A high-throughput method for Saccharomyces cerevisiae (yeast) ionomics†
John M. C.
Danku
a,
Luke
Gumaelius
b,
Ivan
Baxter
b and
David E.
Salt
*ab
aCenter for Plant Environmental Stress Physiology, 1165 Horticulture Building, Purdue University, West Lafayette IN 47907, USA. E-mail: dsalt@purdue.edu
bBindley Bioscience Center, Discovery Park, Purdue University, West Lafayette IN 47907, USA
First published on 21st October 2008
Abstract
Reliable and rapid analytical methods are the backbone for generating data and deciphering gene functions in the post-genomics era. We describe here a high-throughput method for the rapid profiling of fourteen elements in the 5153 strain gene deletion collection of Saccharomyces cerevisiae. Samples were grown and processed in standard 96-well plate format followed by inductively coupled plasma mass spectrometry (ICP-MS) analysis. Optical densities of the yeast were measured prior to ICP-MS analysis and used for the normalization of the data. The elemental profiling data are stored in an online database for later bioinformatics analysis. This method has the capacity to run 288 yeast samples per day on a single ICP-MS, and has allowed the quantification of the ionome in four replicate cultures of approximately 240 yeast deletion strains per week, along with appropriate wild-type and positive control strains. We identified 400 strains that were outliers from the overall deletion collection in at least one element out of the fourteen that were monitored.
Introduction
Ionomics is the “quantitative and simultaneous measurement of the elemental composition of living organisms, and changes in this composition in response to physiological stimuli, developmental state and genetic modification”.1 The ionome is an expansion on the previous concept of the “metallome”2,3 to include metalloids and non-metals.4 Since the ionome is involved in a host of biologically important phenomena such as osmoregulation, transport, signalling, enzymology and electrophysiology,5 an understanding of its genetic basis, and how it interacts with other cellular systems is paramount. This in turn is predicated on developing reliable and rapid analytical tools capable of handling the large numbers of biological samples required for such an analysis.A recent study characterized the ionome of 4385 Saccharomyces cerevisiae (yeast) mutants strains (homozygous diploid collection) which represent a subset of the complete yeast knockout collection including only non-essential genes.6 However, given the limited sample throughput of the method used by Eide and co-workers only single cultures of the majority of the 4385 yeast mutants were analyzed. Such a limited sampling density for each mutant reduces significantly the resolution of the yeast ionome study published by Eide and co-workers.6 To improve both the sampling density, and to complete analysis of the full collection of yeast knockouts (5153 strains, MATa haploid collection7,8) it is essential to streamline and optimize the ionomic method, to allow the efficient processing of significantly more samples without the loss of analytical precision.
We describe here a high-throughput elemental profiling methodology for the analysis of the yeast ionome that employs a 96-well plate format integrated into the process from sample growth through to the final ICP-MS analysis. This method has the capacity to analyze 288 yeast cultures per day on a single ICP-MS. Application of this methodology has allowed the analysis of the complete yeast gene deletion collection of 5153 strains with four replicate cultures per mutant strain. To enhance downstream data normalization each 96-well plate also contained the same set of four different control lines. Analysis of the complete knockout collection took 6 months and represents the processing of approximately 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 yeast cultures. This represents a 10-fold increase in throughput over the previous study6 while maintaining a high level of precision and improved analytical sensitivity. The availability of such a high-throughput methodology now opens up the possibility of performing other genome-wide ionomics analyses in yeast, including the screening of the complete ORF (open reading frame) over expression collection, and cDNA (complementary DNA) expression libraries from heterologous genomes and metagenomes.
000 yeast cultures. This represents a 10-fold increase in throughput over the previous study6 while maintaining a high level of precision and improved analytical sensitivity. The availability of such a high-throughput methodology now opens up the possibility of performing other genome-wide ionomics analyses in yeast, including the screening of the complete ORF (open reading frame) over expression collection, and cDNA (complementary DNA) expression libraries from heterologous genomes and metagenomes.
