Advantages and limitations of a desolvation system coupled online to HPLC-ICPqMS/ES-MS for the quantitative determination of sulfur and arsenic in arseno-peptide complexes
Katharina
Bluemlein
,
Eva M.
Krupp
and
Jörg
Feldmann
College of Physical Sciences, Chemistry, Trace Element Speciation Laboratories Aberdeen (TESLA), University of Aberdeen, Meston Walk, Old Aberdeen, UK AB24 3UE
First published on 21st October 2008
Abstract
High sulfur detection limits, mainly caused by the interfering O2+ ions, made simultaneous sulfur and arsenic speciation analysis by on-line coupling of RP-HPLC with ES-MS and ICP-MS rather difficult in the past. The application of a desolvation system avoids the formation of the interfering ions in the first place. In this study the Aridus system was tested. The sulfur detection limit of the used ICP-qMS in its standard mode could be improved by a factor 2000 to 50 ng S g−1. But great care had to be taken regarding species independent quantification, the major advantage of ICP-MS, since the analyte transport of certain compounds such as methionine or arsenite to the ICP was affected by the desolvation system. However, sulfate was found to be a suitable calibrant for sulfur containing peptides such as phytochelatins. Hence, this method demonstrates for the first time, the potential to identify and quantify individual phytochelatins without phytochelatin standards. This is of importance, since no reliable and affordable phytochelatin standards are available, which would be necessary when molecular mass spectrometric methods would have been used solely. But a successful quantitative speciation analysis of free phytochelatins and their arsenite complexes was hampered due to peak broadening and more serious, the formation of arsenic-artefacts.
Introduction
Recently, it has been demonstrated that the use of RP-HPLC coupled simultaneously on-line with ES-MS and ICP-MS can offer species independent quantification. This approach has proved invaluable for the identification and quantitative determination of novel arsenic containing phytochelatins by using the arsenic element trace from the ICP-MS while gaining structural information via the ES-MS data.1,2In metallo-phyochelatin complexes, the metal ion is bound to the sulfhydryl function(s) (–SH) of the cysteine residue(s) contained in the peptides. While the metal ion can easily be quantified using ICP-MS, information on the structural binding properties, i.e. the metal–sulfur binding, could only be determined using the molecular structure information gained by the ES-MS data.
However, plant extracts contain a large number of free phytochelatins and other sulfur containing peptides, which could not be quantified using ICP-MS in the past. For this task, HPLC followed by post-column derivatisation with Ellman's reagent (DTNB, 5,5′-dithio-bis(2-nitrobenzoic acid), or pre-column derivatisation with monobromobimane (mBBr) were considered as the standard methods.3–5 Both reagents react specifically with free –SH groups and either the derivatisation product or, in the case of DTNB, the released NTB− (3-carboxylato-4-nitro-thiophenolate) is quantified photometrically.3,5 However, the main disadvantage of both methods is the fact that the derivatisation efficiency is dependent on the number of the –SH groups in the molecules, and therefore on the chain length of the phytochelatins4 (multiple length of γ-glutamyl-cysteine dipeptides chains with or without an end group which can be β-alanine, serine, glutamic acid or glycine). Thus, no species independent sulfur quantification is possible when DTNB or mBBr or any molecular mass spectrometric methods are used. Species independent element quantification, however, is the major advantage of an ICP-MS system.
In principle, ICP-MS can also be used for the quantitative determination of sulfur, but with some limitations. First, sulfur has a relatively high first ionization potential of 10.4 eV, which means that only about 10 % of all sulfur atoms are ionised in the ICP. And second, the main sulfur isotope on m/z 32 with 95% abundance is strongly interfered by 16O16O+ ions, which is simply due to the sample introduction in aqueous solution. Thus sulfur detection limits are high and a direct sulfur measurement with RP-HPLC/ICP-MS is not feasible straightaway.
Different strategies have been developed to date to overcome this problem and allow S measurements with ICP-MS at low detection limits needed for this kind of analysis. The most prominent approach to date is the use of an ICP-MS equipped with a collision or reaction cell, instruments which have been introduced into the market about 10 years ago.
Here, 16O16O+ ions can either be removed by using a heavy collision gas, such as xenon,6–8 or the analyte mass can be shifted from m/z 32 to 48 by collision-induced formation of 32S16O+ when the reaction cell is pressurised with oxygen.9–11 Another approach is the use of HR-ICP-MS, where the different masses of 32S (m/z = 31.9898) and 16O16O+ (m/z = 31.9721) can be separated11 at a resolution of 2000 (m/Δm).
