which results in the photoreduction of the POM. Addition of a solution of HAuCl4 under continuous stirring gives polydisperse POM-capped gold nanoparticles which were separated by dialysis from unreacted POM. The POM-Au particles were again irradiated before addition of an Ag2SO4 solution. The whole procedure results in the synthesis of Au core–Ag shell nanoparticles as confirmed by several analysis techniques. Provisionally, this experiment confirms the capping of the synthesized nanoparticles by POMs. The nanoparticles are polydisperse, with a size ranging from 15 to 70 nm and are of irregular morphology. Also, organized silver nanoparticle assemblies could be obtained by forming first an aqueous colloidal solution of Ag+ particles complexed with phosphotungstate [PW12O40]3− Keggin ions and showing that they may be used as a new class of inorganic scaffolds in the synthesis of silver nanoparticle assemblies.29 Specifically, the Keggin POM served both as a UV-switchable reducing agent for Ag+ and as a support for the resulting organized silver nanostructures. Treatment with alkali results in dissolution of the colloidal Keggin particle template leaving behind the silver nanoparticle network reasonably intact. Another paper describes the synthesis of gold nanosheets in a constrained environment.31 In that case, anthracene anions generated by electron transfer from UV light irradiation reduced [PW12O40]3− were used to reduce chloroaurate ions at a water–chloroform interface. Finally, in another example, advantage was taken of the overall negative charge imparted on Keggin ion capped Pd nanoparticles to change their environment and fabricate a truly multifunctional catalyst dispersible in organic solvents.30 In short, Pd nanoparticles were synthesized in aqueous medium by reduction of Pd(NO3)2 by photoexcited phosphotungstate anion and then transferred into non-polar organic solvents such as toluene by electrostatic complexation with cationic surfactants such as octadecylamine at the liquid–liquid interface. The Pd nanoparticles are irregularly shaped and rather polydisperse in size (average diameter: 9 ± 2 nm). However, the variety of achievements realized solely with the phosphotungstate Keggin anion suggests a wealth of possibilities which could be associated with the wealth and variety of POMs.
A POM-based reduction technique for the preparation of noble metal supported carbon was also devised.32 Specifically, a gel made with polyvinyl alcohol, water, tetraethylorthosilicate and containing silicotungstic acid was coated on a glass plate and exposed to sunlight for 10–30 min. Photoreduction of the POM ensues, as evidenced by the color change from colorless to blue. The reduced composite was soaked in a medium containing H2PtCl6 and Vulcan XC 72 carbon. The POM was reoxidized to its colorless form with concomitant reduction of platinum to the zero valent state and fixation of the nanoparticles on the carbon powder. The TEM image shows triangular, hexagonal, pentagonal, square, fused pentagonal and star like large size particles. The Pt/C composite was evaluated for methanol oxidation in perchloric acid and in sulfuric acid media and found to exhibit higher activity and stability in the former medium.
A series of papers by Ershov et al. are also devoted to the synthesis of metal nanoparticles using γ-irradiation in the presence of two Keggin-type [PW12O40]3−, [PW11O39]7− and two Dawson-type [P2W18O62]6− or [P2W17O61]10− POMs.33–35 The reaction scheme follows the same basic lines as those for UV irradiation, i.e. in the presence of sodium formate or propan-2-ol, the POMs are reduced into the corresponding blues. Subsequently and depending on their respective reduction potentials, these blues reduce a variety of metal cations, including silver, copper, cadmium, thallium, lead, cobalt and nickel, to form nanoparticles stabilized by the presence of the POMs. It is noteworthy that the lacunary species [PW11O39]7− and [P2W17O61]10− proved also to be efficient stabilizing agents.33 Typically, coarse silver nanostructures with an average size about 40–60 nm were obtained.35 The effect of adding NaClO4 on the stability of the POM-stabilized colloidal silver in an evacuated solution was studied. The attenuation and broadening of the silver spectrum was interpreted as likely due to gradual particle flocculation upon stepwise addition of the electrolyte. Subsequent introduction of air in a solution containing 2 M NaClO4 resulted in the coagulation of the colloids.33 This high energy radiation synthesis features particularly harsh conditions as evidenced by the following observations. Specifically in the case of Keggin anions [PW12O40]3− and [PW11O39]7−, the blues contain up to six additional electrons upon γ-irradiation. Upon continued irradiation of the acidic solutions (pH 2.0), a maximum concentration of the blues was attained. Thereafter, the blues decomposed as a result of the deep irreversible reduction of the POMs. The blues formed in a solution with a pH 4.7 were stable to irradiation.
A feature noticeable from the results described so far is that regularly shaped, small size, monodisperse nanostructures are uncommon.