Experimental section
Instrumentation
A quadrupole inductively coupled plasma mass spectrometry, ICP-MS, (Elan DRC II, Perkin Elmer, Shelton, CT, USA) coupled with an SC-2 autosampler and an Apex Q sample introduction system (Elemental Scientific Inc., Omaha, NE, USA) was used for analysis of the yeast cultures. A liquid handling robot, MultiPROBE II PLUS HT EX, (Perkin Elmer) was used to perform the initial yeast inoculation into 96-well square deep-well plates. A shaking incubator (Shel Lab SI6, Sheldon Mfg. Inc., Cornelius, OR, USA), which was custom fitted to accommodate up to 15 deep-well plates (Metalhead LLC, Salem, IN, USA), was employed for yeast cultivation. Yeast optical densities (OD) were measured using an OpsysMR plate reader (DYNEX Technologies, Chantilly, VA, USA). Digestion of yeast samples was performed using a multi-block heater (Lab Line Instruments, Melrose Park, IL, USA).Materials
Custom made AcroPrep 96 PVDF (polyvinylidene fluoride) filter membrane (0.45 µm, 350 µL) micro-well plates (Pall Life Sciences, Ann Arbor, MI, USA) were used for processing yeast samples. Clear View micro-plates were used for yeast optical density measurements. 96-well square deep-well (2 mL) plates (Axygen Scientific, Union City, CA, USA) were used both for yeast growth and after processing for ICP-MS analysis (using the SC-2 autosampler). Polypropylene lids (Axygen) were used to cover the deep-well plates during sample storage and digestion, while Axymat (Axygen) chemically resistant and flexible lids were used to cover the plates during mixing. Adhesive breathable sealing film (AeroSeal, Dot Scientific Inc., Burton, MI, USA) was used to seal the deep-well plates during yeast growth. Three multi-channel pipettors with volume ranges of 2 to 20 µL, 20 to 300 µL and 100 to 1200 µL were used to dispense yeast cultures, reagents and solutions.Standards and reagents
AR Select grade concentrated nitric acid from Mallinckrodt (Phillipsburg, NJ, USA) was used for sample digestion. Single element standard stock solutions for the calibration procedure and for spiking yeast growth media were obtained from ULTRA Scientific (Kingstown, RI, USA). Deionized water (18 MΩ) for all dilutions was from a NANOpure Diamond (Barnstead International, Dubuque, IA, USA) water purifier. Triton X-100 was obtained from Sigma (St. Louis, MO, USA), and was added to both the processed samples and calibration standards to enable smooth self aspiration up the micro-nebulizer. Sodium chloride, sodium ethylenediaminetetraacetate (EDTA), methanol and ethanol were purchased from Mallinckrodt (Phillipsburg, NJ, USA). Synthetic defined minimal medium components for yeast culture were from the following vendors: Yeast Nitrogen Base with nitrogen (YNB) and CSM-Ura (complete supplement mixture minus uracil), Sunrise Science Products (obtained from MIDSCI, St. Louis, MO, USA); uracil from BIO 101 (Vista, CA, USA), and D-Glucose monohydrate from Research Products International (Mt. Prospect, IL, USA).Sample preparation
The yeast knock-out (YKO) collection used in this work was from the yeast MATa collection generated from the BY4741 background—MATa his3△1 leu2△0 met15△0 ura3△0.7,8 Most of the collection was obtained from Dr Tony Hazbun of Purdue University and the rest purchased from Open Biosystems (Huntsville, AL, USA). The stock YKO lines came in 96-well micro-plates and were maintained at −80 °C.Growth of yeast was carried out in two stages. The first stage, preliminary growth or pre-growth, involved bulking up from the stock collection; the second, growth for analysis or simply growth, was for both the ICP-MS analysis and the corresponding optical density measurement. The yeast culture medium used was synthetic defined minimal (YNB + CSM − Ura + uracil) medium. The minimal growth medium was supplemented with the following elements for the second stage of growth: 2 ppb Co, 20 ppb Cd, 50 ppb Mo, 100 ppb Ni and 200 ppm Na. This was to either compensate for the elements lacking (Co, Cd, Ni) in the synthetic medium or else to increase the levels (Mo, Na) for better ICP-MS detection. Note that the same lots or mix of lots, of medium components were used throughout this work to ensure consistent growth conditions. In all cases growth was carried out in 96-well square deep-well plates.