However, when neither collision/reaction cell nor HR-ICP-MS is available, another approach is to avoid the formation of the interfering ion in the first place. This can be achieved by removing the solvent before the analytes enter the plasma using a membrane desolvation system, such as the Aridus system (CETAC Technologies, Omaha, US)12 (Fig. 1). Even though this approach did not result in sulfur detection limits obtained with SEC-ICP-DRC-MS (4.3 ng g−1; absolute LOD: 85 pg13) or LC-HR-ICP-MS (0.6–2.0 ng g−1; absolute LOD: 6–20 pg14), an improvement by a factor of 2000 to 50 ng g−1 (absolute LOD: 5 ng) compared to an ICP-qMS in its standard mode11 was achieved.
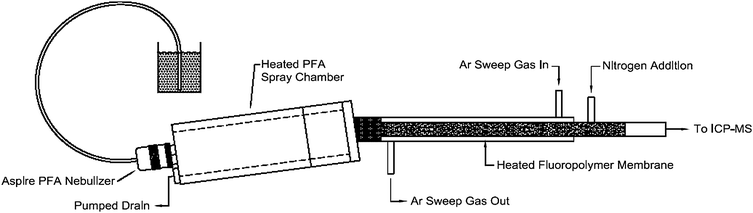 | ||
| Fig. 1 Schematic description of the Aridus system. The sample is introduced through a concentric PFA nebuliser into a heated PFA spray chamber (T = 383 K). The sample aerosol is then carried through a heated fluoropolymer membrane (recommended temperature 433 K) by the carrier gas argon. The solvent vapour evaporates through the membrane pores and is removed from the system by a counter flow of argon, whereas the analyte molecules are transported into the ICP. | ||
In this article, the use and suitability of a membrane desolvation system (Aridus, CETAC Technologies, Omaha, US) in combination with an ICP-qMS (Agilent 7500c) is reported and discussed for the quantitative speciation analysis of free phytochelatins and their arsenite complexes.
Experimental
Chemicals
All chemicals used were of analytical grade or better. Ultrapure water (Elga UK) was used throughout. The following chemicals were used: formic acid (ThermoFisher, UK); methanol, HPLC-grade (Rathburn UK); sodium-dimethylarsinic acid (98 %, DMAV) and sodium arsenite (AsIII) (ChemService, US); hydrogen peroxide (H2O2, 32 %, Fluka, UK); indium (ICP-MS standard, high purity standards, Charleston, US); disodiumhydrogenarsenate heptahydrate (AsV, BDH.); L-methionine and reduced glutathione (Sigma Aldrich, UK), phytochelatin standards PC2 and PC3 (Clonestar Biotech, Czech Republic).Synthesis of a calibration standard for sulfur containing peptides
For the synthesis of a suitable sulfur calibration standard 95 mg free glutathione (GSH) was dissolved in 1 mL ultrapure water. After addition of 0.5 mL hydrogen peroxide the solution was left for 12 h at room temperature, in order to achieve complete oxidation to the stable GSSG. The product was precipitated by addition of methanol, centrifuged and dried under vacuum. RP-HPLC-ES-MS analysis revealed the presence of two oxidation products (Fig. 2). One was identified as oxidised glutathione (GSSG) ([M-H]+: m/z 613; MS2: m/z 595; 484 and 355) and the second one as oxidised glutathione with one sulfoxy group in the molecule ([M-H]+: m/z 629; MS2: m/z 611; 500 and 324). The synthesised sulfur compound was used to make up standards, applied in the quantitative speciation analysis, in a concentration range of 50 to 700 ng S g−1.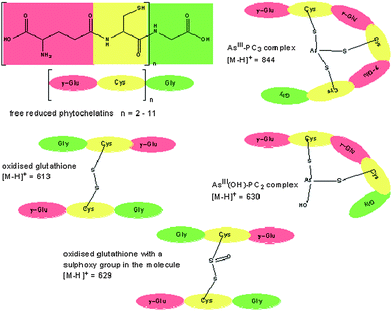 | ||
| Fig. 2 General structure of free reduced phytochelatins and their two arsenite complexes AsIII(OH)-PC2 and AsIII-PC3. The synthesised sulfur-standard contained the two forms of oxidised GSH shown in this figure. | ||
Standards: AsIII-PC2 and AsIII-PC3 mixture
The free reduced phytochelatins were dissolved in deionised water. For the synthesis of AsIII-PCs, an AsIII solution containing approx. 1.1 µmol As g−1 was made up in ultrapure water using NaAsO2. The formation of the AsIII-PC complexes was achieved by adding 0.9 g of the AsIII solution to 0.1 g of the PC stock solutions. The preparation of these standards was performed under nitrogen atmosphere to avoid oxidation of the PCs.Total analysis (ICP-qMS [Aridus])