An example of “giant” silver structures obtained in acetonitrile
Finally, an example of “giant” silver structures obtained in acetonitrile in a one-step synthesis by POMs36 without UV irradiation switching was published recently. Three polyoxomolybdovanadates were shown to generate silver crystalline ribbons and saws upon addition of silver cation salts in acetonitrile solutions. The three types of POMs differ in the location and the number of vanadium centres: H(VO)[PMo12O40] (denoted VOPMo12) containing one vanadyl counter-cation, [N(Bu)4]5[PMo11VO40] (denoted PMo11V) in which the vanadium(IV) centre is part of the Keggin structure, and a mixture of the lacunary Keggin ion Na3[H6PMo9O34] (denoted PMo9) with three equivalents of vanadyl acetylacetonate in order to form in situ the vanadium trisubstituted Keggin anion. Following the mixing of silver salts with POMs/vanadium (iv) systems in acetonitrile, the precipitate that formed rapidly was filtered off, and the filtrate was allowed to stay undisturbed for 2–3 days. Silver 1D nano- and micro-structures appear in the solution. The presence of both vanadium(IV) and of the POM structure is necessary for reduction and for orienting toward 1D structures. With VOPMo12, the metallic 1D microstructures were visible to the naked eye, which underscores their size. SEM analysis revealed 1D microribbons with a width of about 4 µm and an irregular surface covered with a thin skin. In the case of [N(Bu)4]5[PMo11VO40], TEM analysis revealed the formation of ribbons by coalescence of nanoparticles and several fibrous structures containing metallic nanoparticles and POMs. Finally, no noticeable modification of the final structure was observed in the synthesis with VOPMo12 or PMo11V. In short, 1D structures with micrometer length and nanometer thickness were obtained. The third system contains a higher proportion of vanadyl per POM. Only ribbons were obtained, with length and width similar to those observed in the two previous systems. EDX cartography (Ag,Mo,V) showed the presence of POM on the silver 1D structure. Crystallization of these systems in the dark modified noticeably their morphologies as, in this case, only saws were observed. With an excess of silver acetate, bundles of ribbons with saws on both sides of the ribbons were obtained. If crystallization was realized at 4 °C, only silver nanoparticles (NPs) were obtained, with a diameter between 3 and 12 nm. This last experiment showed the crucial role of the temperature on the formation of these 1D nanostructures. Obviously, the solvent influences heavily the observed silver nanostructures.Energetic considerations
In summary, for practical purposes and due to widespread availability of appropriate lamps, UV-near visible irradiation of POMs for the synthesis of metal nanostructures might be preferred to γ-irradiation. Among lessons learnt from the use of POMs as UV-switchable reductants, special attention must be devoted to the important influence of ionic strength, which must be kept at a very low level for a good limitation of particle size to the nanoscale to be obtained. Then, a difficult compromise should be found as concerns the reaction media to ensure also the stability of the specific POM in its oxidized and reduced forms. Even so, to the best of our knowledge, very small size nanostructures obtained in the presence of photoreduced classical POMs were not described. However, an exception might be made for cases such as silver for which very small particles might be re-oxidized by dioxygen. Another general drawback concerns the energetics of the primary photoreduction step of the POM in its excited state:| hν + (oxidized POM) + S → (reduced POM) + Sox |
As a matter of fact, the redox potentials of POMs can be evaluated in the excited state simply by adding the light energy that corresponds to the 0–0 energy transition, that is ca. 350 nm,26d at the redox potential of the ground state POM. As examples, the following values were obtained for usual POMs: POM* [E0((PW12O40)3−*/(PW12O40)4−*) = +3.52V, E0((SiW12O40)4−*/(SiW12O40)5−*) = +3.36 V and E0((H2W12O40)6−*/(H2W12O40)7−*) = +2.96 V versus SCE]. These values are more positive than that necessary to obtain OH radicals from water-solvent molecules [E0(OH/H2O) = +2.56 V versus SCE].37 Altogether, these potentials indicate that the primary photoreduction step of POMs involves highly strong oxidants. The comparison with the much lower redox potentials which trigger the subsequent thermal reaction of reduced POMs with metal cations clearly indicates that energy-saving routes should be looked for. In a complementary line of reasoning, it should be noted that even POM photoreductions driven by sunlight utilize only a small fraction of the available energy.
One step “green chemistry-type” synthesis of metal nanostructures
A few guidelines
In essence, the problem is to devise energy-saving experimental conditions which permit the preparation of stable colloidal metal nanostructures through the overall following thermal reaction:| (reduced POM) + M+ → (oxidized POM) + M0 |
Loosely speaking, these few simple guidelines might be considered to constitute the basis for introducing a green chemistry-type synthesis of metal nanoparticles. The aim to save energy guides toward the synthesis or selection of POMs with “built-in” reduction capabilities. In other words, efforts will be directed toward the synthesis and/or selection of POMs in which one or several addenda atoms or substituent centers are not in their highest oxidation state and could thus participate in electron donating events. A complementary beneficial outcome of this strategy is that such species are likely to be used in pure water, in agreement with the known propensity of reduced POMs to be stable in higher pH media than their oxidized forms.19i,21 The absence of any additional electrolyte in the synthesis medium will be very useful for circumventing part of the ionic strength issue. In favorable cases, the generation of few or no byproducts is another advantage. Finally, it is also desirable that no blocking effect toward expected catalytic processes be exerted by the capping layer on the nanoparticle surface. In favorable cases, even a synergistic effect can be expected.
Synthesis and characterization of metal nanostructures
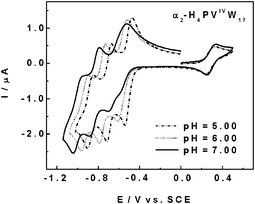 | ||
| Fig. 1 Cyclic voltammograms (CVs) of 2 × 10−4 M α2-H4PVIVW17 as a function of the pH of the supporting electrolyte. The scan rate was 2 mV s−1, the working electrode was glassy carbon, and the reference electrode was SCE. For further details, see text (from Fig. 2, ref. 40). | ||
Typically for nanoparticle synthesis, 0.5 mM α2-[H4PVIVW17O62]9− (E0′ = 0.291 V vs SCE at pH 7) was coupled with K2PdCl4 (E0′([PdCl4]2−/Pd0) = 0.350 V vs SCE)42 in aqueous solutions. The synthesis was monitored by UV-Vis spectroscopy as sketched in Fig. 2. The large dot line represents the spectrum of 0.5 mM α2-[H4PVVW17O62]8− in water. It is worth noting that no significant absorption was observed for this VV form for a wavelength larger than 500 nm. The small dot line represents the spectrum of 0.5 mM α2-[H4PVIVW17O62]9− in water. Upon addition of the amount of [PdCl4]2− appropriate to prepare a solution 0.9 mM in palladium salt, the formerly blue color of the POM solution turned immediately to dark brown. The full line curve showing significant absorption increase at longer wavelengths corresponds to complete consumption of α2-[H4PVIVW17O62]9−. This final spectrum is assigned, in most of its long wavelength region, to the plasmon resonance absorbance of Pd0 nanoparticles43 and in its shorter wavelength part to a superposition of the spectra of the remaining [PdCl4]2− and the generated α2-[H4PVVW17O62]8−. From the TEM image in Fig. 3, a narrow distribution of nanoparticle sizes around 3 nm was determined. These observations indicate a very efficient stabilization of the metal nanoparticles by the POM. Several cross-check experiments by coupling a variety of POMs from Table 2 with different metal salts suggest that the electron transfer process follows thermodynamics.