For pre-growth 5 µL of starter yeast stock (inoculate) was added to 500 µL of medium per well. The plate was sealed with breathable sealing film and incubated at 30 °C and 400 rpm for 48 h. Usually two and a half 96-well plates of the primary yeast collection were utilized per week for analysis, along with background, wild-type and positive controls strains. During the growth stage 20 µL of yeast inoculate was added to 750 µL of supplemented medium per well. This stage required the use of a liquid handling robot. A total of twelve 96 deep-well plates were usually generated from the two and a half bulked pre-growth yeast plates. Each plate had 20 yeast lines transferred from a pre-growth plate with four replicates per line. These covered 10 of the 12 columns of the plate (two lines per column) with the background and wild-type, and the two positive control lines occupying the other two columns. The plates were sealed as before. Three plates per day were grown with the rest kept in the refrigerator (4 °C) and grown on successive days. The incubation conditions in this case were 30 °C and 400 rpm for 36 h. The yeast cells are at the post-diauxic growth period before harvesting for both of these growth conditions. The cells grow rather slowly during this growth phase.9 Eide and co-workers6 harvested yeast cells at a similar growth phase.
Sample processing for ICP-MS analysis, until the final step, was performed in AcroPrep 96 PVDF filter membrane micro-plates. The hydrophobic membrane of the plate was wetted with methanol and then rinsed with deionized water. Yeast cultures (200 µL well−1) were transferred from the growth plates into filter plates using multi-channel pipettes. The same amounts were concurrently transferred into Clear View microtiter plates and the optical densities measured with a plate reader. The cells in the filter plates were washed and rinsed in situ, respectively, with EDTA (1 µM, pH 8) and deionized water, using a vacuum manifold. Four separate washes and rinses were performed (350 µL well−1 each). The filter plates were dried (∼88 °C for 2 h) to restore the membranes hydrophobicity. Washed yeast cells were digested directly inside the filter plates (100 µL well−1nitric acid, ∼88 °C for 40–45 min) using a heating block. The yeast digests were drawn through the filter and into 96 deep-well collection plates containing Triton X-100 (0.025% v/v, 300 µL well−1) with Ga (6.67 ppb) internal standard solution. The final dilution volume was 1.6 µL well−1 including Ga (5 ppb) internal standard and Triton X-100 (0.005% v/v).
ICP-MS analysis
The processed yeast samples were run on an Elan DRC II ICP-MS equipped with ESI SC-2 autosampler unit that could accommodate 96 deep-well plates, and an Apex Q sample introduction system. Triton X-100 (0.005% v/v) was also added to the calibration standards as well as the wash solution to reduce surface tension and enable smoother self aspiration of the PFA micro-nebulizer. Fourteen elements (Na, Mg, P, S, K, Ca, Mn, Fe, Co, Ni, Cu, Zn, Mo, Cd) were monitored in the yeast samples.A flowchart representation of the yeast ionomics high-throughput workflow is shown in Fig. 1.
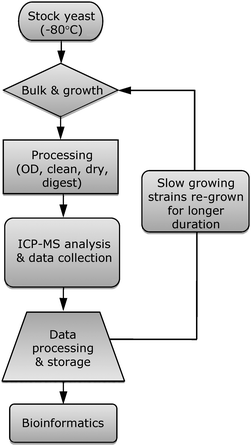 | ||
| Fig. 1 Yeast ionomics high-throughput method workflow. OD—optical density. | ||
Results and discussion
The method reported here is a low cost, rapid, robust and sensitive method for the elemental analysis of yeast, allowing for the precise analysis of many thousands of samples with the capacity to resolve small ionomic differences between samples. Three 96-well plates of yeast cultures can be processed per day using a single ICP-MS instrument. Currently, we analyze approximately 240 yeast deletion strains per week, each in replicates of four cultures, along with background, wild-type and positive control strains. This translates into a throughput of about 30000 samples in 6 months. In comparison Eide and co-workers6 analyzed approximately 10000 samples in 24 months. Fourteen biologically essential or potentially toxic elements (Na, Mg, P, S, K, Ca, Mn, Fe, Co, Ni, Cu, Zn, Mo, Cd) are quantified in the yeast samples, and the elemental profiling data stored in an online ionomics database, based on the Purdue Ionomics Information Management System (PIIMS).10Boron, lithium, arsenic and selenium were also monitored during the preliminary stages but later dropped due to low detection of these elements in the yeast samples. Even after supplementation of the culture medium with these elements there was not an appreciable uptake by the yeast cells. ICP-MS is needed for the method described here. However, ICP-OES (optical emission spectroscopy) could also be used but would require minor modifications of the protocol. For instance, a larger amount of yeast culture would need to be digested to increase the elemental concentrations in the sample, or a larger flow PFA micro-nebulizer could be used with the Apex sample introduction system in order to increase the amount of sample reaching the plasma. Assuming one already has the necessary equipment and personnel in place, the cost for sample analysis in the method reported here is under US$200 per 96-well plate (or ∼$2 per sample; 96 samples per plate), making this a truly low cost, high throughput method.