The experimental set-up described below, was used to test the desolvation system (Aridus, CETAC Technologies) for its suitablity in species unspecific quantification. This was necessary since certain species might be lost within the desolvation unit either due to interaction with the membrane or due to volatiliy. Therefore, arsenite, arsenate and DMAV solutions, as well as sulfate, methionine and the sulfur-peptide standard were analysed for total content. All standards were made up in 0.1% formic acid in a concentration range of 100 to 500 ng S or As g−1. The sample uptake was achieved by self-aspiration. To exclude the possibility that varying sample uptake falsified the results every standard contained indium as internal standard. The applied settings for the desolvation unit can be found in table 1.| Parameters | ICP-qMS (Aridus) | ICP-qMS (oxygen) |
|---|---|---|
| a Using a xenon flow meter. b Under consideration of the ES-MS back-pressure. c During speciation analysis desolvator temperature was always set to 383 K. | ||
| ICP-qMS | ||
| RF-power/W | 1570 | 1570 |
| Carrier gas/L min−1 | 0.8–0.95 | 0.8 |
| Optional gas: 20% O2 in argon (%) | — | 5 |
| OctP bias/V | −6.2 | −4.0 |
| QP Bias/V | −3.5 | −3.5 |
| Reaction gas: O2 (%)a | — | 20 |
| (Integration time/isotope)/s | As (75) 0.6 | AsO (91) 0.7 |
| S (32) 0.9 | SO (48) 1 | |
| ICP-qMS : ES-MS split ratiob | 1 : 9 | 2 : 8 |
| HPLC | ||
| Column | Agilent, Eclipse XDB-C18, 5 µm, 4.6 × 150 mm | Agilent, Eclipse XDB-C18, 5 µm, 4.6 × 150 mm |
| Autosampler temperature/K | 277 | 277 |
| Mobile phase | Gradient of 0.1% formic acid and 0.1% formic acid in methanol, up to 20% methanol within 20 min | Gradient of 0.1% formic acid and 0.1% formic acid in methanol, up to 20% methanol within 20 min |
| Flow rate/mL min−1 | 1 | 1 |
| Column temperature/K | 293 | 293 |
| Aridus | ||
| Sweep gas/L min−1 | 3–4 | |
| Spray chamber temperature/K | 383 | |
| Desolvator temperature/K | 383c and 433 | |
HPLC-(ICP-qMS [Aridus]/ES-MS)
For the separation of the As- and S-species present in the AsIII-PC standard solutions a reversed phase column (Eclipse XDB-C18, 4.6 × 150 mm, Agilent) was used. Throughout all experiments the HPLC was coupled to an ES-MS (Agilent 1100, MSD, Agilent, USA) and an Agilent 7500c quadrupole ICP-MS with an octopole-collision cell system (Agilent, USA). In order to avoid the formation of 16O16O+ interferences the desolvation system replaced the concentric nebuliser of the ICP-qMS. The eluent-flow was split after the column 1 : 9 (Table 1). All quantification was done using the ICP-qMS signals on m/z 75 and m/z 32, for speciation arsenate and the synthesised S-standard (peak areas vs. concentration) were used as calibration species. The methanol gradient, necessary to separate the compounds of interest by RP-HPLC, led to changing analyte sensitivity for the analytes in the ICP-qMS in dependence on the methanol gradient. Hence, the quantification had to be adjusted correspondingly, as reported elsewhere.11HPLC-(ICP-qMS [with oxygen]/ES-MS)
For the separation of the As-species present in the AsIII-PC standard solutions a reversed phase column (Eclipse XDB-C18, 4.6 × 150 mm, Agilent) was used. Throughout all experiments the HPLC was simultaneously coupled to an ES-MS (Agilent 1100, MSD, Agilent, USA) and an Agilent 7500c quadrupole ICP-MS with an octopole-collision cell system (Agilent, USA). The reaction cell was pressurised with oxygen, in order to shift the interfered analytem/z 32 (32S+) to 48 (32S16O+). In order to prove artefact formation within the desolvation system, as shown and discussed later on, the ICP-qMS was used in its oxygen reaction cell mode. During this study sulfur was monitored on m/z 48 and arsenic on m/z 75. The eluent-flow was split after the column 2 : 8.For the quantitative analysis of the used AsIII-PC standards both elements, sulfur and arsenic, were monitored on their oxide-ions m/z 48 and 91. All quantification was done using the ICP-qMS signals on m/z 48 and m/z 91. The calibrants DMA and methionine were used. The quantification was adjusted corresponding to the changes caused by the applied methanol gradient.