![UV-vis absorption spectra of the following solutions: Curve 1: 0.5 mM α2-H4PVVW17 in water; Curve 2: 0.5 mM α2-H4PVIVW17 in water; Curve 3: 0.5 mM α2-H4PVIVW17 in water after addition of 0.9 mM of [PdCl4]2− and completion of the redox process (from Fig. 3, ref. 40).](/image/article/2009/JM/b813303d/b813303d-f2.gif) | ||
| Fig. 2 UV-vis absorption spectra of the following solutions: Curve 1: 0.5 mM α2-H4PVVW17 in water; Curve 2: 0.5 mM α2-H4PVIVW17 in water; Curve 3: 0.5 mM α2-H4PVIVW17 in water after addition of 0.9 mM of [PdCl4]2− and completion of the redox process (from Fig. 3, ref. 40). | ||
 | ||
| Fig. 3 Transmission electron micrograph of Pd0 nanoparticles generated in the solution corresponding to curve 3 (full line) of Fig. 2 (from Fig. 4, ref. 40). | ||
| POM (with reduced centers) + Mn+ → POM (with oxidised centers) + M0colloïdal |
![Polyhedral representation of H7[β-P(MoV)4(MoVI)8O40] (1), (NH4)10[(MoV)4(MoVI)2O14(O3PCH2PO3)2(H2OPCH2PO3)2]·15H2O (2) and εP(MoV)8(MoVI)4O36(OH)4{La(H2O)2.5Cl1.25}4·27H2O (3).](/image/article/2009/JM/b813303d/b813303d-s2.gif) | ||
| Scheme 2 Polyhedral representation of H7[β-P(MoV)4(MoVI)8O40] (1), (NH4)10[(MoV)4(MoVI)2O14(O3PCH2PO3)2(H2OPCH2PO3)2]·15H2O (2) and εP(MoV)8(MoVI)4O36(OH)4{La(H2O)2.5Cl1.25}4·27H2O (3). | ||
The selected POMs are very stable in pure Millipore water and all the nanoparticle syntheses were performed in this medium. The choice of pure water allows to generate as small size particles as possible by keeping the ionic strength low.26a,b In a typical experiment, a mixture containing 1 mM of Pt2+ and 0.5 mM of 1 was assembled in water. In this example, the excess parameter γ = [metallic salt]/[POM] is equal to 2 (γ is defined as the ratio of the concentration of metallic salt to that of the relevant POM, irrespective of the number of reduced centers initially present in the POM). The progress of the reaction is followed by UV-visible spectroscopy. The characteristic absorption band of the MoV centers of 1 decreases steadily while the surface plasmon resonance (SPR) spectrum of Pt0 appears. This SPR spectrum is directly observable with most POMs and develops essentially as an increase in the Mie scattering through the visible region toward the UV domain. Finally, the MoV band vanishes completely. The spectral features attributed to the Pt nanoparticles correspond well to those seen experimentally.48 Complete vanishing of the MoV band was encountered when the same experiment was carried out with γ ≥ 2, but not with γ = 1. Therefore, it must be concluded that all the four MoV centres initially present in 1 are actually oxidised, as soon as γ = 2. The same studies with analogous observations were made with 2 and 3 as reductants used with several γ values. Examples of UV-visible spectra evolution are shown in Fig. 4 with the corresponding TEM images. Several operational parameters were tested. The faster reductions were observed for 3, accompanied by a tendency to faster precipitation compared with the other oxometalates. With the absence of precipitate for more than 3 months in nanoparticle solutions prepared from 1, the following stabilisation ability of the present POMs could be deduced: 1 > 2 ≫ 3. In short, 1 and 2 impart considerable stability on Pt0 and Pd0 colloïdal solutions. Table 3 gathers the average diameters measured for nanoparticles prepared using the three oxometalates.
| POM | Metal | Average size/nm |
|---|---|---|
| 1 | Pt | 1.7–2 |
| 2 | Pt | 1.4–2 |
| 3 | Pt | 1.7–2 (γ = 1) |
| 2.5–4 (γ = 2) | ||
| 1 | Pd | 2–5 |
| 2 | Pd | 3–5 |
| 3 | Pd | 12–15 |
![TEM images of Pt0 (left) and Pd0 (right) nanoparticles synthesised in water at room temperature using 1 as the reductant. The associated evolution of the UV-visible spectra run on the solutions as a function of time is also shown during Pt0 and Pd0 synthesis respectively. For further details, see text (from Fig. 1, ref. 44). (A) 1 0.5mM, Pt2+ 1mM. (B) 1 0.5mM [PdCl4]2− 1mM.](/image/article/2009/JM/b813303d/b813303d-f4.gif) | ||
| Fig. 4 TEM images of Pt0 (left) and Pd0 (right) nanoparticles synthesised in water at room temperature using 1 as the reductant. The associated evolution of the UV-visible spectra run on the solutions as a function of time is also shown during Pt0 and Pd0 synthesis respectively. For further details, see text (from Fig. 1, ref. 44). (A) 1 0.5mM, Pt2+ 1mM. (B) 1 0.5mM [PdCl4]2− 1mM. | ||
XPS analysis (Fig. 5) confirms that Pt0 and Pd0 were indeed synthesized. Importantly, in both types of sample, the presence of molybdenum was detected, albeit with a much less important concentration in Pt preparations which have been washed. This observation supports the hypothesis that the polyoxometalate serves both as a reductant and as a protective layer, in agreement with the propensity, particularly of Mo-based POMs, to self-assemble on metal and other solid surfaces.20b,27 Cyclic voltammetry confirmed the XPS observation as follows. Typically, a few µl of Pt0 nanoparticle suspension in water synthesized with 1 as reductant was deposited on a polished glassy carbon (GC) electrode surface and allowed to dry in the air at room temperature. Then the appropriate amount of 5 wt% Nafion solution was added and left to dry in the air. Fig. 6 shows the cyclic voltammetry response in 0.5 M H2SO4 solution (pH 0.33) of the Pt0-modified GC electrode. The scan rate was 50 mV s−1 throughout. In addition to the hydrogen adsorption–desorption characteristics observed between +0.1 V and −0.2 V vs SCE, two redox couples are observed between +0.100 and +0.350 V vs SCE and are associated with the MoVI centers of 1. The presence of these waves, even after three washing and centrifugation cycles of the nanoparticles, is in agreement with XPS analysis. Even though the ratio between the attached Mo-based POM to nanoparticles might vary with both the metal and the starting POM, it is tentatively assumed that the ratio obtained by comparison of the charge of Mo waves with that of hydrogen adsorption–desorption pattern on Pt nanoparticles might allow a qualitative assessment. This hypothesis puts the smallest charge ratios observed around (8 ± 2) %.