In order to develop a high-throughput ionomic method that not only can rapidly characterize many thousands of yeast strains, but can do so in a robust, routine and analytically precise manner, several factors have to be taken into account. Chief among these is to use standardized instrumentation, equipment and materials. The 96-well plate sample format was deemed suitable for this purpose. It allows for parallel handling of large numbers of yeast strains and can fit nicely into most liquid handling robotic systems, and some specially configured ICP-MS autosampler units. The method reported here was therefore designed to allow the use of a 96-well plate format from yeast cultivation through to ICP-MS analysis.
Yeast culture in a 96-well plate format requires the correct amount of agitation, media volume and headspace to allow adequate aeration for uniformity of yeast growth in all wells. The initial conditions were adapted from work performed on bacterial strains.11 Preliminary experiments were performed to establish the ideal orbital shaking frequency of 400 rpm. The media volumes used for the pre-growth and growth stages were selected based on the amount needed to adequately carry out the yeast culture processing, and the fact that there is some evaporative loss during the cultivation. Evaporative loss was found to be practically identical in all wells, and independent of position in the incubator.
The custom ordered AcroPrep 96-well plate with polyvinylidene fluoride (PVDF) hydrophobic filter membrane was chosen because the hydrophobic/hydrophilic nature of the membrane can be switched by wetting with solvent, drying or acid digestion. Once yeast cells had been dispensed into the wells, this membrane property means that all the various sample processing stages, including washing and digestion, can occur in the same plate, minimizing the need for repeated transfers between plates, reducing sample loss associated with transfers, and the potential for contamination.
As part of this high-throughput yeast ionomic methodology, we include 4 control yeast strains in each 96-well plate. These control strains are the parental line for the knockout collection YDL227c, which is derived from BY4741, along with two previously identified yeast ionomic mutants6 YLR396c (deletion of VPS33 involved in vacuolar function) and YPR065w (deletion of ROX1 involved in mineral nutrient homeostasis). YPR065w and YLR396c, known to have elevated or depressed concentrations of several elements (including Na, Mg, S, K, Mn, Co),6 are used as positive controls to confirm that for each 96-well plate analyzed the analytical methodology is working correctly. The ionomic profiles for these positive control lines are displayed in Fig. 2A and B. Here the elements monitored by ICP-MS are plotted against their z-scores, which represent the number of standard deviations away from the mean of the control parental line (YDL227c) grown in the same 96-well plate. Arabidopsis thaliana shoot ionomic data was previously displayed in a similar manner.4 It is clear from this type of z-score plot that both positive control lines, YLR396c and YPR065w, show major differences in several elements when compared to the YDL227c background. YLR396c has elevated levels of Mg, S, Co, Ni and Cd, and reduced Na, K, Mn, Cu and Mo levels. Whereas YPR065w has elevated levels of K, Co, Ni and Cd, and reduced Na, Mg, Ca and Mo levels.
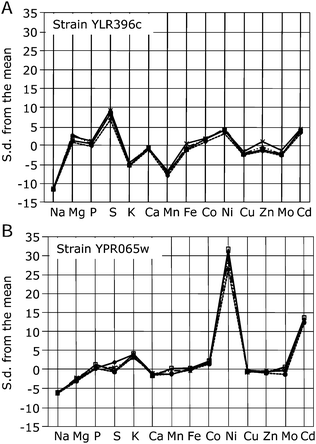 | ||
| Fig. 2 Ion profile data for yeast mutant lines. Standard deviations (s.d.) from the mean for (A) positive control yeast, YLR396c, and (B) positive control yeast, YPR065w. Mean and standard deviations are calculated for each element from the background yeast, YDL227c (n = 4), and used to calculate the number of standard deviations each strain is distant from the mean background value for each element. Same scaling on ordinate used to highlight differences in mutant magnitude. | ||
In any high-throughput analysis platform, where comparisons need to be made between samples, analytical precision is critical. A precise measurement is one that when made repeatedly on the same sample will give the same answer. Similarly, repeated ionomic analyses of two different yeast strains, if made in a precise way, would be expected to produce two sets of similar ionomic profiles. Such profiles, if analyzed using Principal Component Analysis (PCA) would form two clear clusters of points associated with each of the two yeast strains. Using this approach we show that the four control yeast strains BY4741, YDL227c, YLR396c and YPR065w, included in all of the 96-well plates analyzed in this screen, form discrete clusters based on a PCA of their ionomic profiles (Fig. 3). Such clear clustering of strains, based on their ionomic profile measured repeated across 356 96-well plates, clearly establishes the high precision of our method; from yeast cultivation through ICP-MS analysis.