Results and discussion
The performance of the desolvation unit for HPLC coupling applications was systematically investigated. In a first attempt, the linearity of the system was determined by using the desolvation system directly coupled to the ICP-qMS by aspiration of different sulfur and arsenic species, as detailed in the experimental part. This approach proved necessary because of the potential loss of species on the Aridus desolvation membrane or due to volatility. In a second approach sulfur and arsenic detection limits (LOD) were determined when RP-HPLC was coupled to ES-MS and ICP-qMS (Aridus). These LODs were compared with the literature values reported for the same ICP-qMS system in its collision and reaction cell mode. Finally AsIII-PC standards were analysed using on-line coupling of RP-HPLC with ES-MS and ICP-qMS (Aridus). The performance of this system was compared with the results obtained from the use of the oxygen reaction cell mode without prior desolvation.The most important feature is that a species-independent calibration must be possible due to the fact that unknown PC complexes are to be quantified, for which no standards are available.
Investigations regarding a potential loss of certain As- and S-species due to the desolvation system
To our knowledge, the chosen desolvation system has yet not been tested for its suitablity in speciation analysis of phytochelatins and their arsenite complexes. Therefore it had to be verified if species independent sulfur and arsenic quantification by ICP-qMS is still maintained, when the described desolvation system is applied.It could be shown that not for all compounds investigated a species independent quantification could be maintained and therefore great care had to be taken with regards to the calibration (Fig. 3). This was not entirely unexpected since the recommended membrane temperature of 433 K and the sweep gas are very likely to contribute to the removal of volatile compounds from the system. The transport of methionine, normally used for the sulfur calibration, to the ICP was strongly affected by the desolvation system. Running the desolvation unit at the recommended membrane temperature of 433 K did not lead to a linear calibration graph (data not shown). Only after changing the desolvator temperatur to 383 K a linear response was observed. However, the obtained R2 value of 0.9204 was evidence for poor linearity. But more importantly the smaller slope, compared to a sulfate calibration curve, indicated a loss of methionine in the desolvation system. Therefore methionine had to be replaced by a synthesised sulfur standard, containing two oxidised glutathione species (Fig. 2). This standard showed a matching slope with a sulfate calibration graph (Fig. 3) and hence showed its suitablity for species independent sulfur calibration. Following this trend, sulfur containing amino acids and peptides smaller than oxidised glutathione might be lost within the desolvation system. Hence, it was concluded that non-volatile peptides larger than the oxidised glutathione should show the same behaviour than sulfate and can therefore be quantified by using sulfate as a calibrant.
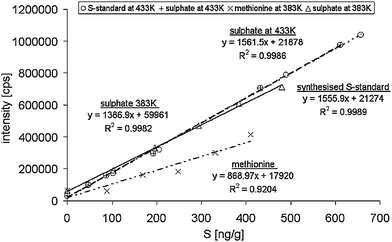 | ||
| Fig. 3 Total analysis of the synthesised S-standard for S-containing peptides and methionine in comparison with sulfate. | ||
From the investigated arsenic species, arsenite, DMAV and arsenate, only arsenate showed a linear response (R2 between 0.99 and 0.999) with increasing concentration. The removal of arsenite and DMAV due to volatility could be excluded since their sodium salts were used. Hence, other effects must be responsible for the partial loss of analyte. For arsenite indirect evidence for its adsorption onto the desolvation membrane was obtained as shown later.
These data showed that the application of the desolvation system has limitations concerning the quantification of certain sulfur and arsenic compounds.