 | ||
| Fig. 5 Deconvolution of the platinum core 4f level XPS spectrum (upper) and of palladium core 3d level XPS spectrum (lower) (from Fig. 2, ref. 44). | ||
 | ||
| Fig. 6 Cyclic voltammogram obtained with the Pt0-modified GC electrode in 0.5 M H2SO4; scan rate: 50 mV s−1; the reference electrode was a SCE; the counter electrode was a platinum gauze of large surface area (from Figure SI2, ref. 44). | ||
 | ||
| Fig. 7 SPR spectra of Ag nanoparticles obtained from different molar ratios between Ag2SO4 and 2 (a), a representative TEM image of Ag nanoparticles obtained from the mixture with γ = 4 (b), size histogram of Ag nanoparticles of about 200 NPs counted from TEM image showing the distribution of Ag NPs (c), a magnified Ag nanoparticle (d) (from Fig. 1, ref. 49). | ||
Silver cation reduction by 1 proceeds relatively slowly and the observed structures strongly depend on the aging time of the mixture. Note the appearance of irregularly shaped and pentagonal symmetry shaped Ag nanoparticles over a period of several hours when the shape evolution continues. In the long run, after roughly 6h aging of the mixture, TEM analysis of the mixture shows that more than 95% of the image represent long nanowires, with an average diameter of 40 nm and a length of several tens of micrometers (Fig. 8(upper)). The aspect ratio of the nanowires ranges from 300 to more than 1000. Observation of nanocrystals with pentagonal symmetry suggests that nanowires grow along the <111> direction. Fig. 8(lower) shows a magnified picture of the nanowires, with a clearly suggested Ag@POM core–shell structure. Upon thorough washing of the nanowires with water, the shell thickness decreases to only about 1nm, just matching the molecular dimension of 1. The electronic diffraction (ED) pattern of the nanowires, shown in the inset of Fig. 8(lower), demonstrates their high crystallinity. XPS analysis indicates unambiguously that silver is present only in the metallic form (Fig. 9). Detection of molybdenum during XPS analysis reinforces the conclusion from TEM images that an Ag@POM core–shell structure is formed. In the case of the present Ag nanowires, a semi-quantitative analysis of the sample, with the respective surface areas of the peaks corrected by the appropriate Scofield sensitivity factors, gives an atomic percentage of 21.7 % for Mo and 78.3 % for Ag. It is worth noting that Mo(VI) and a smaller amount of Mo(V) are simultaneously detected in the shell. Such an example suggests that POMs are likely to induce various shapes and geometries in the synthesis of metal nanostructures, as do classical surfactants.
 | ||
| Fig. 8 (Upper) Large scale TEM image of nanowires got from the reaction mixture of 1 after 4h aging (from Figure 4SI, ref. 49); (lower) magnified nanowires with associated electronic diffraction (from Fig. 2b, ref. 49). | ||
 | ||
| Fig. 9 Deconvolution of the silver core 3d levels XPS spectrum (from Fig. 3, ref. 49). | ||
![Structural representation of [Mo3S4(Hnta)]2− (from Fig. 1, ref. 50).](/image/article/2009/JM/b813303d/b813303d-f10.gif) | ||
| Fig. 10 Structural representation of [Mo3S4(Hnta)]2− (from Fig. 1, ref. 50). | ||
![Evolution, as a function of time, of the UV-visible spectrum of a mixture of Na2[Mo3(µ3S)(µS)3(Hnta)3] (C° = 0.5 mM) and HAuCl4 0.5 mM (γ = 1). The spectra of the individual species are superimposed for comparison. The quartz cell has 2 mm optical path (from Figure S1, ref. 50).](/image/article/2009/JM/b813303d/b813303d-f11.gif) | ||
| Fig. 11 Evolution, as a function of time, of the UV-visible spectrum of a mixture of Na2[Mo3(µ3S)(µS)3(Hnta)3] (C° = 0.5 mM) and HAuCl4 0.5 mM (γ = 1). The spectra of the individual species are superimposed for comparison. The quartz cell has 2 mm optical path (from Figure S1, ref. 50). | ||
![A) TEM images of Au0 nanoparticles synthesized by the reduction of HAuCl4 with Na2[Mo3(µ3S)(µS)3(Hnta)3]. The concentration ratio γ = [HAuCl4]/[Oxothiometalate] was 1. The initial concentration of the oxothiometalate was 0.5 mM. B) XPS analysis: deconvolution of the gold core 4f level XPS spectrum. C) XPS analysis: deconvolution of the Mo core 3d level XPS spectrum and the sulfur 2s level. D) XRD analysis of the Au0 nanoparticles (from Fig. 2, ref. 50).](/image/article/2009/JM/b813303d/b813303d-f12.gif) | ||
| Fig. 12 A) TEM images of Au0 nanoparticles synthesized by the reduction of HAuCl4 with Na2[Mo3(µ3S)(µS)3(Hnta)3]. The concentration ratio γ = [HAuCl4]/[Oxothiometalate] was 1. The initial concentration of the oxothiometalate was 0.5 mM. B) XPS analysis: deconvolution of the gold core 4f level XPS spectrum. C) XPS analysis: deconvolution of the Mo core 3d level XPS spectrum and the sulfur 2s level. D) XRD analysis of the Au0 nanoparticles (from Fig. 2, ref. 50). | ||
A related approach was also described recently. With the aim to avoid the presence of organic catalysts, Maayan and Neumann53 coupled two redox reactions to synthesize Ag, Ru, Rh, Ir and Pt nanoparticles stabilized by H5PV2Mo10O40. The sequence of reactions reads:

The Mn-POM nanoparticles were then characterized and supported by wet impregnation of various supports for use as oxidation catalysts.