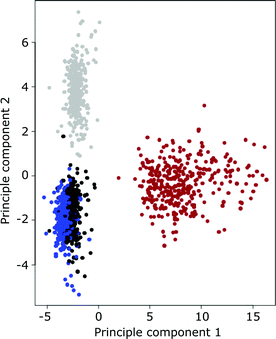 | ||
| Fig. 3 Principal component analysis of the yeast ionome of the four yeast control strains included in the 356 96-well plates analyzed. Black—BY4741, blue—YDL227c, red—YPR065w, light grey—YLR396c. | ||
In this screen the first 850 yeast deletion strains were run with eight replicate cultures, and eight deletion strains per plate. Statistical simulations based on this initial data set established that the high precision of the method would allow the reduction of the number of replicate yeast cultures needed in the analysis. The difference in means between the eight replicates and a randomly selected set of four replicates was calculated for each element across the 850 lines. All of the elements except Ca had a difference <10% for 95% of the strains, whereas the mean difference for Ca of <22%. This was attributed to the low levels of Ca in the wild-type yeast. Given the only minor differences in mean differences between eight and four replicate cultures we adopted four replicate cultures for the rest of the screen. This reduced level of replication allowed an increase in the number of yeast strains that could be analyzed per 96-well plate, producing a significant increase in throughput without loss of sensitivity to detect ionomic differences between yeast strains.
High sensitivity, or the ability to detect small ionomic differences between yeast strains, is another important parameter of this methodology. A major factor limiting sensitivity is the reproducibility or precision of the measuring device.12 Here we express the sensitivity as the average percent relative standard deviation (%RSD) for each element, based on each line across the 356 96-well plates analyzed in the screen. That is, if the %RSD is small then we would expect to be able to detect small differences between yeast strains. Table 1 shows the %RSD for each line analyzed in every 96-well plate run in this screen (n = 6888 individual samples) at different confidence levels. It is apparent from Table 1 that most of the elements monitored have 95% of their %RSD values < 12% (Mg, P, K, Mn, Co, Ni, Cu, Zn and Cd). The corresponding values for Na, Mo, S and Fe are < 20%RSD. Again, Ca shows the highest value of 32%RSD, due to its rather low levels in yeast. Overall, the %RSD values measured across the complete screen are low, allowing detection of small differences in the ionome of the yeast strains in the deletion collection. The %RSDs obtained from yeast are generally lower than those previously observed in a genomic-scale ionomic screen of A. thaliana,4 likely due to the more reproducible method of cultivation and sample analysis afforded by yeast.
| Confidence level | Na | Mg | P | S | K | Ca | Mn | Fe | Co | Ni | Cu | Zn | Mo | Cd |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 50% | 6.0 | 4.3 | 3.8 | 5.4 | 5.1 | 8.5 | 4.5 | 8.2 | 3.9 | 4.6 | 4.4 | 4.2 | 7.3 | 3.8 |
| 75% | 8.9 | 6.1 | 5.4 | 7.9 | 7.2 | 13.9 | 6.1 | 11.8 | 5.2 | 6.6 | 6.2 | 5.9 | 10.7 | 5.1 |
| 90% | 13.2 | 8.4 | 7.6 | 12.2 | 9.5 | 22.4 | 8.2 | 16.2 | 6.6 | 9.3 | 8.1 | 7.7 | 14.8 | 6.7 |
| 95% | 17.6 | 10.8 | 10.8 | 17.4 | 11.3 | 32.1 | 9.7 | 19.3 | 7.8 | 11.7 | 9.7 | 9.2 | 17.6 | 7.8 |
A preliminary analysis of the ionomic data set from the yeast gene deletion collection identified 400 strains that display differences from the overall average of the complete deletion set, with at least one element exceeding three z-scores (out of a total of 4952 lines, excluding repeat lines). Eide et al.6 identified 233 strains using the same criterion (out of 4385 yeast mutants).