Determination of As- and S-detection limits
For parallel arsenic and sulfur speciation analysis by RP-HPLC/ES-MS/Aridus-ICP-qMS arsenate and the synthesised S-peptide standard were chosen as calibrants. The sulfur detection limit of the ICP-qMS (Agilent 7500c) in its standard mode of approximately 100 µg g−1 could be improved by a factor of 2000 to 50 ng g−1 (3σ baseline noise) when the Aridus system was used. Comparable sulfur detection limits could be achieved when the ICP-qMS system was run in its reaction(O2)/collision(Xe) cell mode without prior desolvation,11 and the eluent flow to the ICP-qMS was higher than 200 µL min−1 (Table 2). Using an Aridus, however, required a flow of less than 100 µL min−1 and hence almost all sample was introduced into the ES-MS enhancing molecular information due to the set ES-MS/ICP-qMS flow split of 9 : 1. It was also evident and expected that none of the mentioned ICP-qMS (Agilent 7500c) applications provided sulfur detection limits as low as reported for a HR-ICP-MS system.11 The arsenic detection limits reported for the Agilent 7500c in its collision and reaction cell mode were comparable with the one for the HR-ICP-MS, which were lower than the ones obtained with the ICP-qMS in its standard mode or the application of the described desolvation system11 (Table 2).| Element | ICP-qMS (Aridus) | ICP-qMS | ICP-qMS (oxygen) | ICP-qMS (xenon) | HR-ICP-MS |
|---|---|---|---|---|---|
| As | 7.7 | 3 | 0.6 | 1.4 | 0.38 |
| S | 50 | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
86 | 79 | 4.9 |
The obtained sulfur and arsenic detection limits were promising for a successful application of the desolvation system regarding the parallel sulfur and arsenic speciation analysis in the case of free phytochelatins and their arsenite complexes. This could be proven by analysing a T. alata root extract after 24 h exposure to 1 ng AsV g−1, extracted as described in our previous paper.11 The applied instrumental set-up made not only the detection of the main AsIII-PC species, AsIII-PC3, but also the detection of minor species such as the two forms of AsIII-(PC2)2 possible (Fig. 4).
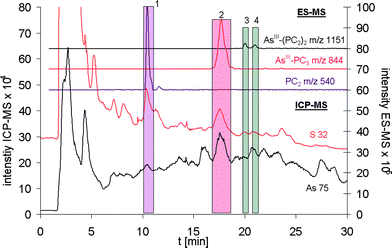 | ||
| Fig. 4 Arsenic and sulfur speciation analysis of a T. alata root extract using on-line coupling of RP-HPLC with ES-MS and Aridus-ICP-qMS (spray chamber: 383 K; desolvator: 383 K). The peak 1 represents free reduced PC2. The application of the desolvation system allowed next to detection of the major AsIII-PC complex, AsIII-PC3 (peak 2), also the detection of minor compounds such as the two forms of AsIII-PC2 (peak 3 and 4) could be detected. | ||
Speciation analysis by RP-HPLC-Aridus-ICP-qMS/ES-MS
In spite of the initial problems concerning the sulfur and arsenic calibration, the relatively low sulfur detection limits proved the suitability of the desolvation system for sulfur speciation analysis. Therefore AsIII-PC2 and AsIII-PC3 standards (Fig. 2) were made up and analysed using on-line coupling of RP-HPLC with ES-MS and ICP-qMS (Aridus). For the AsIII-PC2 standard the formation of an AsIII(OH)-PC2 (m/z 630) with a As : S ratio of 1 : 2 was reported.11 This ratio could not be confirmed when the desolvation system was used. Its application resulted in an As : S ratio of 1 : 2.7 (±0.2; four replicates) for the AsIII(OH)-PC2 complex. This result indicated that either another sulfur compound was co-eluting with the complex or that the complex was destroyed within the desolvation unit and not all of the released arsenite was transported into the plasma. Regarding a co-eluting S-compound seemed rather unlikely since a standard solution was used and the only other sulfur containing compounds present should have been reduced and oxidised PC2 (m/z 540 and 538), which eluted after the AsIII(OH)-PC2 complex (Fig. 5). Another unusual observation which did not affect the sulfur speciation was the appearance of two new arsenic peaks (highlighted with * in Fig. 5). These arsenic peaks co-eluted with those of the sulfur signals. The latter ones were established to be free reduced and oxidised PC2 by their protonated molecular masses in the simultaneously running ES-MS spectra. No other peak has been observed in the ES-MS at