| CH3OH + H2O → CO2 + 6H+ + 6e− |
 | ||
| Fig. 13 Cyclic voltammograms obtained with the Pt0-modified GC electrode in 0.5 M H2SO4; scan rate: 50 mV s−1: dotted line curve obtained in the pure supporting electrolyte; full line curve was obtained in the presence of 0.5 M MeOH; the reference electrode was SCE; the counter electrode was a platinum gauze of large surface area (from Fig. 3, ref. 44). | ||
The voltammogram obtained in the absence of MeOH is superimposed (dotted line curve) for comparison. For the methanol oxidation voltammetric pattern, the current intensity increases between the first and second voltammetric runs and then stabilises. No current decrease is observed for more than 100 cycles between −0.2 V and +0.9 V vs SCE. During this cycling, the peak potential for the forward process remains located between +0.600 and +0.620 V vs SCE while the peak potential for the reverse scan experiences a very slight negative shift, but remains between +0.475 and +0.465 V vs SCE, in favourable comparison with the best literature results.56 In addition, this Mo-stabilised Pt0 was resistant to poisoning during methanol oxidation. Note that thicker films than shown in Fig. 13 could be deposited on the electrode surface without any deleterious consequence on the observed voltammetric characteristics. Ethanol oxidation in alkaline medium with a Pd0-modified glassy carbon electrode was also successful.
| [PdCl4]2− + 2[H4 PVIVW17O62]9− = Pd0 + 2[H4 PVVW17 O62]8− + 4Cl− |
The starting molar ratio of K2PdCl4 to HPVIV (R ≡ γ value) in solution is important for the formation of Pd nanoparticles. When R < 0.6, ∼20 nm radius Pd0 colloidal nanocrystals are formed. When R ≥ 0.6, the HPV-capped (therefore negatively charged) 3 nm radius Pd0 nanoparticles are formed.
Interestingly, the POM-capped Pd0 nanoparticles, although quite soluble in aqueous solution, do not exist as discrete macroions. Instead, they tend to further self-assemble into stable, large, spherical 30–50 nm radius supramolecular structures in solution, as confirmed by laser light scattering and TEM studies (Fig. 14).57 Static light scattering (SLS) studies indicate that the POM-capped Pd nanoparticles slowly form large structures with an average radius of gyration (Rg) of 50.2 ± 2.0 nm. Meanwhile, dynamic light scattering (DLS) measurements indicate that the average hydrodynamic radius (Rh) of the large structures is ∼48.5 ± 1.5 nm. Therefore, a Rg /Rh ratio of 1.03 ± 0.05 is obtained. For a solid, homogeneous sphere, Rg/Rh = 0.778. This ratio will increase if more mass distributes closer to the surface of the sphere. If a spherical object has all its mass distributed on the surface, then Rg/Rh = 1, which is the current case. This is a typical model for a hollow, vesicle-like structure (Fig. 15). SLS measurements also show that each assembly is made up of ∼900 small 3 nm radius Pd0 nanoparticles. If they were solid, each spherical assembly would contain 6100 nanoparticles. This large discrepancy strongly suggests that the assemblies cannot be solid. The TEM images (Fig. 14) clearly show that the large Pd0 assemblies are composed of 3 nm radius small particles. There is still a certain amount of space between the single Pd nanoparticles in the assemblies, indicating that the nanoparticles interact weakly with each other, very possibly due to the electrostatic repulsion introduced by the absorbed anionic HPV. The relatively low electron densities at the center of the assemblies support our previous argument that the spherical assemblies are not solid. The hollow structure is a more stable state than the single 3 nm radius palladium nanoparticles in solution.
 | ||
| Fig. 14 A typical TEM image of the supramolecular structures formed by HPV-capped 3 nm radius palladium nanoparticles at R = 1.0 (from Fig. 7b, ref. 57). | ||
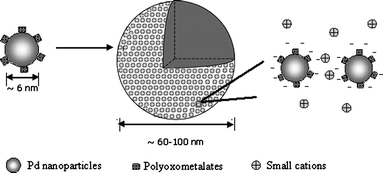 | ||
| Fig. 15 Schematic representation of hollow suprastructures of palladium aggregates. 3 nm radius Pd nanoparticles with a layer of POM HPVIV or HPVV on the surface self-organized into large hollow aggregates with the size of 60–100 nm. Small counter-ion cations might be incorporated into the large hollow structures (from Scheme 1, ref. 57). | ||
Considering that the POM-capped Pd nanoparticles are hydrophilic and have no obvious hydrophobic part, it is unlikely that they form bilayer vesicular structures like surfactants. Instead, we expect that the supramolecular structure should resemble the phenomenon in some macroionic solutions, which was first observed in hydrophilic polyoxometalate macroanionic solutions. Various highly soluble POM macroions tend to self-assemble into such unique structures (which we refer to as “blackberry” structures) in polar solvents.58 The blackberry size can be accurately controlled by tuning the macroionic charge density, solvent quality or solution pH.59 The driving forces of the blackberry formation are different from other types of self-assembly behavior such as the formation of micelles/vesicles (due to hydrophobic interactions) or colloid crystals (due to van der Waals forces). Instead, the size disparity between macroanions and small cations results in counter-ion association around macroions.59,60 Therefore, the counter-ion-mediated attraction and hydrogen bonds are important for the blackberry formation (Fig. 15). Recently, blackberry structures were identified in cationic metal–organic nanocage solutions, indicating that this type of self-assembly is quite universal for macroions.61
This result suggests that blackberry-type structure formation is most likely a general phenomenon for hydrophilic macroions with suitable size and moderate amount of charges in polar solvents, and not a specific feature of giant POM clusters. Work in progress will check the ability of this Pd0-based blackberry-type structure to function as a better hydrogen reservoir than isolated Pd0 nanoparticles.