Conclusions
In the present study, a low cost, robust, precise, sensitive and high-throughput method for profiling the yeast ionome was developed. The method involves a novel integration of 96-well plate sample formats for yeast cultivation, sample preparation and ICP-MS analysis, with the power of simultaneous determination of a broad range of elements using ICP-MS. This analysis platform was coupled to an online ionomics database (YeastPIIMS) for data management. The value of this system has been established by using it to profile fourteen elements across the entire set of 5153 mutant strains in the yeast gene deletion collection, representing the analysis of approximately 30000 yeast cultures in a 6 month period. Bioinformatics analysis of this dataset is currently ongoing to identify the genes and gene networks involved in regulating the yeast ionome.
Acknowledgements
We thank Brett Lahner for developing the original spreadsheet for analyzing the raw ICP-MS data. We are grateful to Tony Hazbun for supplying most of the stock yeast for this work and his helpful discussion concerning growing of the yeast, and we thank Brad Kennedy of the Purdue University Discovery Park Cyber Center for help developing the YeastPIIMS information management system. The study was supported by the National Institutes of Health (4 R33 DK070290-02).References
- D. E. Salt, I. Baxter and B. Lahner, Annu. Rev. Plant Biol., 2008, 59, 709 Search PubMed.
- C. E. Outtern and T. V. O'Halloran, Science, 2001, 292, 2488 CrossRef CAS.
- R. J. P. Williams, Coord. Chem. Rev., 2001, 216, 583 CrossRef.
- B. Lahner, J. Gong, M. Mahmoudian, E. L. Smith, K. B. Abid, E. E. Rogers, M. L. Guerinot, J. F. Harper, J. M. Ward, L. McIntyre, J. I. Schroeder and D. E. Salt, Nat. Biotechnol., 2003, 21, 1215 CrossRef CAS.
- D. E. Salt, Plant Physiol., 2004, 136, 2451 CrossRef CAS.
- D. J. Eide, S. Clark, T. M. Nair, M. Gehl, M. Gribskov, M. L. Guerinot and J. F. Harper, Genome Biol., 2005, 6, R77 CrossRef.
- C. B. Brachmann, A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter and J. D. Boeke, Yeast, 1998, 143, 115 CrossRef.
- E. A. Einzeler, D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B. Andre, R. Bangham, R. Benito, J. D. Boeke, H. Bussey, A. M. Chu, C. Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury, S. H. Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M. Laub, H. Liao, N. Liebundguth, D. J. Lockhart, A. Lucau-Danila, M. Lussier, N. M'Rabet, P. Menard, M. Mittmann, C. Pai, C. Rebischung, J. L. Revuelta, L. Riles, C. J. Roberts, P. Ross-MacDonald, B. Scherens, M. Snyder, S. Sookhai-Mahadeo, R. K. Storms, S. Véronneau, M. Voet, G. Volckaert, T. R. Ward, R. Wysocki, G. S. Yen, K. Yu, K. Zimmermann, P. Philippsen, M. Johnston and R. W. Davis, Science, 1999, 285, 901 CrossRef CAS.
- P. K. Herman, Curr. Opin. Microbiol., 2002, 5, 602 CrossRef CAS.
- I. Baxter, M. Ouzzani, S. Orcun, B. Kennedy, S. S. Jandhyala and D. E. Salt, Plant Physiol., 2007, 143, 600 CAS.
- W. A. Duetz, L. Rüedi, R. Hermann, K. O'Connor, J. Büchs and B. Witholt, Appl. Environ. Microbiol., 2000, 66, 2641 CrossRef CAS.
- D. A. Skoog and J. J. Leary, Principles of Instrumental Analysis, Saunders College Publishing, Philadelphia, USA, 4th edn, 1992 Search PubMed.
Footnote |
| † Presented at the 2008 Winter Conference on Plasma Spectrochemistry, Temecula, CA, USA, January 7–12, 2008. |
| This journal is © The Royal Society of Chemistry 2009 |