those retention times. This observation gave reason to suggest the formation of artefacts within the desolvation system before the sample enters the ICP-qMS. To prove this assumption exactly the same AsIII-PC2 standard was analysed after the Aridus was removed from the system and the ICP-qMS was set into its oxygen reaction cell mode. It was evident that no arsenic was co-eluting neither with free oxidised PC2 nor with free reduced PC2 (Fig. 5). Thus it was concluded that the arsenite eluting in the dead volume of the RP-HPLC method was first adsorbed within the desolvation unit and then mobilised by the later eluting free reduced and oxidised PC2. This possibility was supported by the fact that arsenite showed a smaller response and large variability than arsenate and was therefore likely to be lost in the desolvation system. Another contribution to the unexpected co-elution might have been derived from the relative unstability of AsIII-PC complexes. Their disintegration was reported to occur under the release of arsenite which again might have been adsorbed on the desolvating membrane.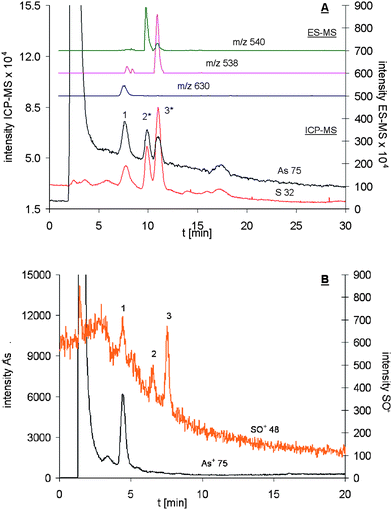 | ||
| Fig. 5 Arsenic and sulfur speciation analysis of an AsIII-PC2 mixture using on-line coupling of RP-HPLC with ES-MS and A: Aridus-ICP-qMS (spray chamber: 383 K; desolvator: 383 K and B: ICP-qMS (oxygen reaction cell). In both chromatograms peak 1 represents AsIII(OH)-PC2 for which the expected co-elution of arsenic and sulfur was observed. For free reduced (peak 2) and oxidised PC2 (peak 3) only a sulfur signal was expected and observed with the ICP-qMS (oxygen reaction cell) set up. The application of the desolvation system resulted in the formation of artefacts as the co-elution of sulfur and arsenic for both free reduced (peak 2*) and oxidised PC2 (peak 3*) showed. | ||
For an AsIII-PC3 standard an As : S ratio of 1 : 2.0 (±0.24; four replicates) instead of the theoretical one of 1 : 3 was found. The obvious reason for the discrepancy was peak overlapping between the sulfur signals of free PC3 and the arsenite complex. The lack of chromatographic resolution could have had two reasons, either column degradation and the chromatographic method itself or the large volume of the Aridus in comparison to the normal spray chamber (Scott-type). Since peak broadening was already observed as a side effect of the chosen desolvation system by Zheng and Hintelmann,15 it seemed very unlikely that column degradation was the reason for the insufficient peak separation. The same method was now used with and without the desolvation unit and the peak broadening reported in Table 3 was the main cause for the lack of chromatographic resolution. Using the RP-HPLC-ICP-qMS (oxygen) method resulted in base line separated peaks for free PC3 and the AsIII-PC3 complex (Fig. 6).
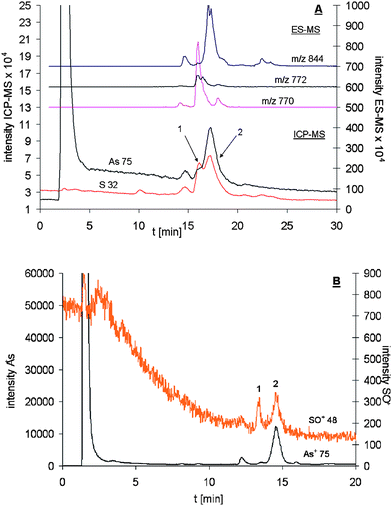 | ||
| Fig. 6 Arsenic and sulfur speciation analysis of an AsIII-PC3 mixture using on-line coupling of RP-HPLC with ES-MS and A: Aridus-ICP-qMS (spray chamber: 383 K; desolvator: 383 K) and B: ICP-qMS (oxygen reaction cell). Here, the free reduced and oxidised PC3 are represented as peak 1 whereas the AsIII-PC3 complex was labelled as peak 2. Both are shown as baseline separated peaks when the ICP-qMS was run in its oxygen reaction cell mode. Insufficient peak separation was observed when the desolvation unit was coupled to the ICP-qMS system. | ||
To prove that the discrepancy in As : S was due to the use of the Aridus system, the RP-HPLC/ES-MS/ICP-qMS (oxygen) set-up was optimised (Table 1) and the standards were analysed again. For the described AsIII(OH)-PC2 complex an As : S ratio of 1 : 1.9 (duplicates) was found. For the AsIII-PC3 complex a ratio of 1 : 3.2 (±0.1; triplicates) was observed. This slight discrepancy, in the later case, from the expected value was due to a not complete base-line separation between the free PC3 and the AsIII-PC3 complex (data not shown) during this analysis. The As : S ratios found with the ICP-qMS oxygen set-up varied only slightly from the expected values. Hence, it was concluded that the mismatching results obtained with the desolvation system was due to analyte interaction with the desolvation unit.