Perspectives
This survey of the state of the art as concerns the synthesis of metal nanostructures by POMs and their characterization opens new research perspectives in this area. Mainly, it reveals the tremendous amount of work remaining to be done. Such a view is based on the following reasons: i) the wealth and variety of situations encountered so far in this research domain, by using only a limited number of POMs; ii) the potentially enormous number of POMs which are easy to synthesize and stable in water; iii) the number of issues which deserve detailed examination to obtain a rational view of nanostructure synthesis by POMs. The challenges include size, shape and geometry stabilization problems in the framework of the competition between various operational parameters. It is also necessary to keep in mind that energy saving and green chemistry-type processes are desired. In addition, even though synthesis of these nano-objects might be valuable in its own right, such nanostructures are mostly expected to display physical, chemical, medicinal, catalytic and electrocatalytic properties for further studies and uses. As a consequence, their surface must remain accessible and even synergistic behavior with the capping layer might be expected. The electrocatalytic behaviors observed so far for these nanostructures suggest that the surface remains accessible, despite the presence of the POM confirmed through the protection of Pt0 nanoparticles against poisoning during methanol oxidation. Such observations call for further high resolution study of the structure of these nanoparticles. Also, the usefulness of as-prepared metal nanostructures must be developed in several directions, including optical properties, catalytic and electrocatalytic behavior. Finally, the self-assembly of polyoxometalate macroanion capped Pd0 nanoparticles in aqueous solutions should be extended to several other examples and open new avenues for physical chemistry studies and, hopefully, applications.Acknowledgements
This work was supported by the University Paris-Sud 11 and the CNRS (UMR 8000). We thank our colleagues who have participated in various aspects of the original papers published on this topic by our group: Dr R. Contant (POM synthesis, Université Paris VI); Prof. P. Berthet (XRD characterization, ICMMO, Université Paris Sud 11, Orsay); Groupe de Physicochimie des Solides Moléculaires of Prof. F. Sécheresse (synthesis and characterization of POMs, Université de Versailles Saint Quentin, Versailles); Dr F. Miserque (XPS, CEA, Saclay), Miss R. N. Biboum for some syntheses, Dr L. R. Brudna Holzle (UMR 8000) and Mrs D. Jaillard (help with TEM characterization, UMR 8080, Université Paris Sud 11, Orsay). TL thanks support from NSF (CHE-0545983).References
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaf, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098 CrossRef CAS.
- K. J. Klabunde, Ed., Nanoscale Materials in Chemistry, Wiley Interscience, New York, 2001 Search PubMed.
- G. Schmid, M. Baumle, M. Geerkens, I. Heim, C. Osemann and C. Saviatowski, Chem. Soc. Rev., 1999, 28, 179 RSC.
- J. H. Fendler, Ed. Nanoparticles and Nanostructured Films, Wiley-VCH, Weinheim, 1998 Search PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS.
- J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228 CrossRef CAS.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem. Int. Ed., 2007, 46, 1222 CrossRef CAS.
- Y. Xia and N. J. Halas, MRS Bull., 2005, 30, 338 CAS.
- L. M. Liz-Marzan, Langmuir, 2006, 22, 32 CrossRef CAS.
- U. Simon, G. Schön and G. Schmid Angew, Chem. Int. Ed. Engl., 1993, 32, 250 Search PubMed.
- J. Glanz, Science, 1995, 269, 1363 CrossRef CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078 CrossRef CAS.
- M. Antonietti and C. Göltner, Angew. Chem., Int. Ed. Engl., 1997, 36, 910 CrossRef.
- (a) J. Stein, L. N. Lewis, Y. Gao and R. A. Scott, J. Am. Chem. Soc., 1999, 121, 3693 CrossRef CAS; (b) M. T. Reetz, R. Breinbauer, P. Wiedemann and P. Binger, Tetrahedron, 1998, 54, 1233 CrossRef CAS; (c) T. J. Schmidt, M. Noeske, H. A. Gasteiger, R. J. Behm, P. Britz, W. Brijoux and H. Bönnemann, Langmuir, 1997, 13, 2591 CrossRef CAS; (d) S. Ahmadi, Z. L. Wang, T. C. Grean, A. Henglein and M. A. El-Sayed, Science, 1996, 273, 1924 CrossRef.
- (a) Science, Special Issue on Nanotechnology, 2000, vol. 290, pp. 1523–1558 Search PubMed; (b) T. Kyotani, T. Li-Fu and A. Tomita, Chem. Commun., 1997, 301 RSC; (c) B. Rajesh, K. Ravindranathan Thampi, J. M. Bonard, N. Xanthopoulos, H. J. Mathieu and J. Viswanathan, J. Phys. Chem. B, 2003, 107, 2701 CrossRef CAS.
- R. J. Hunter, Foundations of Colloid Science; Oxford University Press: New York, 1987; Vol.1, pp 316–492 Search PubMed.
- (a) E. J. W. Verwey, J. Th. G. Overbeek, Theory of the stability of Lyophobic Colloids, Dover, Mineola, New York 2000 Search PubMed; (b) R. J. Hunter, Foundations of Colloid Science, Clarendon Press, Oxford, UK, 1992 Search PubMed.
- (a) S. Özkar and R. G. Finke, J. Am. Chem. Soc., 2002, 124, 5796 CrossRef; (b) R. G. Finke and S. Özkar Coord, Chem. Rev., 2004, 248, 135 CAS.