These data revealed that simultaneous sulfur and arsenic speciation analysis in the case of free phytochelatins and their arsenite complexes was accompanied by severe disadvantages, such as artefact formation and peak broadening, when the Aridus system is used. These disadvantages can give reason to wrong interpretation of obtained results, as the As : S ratios showed. Nevertheless, in general the desolvation system seems to be suitable for sulfur speciation analysis, of sufficiently separated and stable non-volatile compounds, which do not show interactions, such as adsorption, with the desolvation unit.
Conclusion
The improvement of the sulfur detection limit of the used ICP-qMS by a factor of 2000 to 50 ng g−1 demonstrated the general suitablity of the desolvation system for sulfur analysis. However, in the special case of the speciation analysis of phytochelatins and their arsenite complexes this approach failed. This was due to instablity of the arsenite complexes and the adsorption of free AsIII on the membrane, as well its subsequent mobilisation by free PCs. Next to this artefact formation, the analyte transport of certain arsenic and sulfur species to the ICP-qMS was affected and hence, species independent analysis was restricted to selected compounds.Acknowledgements
The authors thank the late Dr Petra Krause and Fred Smith (both CETAC) for using the Aridus system and the College of Physical Sciences for financial support.References
- A. Raab, J. Feldmann and A. Meharg, Plant Physiol., 2004, 134, 1113 CrossRef CAS.
- M. Quaghebeur and Z. Rengel, Microchim. Acta, 2005, 151, 141 CrossRef CAS.
- M. Berlich, S. Menge, I. Bruns, J. Schmidt, B. Schneider and G.-J. Krauss, Analyst, 2002, 127, 333 RSC.
- F. E. C. Sneller, L. M. van Heerwaarden, P. L. M. Koevoets, R. Vooijs, H. Schat and J. A. C. Verkleij, J. Agric. Food Chem., 2000, 48, 4014 CrossRef CAS.
- S. Döring, S. Korhammer, M. Oetken and B. Markert, Fresenius’ J. Anal. Chem., 2000, 366, 316 CrossRef.
- D. Pröfrock, P. Leonhard and A. Prange, Anal. Bioanal. Chem., 2003, 377, 132 CrossRef CAS.
- J. T. Rowan and R. S. Houk, Appl. Spectrosc., 1989, 43, 976 CAS.
- P. R. D. Mason, K. Kaspers and M. J. van Bergen, J. Anal. At. Spectrom., 1999, 14, 1067 RSC.
- B. P. Jensen, C. Smith, I. D. Wilson and L. Weidolf, Rapid Commun. Mass Spectrom., 2004, 18, 181 CrossRef CAS.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Anal. Chem., 2002, 74, 1497 CrossRef CAS.
- K. Bluemlein, A. Raab, A. A. Meharg, J. M. Charnock and J. Feldmann, Anal. Bioanal. Chem., 2008, 390, 1739 CrossRef CAS.
- C.-F. You and Li , Mon-Dar, J. Anal. At. Spectrom., 2005, 20, 1392 RSC.
- S. Hann, G. Koellensperger, C. Obinger,, P. G. Furtmueller and G. Stingeder, J. Anal. At. Spectrom., 2004, 19, 74 RSC.
- E. H. Evans, J.-C. Wolff and C. Eckers, Anal. Chem., 2001, 73, 4722 CrossRef CAS.
- J. Zheng and H. Hintelmann, J. Anal. At. Spectrom., 2004, 19, 191 RSC.
| This journal is © The Royal Society of Chemistry 2009 |