- (a) Representative examples include the following and references therein: M. T. Pope,Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 Search PubMed; (b) B. Keita and L. Nadjo, Current Topics in Electrochemistry, 1993, 2, 77 Search PubMed; (c) C. L. Hill (Guest Ed.), Chem Rev, 1998, vol. 98, pp. 1–389 Search PubMed; (d) M. T. Pope, A. Müller, Eds., Polyoxometalates: From Platonic Solids to Antiretroviral Activity, Kluwer Academic Publications, Dordrecht, 1994 Search PubMed; (e) M. T. Pope, Polyoxo Anions: Synthesis and Structure. In Comprehensive Coordination Chemistry II: Transition Metal Groups 3–6; (A. G. Wedd, Ed.) Elsevier Science: New York, 2004; Vol. 4, Chapter 4.10, pp 635–678 Search PubMed; (f) C. L. Hill, Polyoxometalates: Reactivity. In Comprehensive Coordination Chemistry II: Transition Metal Groups 3–6; (A. G. Wedd, Ed.) Elsevier Science: New York, 2004; Vol. 4, Chapter 4.11, pp 679–759 Search PubMed; (g) M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS; (h) B. Keita, L. Nadjo, Electrocatalysis of Inorganic Chemicals by Chemically Modified Electrodes, Encyclopedia of Electrochemistry, Vol 10, pp. 705–728, Bard A. J.and Stratmann M. Eds, Wiley-VCH, 2007 Search PubMed; (i) B. Keita, L. Nadjo, Electrochemistry of Isopoly and Heteropoly Oxometalates, Encyclopedia of Electrochemistry, Vol 7, pp. 607–700, Bard A. J. and Stratmann M. Eds, Wiley-VCH, 2006 Search PubMed.
- (a) L. S. Ott and R. G. Finke, Chem. Mater., 2008, 20, 2592 CrossRef CAS; (b) N. T. Flynn and A. A. Gewirth, Phys. Chem. Chem. Phys., 2004, 6, 1310 RSC; (c) Y. V. Geletii, A. Gueletii and I. A. Weinstock, J. Mol. Catal. A: Chemical, 2007, 262, 59 CrossRef CAS; (d) A. Z. Ernst, S. Zoladek, K. Wiaderek, J. A. Cox, A. Kolary-Zurowska, K. Miecznikowski and P. J. Kulesza, Electrochimica Acta, 2008, 53, 3924 CrossRef CAS and references therein; (e) C. R. Mayer, S. Neveu, C. Simonnet-Jégat, C. Debiemme-Chouvy, V. Cabuil and F. Sécheresse, J. Mater. Chem., 2003, 13, 338 RSC; (f) G. M. Maksimov, V. I. Zaikovskii, K. I. Matveev and V. A. Likholobov, Kinetics and Catalysis, 2000, 41, 844 Search PubMed; (g) G. M. Maksimova, A. L. Chuvilin, E. M. Moroz, V. A. Likholobov and K. I. Matveev, Kinetics and Catalysis, 2004, 45, 870 Search PubMed.
- (a) G. Hervé, Ann. Chim. Paris, 1971, 6, 219 Search PubMed; (b) G. Hervé, Ann. Chim. Paris, 1971, 6, 287 Search PubMed; (c) G. Hervé, Ann. Chim. Paris, 1971, 6, 337 Search PubMed; (d) G. Hervé, Thesis, 1970, Université Paris VI; (e) J. P. Launay, Thesis, 1974, Université Paris VI; (f) J. P. Ciabrini, Thesis, 1982, Université Paris VI; (g) M. Abbessi, Thesis, 1989, Université Paris VI.
- (a) E. Papaconstantinou, A. Hiskia, in: J. J. Borrás-Almenar, E. Coronado, A. Müller, M. Pope (Eds.) Polyoxometalate Molecular Science, Kluwer Academic Publishers, The Netherlands, 2003, p. 381 Search PubMed; (b) A. Hiskia, A. Mylonas and E. Papaconstantinou, Chem. Soc. Rev., 2001, 30, 62 RSC; (c) E. Papaconstantinou, Chem. Soc. Rev., 1989, 16, 1 RSC; (d) T. Yamase, Catal. Surv. Asia, 2003, 7, 203 CrossRef CAS; (e) C. L. Hill, C. M. Prosser-McCartha, in: K. Kalyanasundaram; M. Grätzel (Eds.), Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds, Kluwer Academic Publishers, The Netherlands, 1993, p. 307 Search PubMed.
- E. Papaconstantinou, J. Chem. Soc., Farad. Trans, 1982, 78, 2769 Search PubMed.
- M. Rindl, S. Afr. J. Sci., 1916, 11, 362 Search PubMed.
- L. Chalkley, J. Phys. Chem., 1952, 56, 1084 CrossRef.
- (a) A. Troupis, E. Gkika, A. Hiskia and E. Papaconstantinou, C.R. Chimie, 2006, 9, 851 Search PubMed; (b) A. Hiskia, E. Papaconstantinou and A. Troupis-Koukoutsis, PCT Int. Appl., 2006 Search PubMed , PIXXD2 WO 2006038045 A1 20060413; (c) E. Papaconstantinou, J. Chem. Soc., Farad. Trans., 1982, 78, 2769 Search PubMed; (d) A. Troupis, A. Hiskia and E. Papaconstantinou, N.J. Chem., 2001, 25, 361 Search PubMed; (e) A. Troupis, E. Hiskia and E. Papaconstantinou, Environ. Sci Technol., 2002, 36, 5355 CrossRef CAS; (f) A. Troupis, A. Hiskia and E. Papaconstantinou, Appl. Catal. B: Environ., 2003, 42, 305 CrossRef CAS; (g) A. Troupis, A. Hiskia and E. Papaconstantinou, Appl. Catal. B: Environ., 2004, 52, 41 CrossRef CAS; (h) A. Hiskia, A. Troupis and E. Papaconstantinou, Int. J. Photoenergy, 2002, 4, 35 CrossRef CAS; (i) A. Hiskia and E. Papaconstantinou, Inorg. Chem., 1992, 31, 163 CrossRef; (j) A. Troupis, A. Hiskia and E. Papaconstantinou, Angew. Chem. Int. Ed. Engl., 2001, 41, 1911.
- W. G. Klemperer and C. G. Wall, Chem. Rev., 1998, 98, 297 CrossRef.
- S. Mandal, P. R. Selvakannan, R. Parischa and M. Sastry, J. Am. Chem. Soc., 2003, 125, 8440 CrossRef CAS.
- S. Mandal, D. Rautaray and M. Sastry, J. Mater. Chem., 2003, 13, 3002 RSC.
- S. Mandal, A. Das, R. Srivastava and M. Sastry, Langmuir, 2005, 21, 2408 CrossRef CAS.
- A. Sanyal and M. Sastry, Chem. Commun., 2003, 1236 RSC.
- S. Shanmugam, V. Viswanathan and T. K. Varadarajan, J. Mol. Catal. A, 2005, 241, 52 CrossRef CAS.
- A. V. Gordeev, N. I. Kartashev and B. G. Ershov, High Energy Chem., 2002, 36, 75 CrossRef CAS.
- B. G. Ershov and A. V. Gordeev, Mendeleev Commun., 2001, 4, 147 Search PubMed.
- A. V. Gordeev and B. G. Ershov, High Energy Chem., 1999, 33, 218 CAS.
- C. Marchal-Roch, C. R. Mayer, A. Michel, E. Dumas, X. F-Liu and F. Sécheresse, Chem. Commun., 2007, 3750–3752 RSC.
- W. M. Latimer, Oxidation Potentials, Prentice-Hall, Englewood Cliffs, NJ, 2nd ed., 1952 Search PubMed.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939 CrossRef CAS.
- (a) T. Kim, K. Lee, M.-S. Gong and S.-W. Joo, Langmuir, 2005, 21, 9524 CrossRef CAS; (b) T. Kim, C.-H. Lee, S.-W. Joo and K. Lee, J. Colloid Interf. Sci., 2008, 318, 238 CrossRef CAS.
- B. Keita, I. M. M. Mbomekalle, L. Nadjo and C. Haut, Electrochem. Commun., 2004, 6, 978 CrossRef CAS.
- I. M. Mbomekalle, B. Keita, Y. W. Lu, L. Nadjo, R. Contant, N. Belai and M. T. Pope, Eur. J. Inorg. Chem., 2004, 276–285 CrossRef CAS.
- Handbook of Chemistry and Physics, 66th ed., 1985–1986, CRC Press. Inc., Boca Raton, FL, p. D-154 Search PubMed.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881 RSC.
- B. Keita, G. Zhang, A. Dolbecq, P. Mialane, F. Sécheresse, F. Miserque and L. Nadjo, J. Phys. Chem. C, 2007, 111, 8145 CrossRef CAS.
- E. Ishikawa and T. Yamase, Bull. Chem. Soc. Jpn., 2000, 73, 641 CrossRef CAS.
- A. Dolbecq, L. Lisnard, P. Mialane, J. Marrot, M. Bénard, M.-M. Rohmer and F. Sécheresse, Inorg. Chem., 2006, 45, 5898 CrossRef CAS.
- P. Mialane, A. Dolbecq, L. Lisnard, A. Mallard, J. Marrot and F. Sécheresse, Angew. Chem. Int. Ed., 2002, 41, 2398 CrossRef CAS.
- A. Henglein, B. G. Ershov and M. Malow, J. Phys. Chem., 1995, 99, 14129 CrossRef CAS.
- G. Zhang, B. Keita, A. Dolbecq, P. Mialane, F. Sécheresse, F. Miserque and L. Nadjo, Chem. Mater, 2007, 19, 5821 CrossRef.
- B. Keita, R. N. Biboum, I. M. Mbomekalle, S. Floquet, C. Simonnet-Jégat, E. Cadot, F. Miserque, P. Berthet and L. Nadjo, J. Mater. Chem., 2008, 18, 3196 RSC.
- F. A. Cotton, R. Llusar, D. O. Marler, W. Schwotzer and Z. Dori, Inorg. Chim. Acta, 1985, 102, L25 CrossRef CAS.
- T. Shibahara, M. Yamazaki, G. Sakane, K. Minami, T. Yabuki and A. Ichimura, Inorg. Chem., 1992, 31, 640 CrossRef CAS.
- G. Maayan and R. Neumann, Chem. Commun., 2005, 4595 RSC.
- H. Liu, N. Bayat and E. Iglesia, Angew. Chem. Int. Ed., 2003, 42, 5072 CrossRef CAS.
- A. Jimtaisong and R. L. Luck, Inorg. Chem., 2006, 45, 10391 CrossRef CAS.
- C. M. Welch and R. G. Compton, Anal. Bioanal. Chem., 2006, 384, 601 CrossRef CAS.
- J. Zhang, B. Keita, L. Nadjo, I. M. Mbomekalle and T. Liu, Langmuir, 2008, 24, 5277 CrossRef CAS.
- (a) T. Liu, J. Am. Chem. Soc., 2002, 124, 10942 CrossRef CAS; T. Liu, J. Am. Chem. Soc., 2004, 126, 406 CrossRef CAS (Addition/Correction); (b) T. Liu, E. Diemann, H. Li, A. Dress and A. Müller, Nature, 2003, 426, 59 CrossRef CAS.
- (a) T. Liu, B. Imber, E. Diemann, G. Liu, K. Cokleski, H. Li, Z. Chen and A. Müller, J. Am. Chem. Soc., 2006, 128, 15914 CrossRef CAS; (b) M. L. Kistler, A. Bhatt, G. Liu, D. Casa and T. Liu, J. Am. Chem. Soc., 2007, 129, 6453 CrossRef CAS; (c) L. Verhoeff, M. L. Kistler, A. Bhatt, J. Pigga, J. Groenewold, M. Klokkenburg, S. Veen, S. Roy, T. Liu and W. K. Kegel, Phys. Rev. Lett., 2007, 99, 066104 CrossRef.
- G. Liu and T. Liu, J. Am. Chem. Soc., 2005, 127, 6942 CrossRef CAS.
- D. Li, J. Zhang, K. Landskron and T. Liu, J. Am. Chem. Soc., 2008, 130, 4226 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |