Selective aerobic oxidative dibromination of alkenes with aqueous HBr and sodium nitrite as a catalyst†
Ajda
Podgoršek
a,
Marco
Eissen
b,
Jens
Fleckenstein
b,
Stojan
Stavber
a,
Marko
Zupan
ac and
Jernej
Iskra
*a
aLaboratory of Organic and Bioorganic Chemistry, “Jožef Stefan” Institute, Jamova 39, 1000, Ljubljana, Slovenia. E-mail: jernej.iskra@ijs.si; Fax: +386 1 4773 822; Tel: +386 1 4773 631
bGymnasium Ganderkesee, Am Steinacker 12, 27777, Ganderkesee, Germany
cFaculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerčeva 5, 1000, Ljubljana, Slovenia
First published on 17th November 2008
Abstract
Various alkenes (internal, terminal, aryl and alkyl substituted) and 1,2-diphenylethyne were efficiently and selectively dibrominated using 2 equivalents of 48% aqueous hydrobromic acid, with air as an oxidant and sodium nitrite as a catalyst. Despite the presence of water, only trans dibromination occurred producing anti-1,2-dibromoalkanes and (E)-1,2-dibromo-1,2-diphenylethene. A comparison of resource demand, waste production and environmental, health and safety issues of the NaNO2 catalyzed aerobic bromination with molecular bromine and other oxidative bromination methods revealed that the proposed method is not only selective and effective but has a better environmental profile.
Introduction
Bromination of alkenes and alkynes has garnered a significant amount of attention among those studying bromination reactions. Dibrominated derivatives are useful precursors for organometallic reagents and are widely applicable as intermediates in synthetic organic chemistry.1,2 However, the use of molecular bromine in chlorinated solvents remains the preferred choice in the dibromination of organic molecules in spite of its toxic and corrosive nature. Moreover, the reactive nature of bromine results in highly exothermic reactions that makes performing bromination in a selective manner difficult and leads to its substitution by different pyridinium and quarternary ammonium tribromides3 or N-bromosuccinimide in combination with lithium bromide.4 The negative impact of such brominating protocols on the environment includes the use of environmentally unfriendly solvents, significant waste production and the need to remove residual reagents.5 Nevertheless, bromine is still needed to prepare the reagents and when compared to alternative protocols, this classical approach of using molecular bromine remains resource efficient.5b Alternatively, nature has developed a molecular halogen-free strategy for halogenation using hydrogen peroxide (haloperoxidase) or oxygen (flavin dependent halogenase) and X− to produce “X+” at the enzyme's active site.6 The oxidative halogenation with the in situ formation of an active brominating species via the oxidation of bromide in a more suitable solvent could be a promising way of overcoming the current drawbacks, especially when either hydrogen peroxide or oxygen, as the most environmentally benign of the oxidants, are involved.7–10Alkenes are useful substrates for investigating the nature of active halogenating agents11 and are used many times in biomimetic studies of haloperoxidation, mainly with a metal catalyst.12 Research has focused on the selectivity of bromohydrin and dibromide formation in a vanadium(V) or molybdenium(VI) catalyzed oxybromination of alkenes using potassium bromide and hydrogen peroxide. Performing these reactions in water gave bromohydrin as the main product, despite a large excess of bromide ions.13 In a two-phase system using either H2O-CHCl39d,12c,d,13 or H2O-CH2Cl2,12d,e more dibrominated product together with bromohydrin was formed. Only when tetrabutylammonium bromide together with H2O2 and catalytic V2O5 in H2O-CH3CN was used as a tribromide precursor was the formation of dibromides from cyclohexene and styrene observed.9c A non-catalyzed oxidative bromination with H2O2/HBr in a two-phase system with either CCl4 or an ionic liquid also gave dibromides selectively.8a,14
In contrast with hydrogen peroxide, oxygen has seldom been used for oxidative halogenation despite it being the most abundant, cheapest and atom economical oxidant. Only a few examples of aerobic oxybrominations of aromatic compounds catalyzed by various metal catalysts are reported.10b,f,g Zhang et al. describe a metal-free catalyzed oxidative bromination of aromatic compounds and aryl ketones utilizing a combination of aqueous hydrobromic acid, molecular oxygen and sodium nitrite as the catalyst.10c A similar system was also used for aerobic iodination.10d To the best of our knowledge, there are only two examples of aerobic oxidative halogenation that includes the oxidative halogenation of alkenes using a metal species as a catalyst. Bromination of 1-octene10g and chlorination of cyclododecene and trans-5-decene15 resulted in the non-stereoselective formation of dihaloalkanes accompanied by products of addition of hydrohalic acid to the double bond.
This report presents a study on the selective and stereoselective aerobic dibromination of alkenes and alkynes using air at near ambient pressure as the oxidant, an equimolar amount of aqueous hydrogen bromide and sodium nitrite as a catalyst. Environmental aspects, such as resource demands and waste production together with health and safety issues, will be evaluated in comparison to Br2 addition and other oxidative bromination. Furthermore, the effect of the slow generation of the “active brominating agent” on the selectivity of bromination is discussed.
Results and discussion
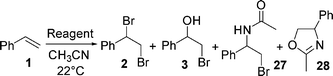
To define optimum reaction conditions for oxidative bromination of alkenes in an HBr/air/NaNO2(cat.) system we initially studied the reaction of styrene (1) in different solvents (acetonitrile, ethyl acetate, dichloromethane, hexane, methanol and water). Styrene (1) makes an excellent test substrate since its transformation is particularly sensitive to reaction conditions, eg.polymerization, incorporation of various nucleophiles from the milieu into the first-formed carbocation. First, 1 mmol of styrene (1) was dissolved in 10 mL of MeCN, then 2 mmol of 48% aqueous HBr (226 μL) and 5 mol% of NaNO2 were added. The flask was immediately covered with an air filled balloon and stirred at 22 °C for 5 hours. On completion, ethyl acetate was added. The organic phase was discoloured by adding a few drops of 37% aq. NaHSO3, neutralized by NaHCO3 and dried over anhydrous Na2SO4. After removing the solvent, the 1,2-dibromo-1-phenylethane (2) was obtained. The role of solvent on functionalization of 1 is shown on Scheme 1. The best yield of 2 is obtained in acetonitrile, where only a trace amount of bromohydrin 3 was observed in the crude reaction product. Also, no excess hydrogen bromide was necessary for the complete conversion of styrene (1) into dibromide 2. Interestingly, although 10 equivalents of water were present in the reaction mixture from using an aqueous solution of HBr, it did not interfere in the bromination process by forming bromohydrin 3. In ethyl acetate, 78% of the dibrominated product 2 was formed non-selectively, the reaction in hexane gave a complex mixture of products, while in dichloromethane only a trace amount of 2 was obtained. Moreover, the use of either methanol or water as a reaction medium completely suppressed the oxidative bromination of styrene (1).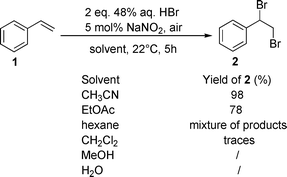 | ||
| Scheme 1 | ||
Next, the effect of the mode of addition of sodium nitrite on conversion was investigated. Five mol% of solid NaNO2 was added in one portion (Method A), in three portions every 2 hours (Method B) and as a 20 μL aqueous solution of 2.5 M NaNO2 (Method C). Table 1 shows that there is no significant difference when sodium nitrite is added either all at once or in three discrete portions. However, a slightly higher yield of dibrominated product 2 was obtained when sodium nitrite was added as a 2.5 M aqueous solution, possibly because the sodium nitrite was uniformly distributed in the reaction mixture. Interestingly, although additional water was present in the system less bromohydrin was formed. Therefore, in all subsequent experiments an aqueous solution of sodium nitrite was added.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Methoda | Distribution (%)b | ||||||
| 1 | : | 2 | : | 3 | : | 4 | ||
| a Method A: 5 mol% of solid NaNO2 was added in a single portion, Method B: 5 mol% of solid NaNO2 was added in three portions every 2 hours, Method C: 20 μL of 2.5 M aq. NaNO2 was added; b Determined by 1H NMR spectroscopy. | ||||||||
| 1 | A | / | : | 94 | : | 6 | : | / |
| 2 | B | / | : | 95 | : | 5 | : | / |
| 3 | C | / | : | 98 | : | 2 | : | / |
| 4 | C, without addition of NaNO2 | 66 | : | / | : | / | : | 34 |
| 5 | C, without an air balloon | 18 | : | 34 | : | / | : | 48 |
To highlight the role of sodium nitrite and the presence of air on the course of oxidative bromination of styrene (1), blank experiments both without sodium nitrite and air were performed. In the absence of sodium nitrite, the addition of HBr to styrene (1) is the sole reaction with 34% of (1-bromo-ethyl)-benzene (4) being formed (Table 1). In the absence of air only 34% of the dibrominated product 2 was formed and the addition of HBr on the double bond was the main reaction resulting in the formation of 4 in a 48% yield. From this we can conclude that catalytic amounts of sodium nitrite under aerobic conditions prevent the addition of hydrobromic acid to the alkene, while air as an oxidant enables the “in situ” formation of the brominating species and therefore the dibromination of the alkene.
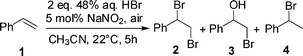
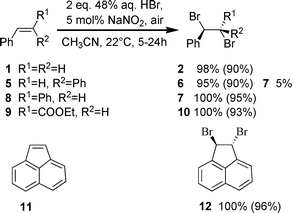
To evaluate the scope of this catalytic system, optimum reaction conditions: 1 mmol of substrate, 2 mmol of 48% aqueous solution of HBr, 20 μL of 2.5 M aq. NaNO2 (5 mol%) and 10 mL of acetonitrile, were used to oxidatively brominate various internal and terminal aryl and alkyl substituted alkenes and 1,2-diphenylethyne. The results are represented in Schemes 2–5. Yields were determined by 1H NMR spectroscopy and are based on the starting compound, while the yields given in parentheses refer to isolated yields. Using this method, β-substituted styrenes were quantitatively and diastereoselectively converted to dibrominated products (Scheme 2). Bromination of trans-stilbene (8), ethyl trans-cinnamate (9) and acenaphthylene (11) gave anti-brominated products diastereo-specifically. cis-Stilbene (5) was also trans brominated under the above conditions yielding syn-1,2-dibromo-1,2-diphenylethane (6) accompanied by 5% of the anti isomer 7.
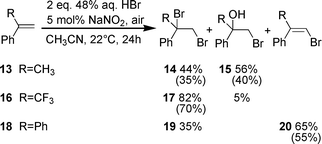
The introduction of a second substituent in the α position in styrene (1) modifies the stability of the carbocation intermediate so that besides the competitive addition of a nucleophile, deprotonation resulting in an addition–elimination process could become an important reaction channel. With α-methylstyrene (13), only an addition reaction occurred with the competitive formation of the dibromide 14 and the bromohydrin 15 in a ratio of 44/56. (Scheme 3). α-(Trifluoromethyl)styrene (16), that forms a more deactivated bromocarbonium ion, was brominated in a lower yield, nevertheless dibromoalkane 17 was the main product. Bromination of 1,1-diphenylethene (18) follows an addition-elimination mechanism to give 2-bromo-1,1-diphenylethene (20) and 35% of the addition product 19.
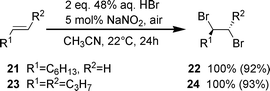
The aliphatic terminal alkene 21 and internal alkene 23 were quantitatively and selectively brominated to give 1,2-dibromooctane (22) and anti-4,5-dibromooctane (24), respectively (Scheme 4).
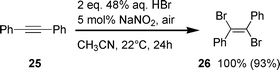
On completion, aerobic oxidative bromination was tested for bromination of the alkyne 25. Similarly to reactions with alkenes, 25 was also trans-brominated to (E)-1,2-dibromo-1,2-diphenylethene (26) stereospecifically with an isolated yield of 93% (Scheme 5).
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
In a small scale experiment, extraction or dilution with an appropriate organic solvent has to be applied in a work-up procedure. This method has the potential of producing a simpler work-up procedure due to the absence of any organic by-products and residues. On a larger scale, 5-times less MeCN is required for the reaction and a different work-up procedure, depending on the aggregate state of the formed product, was employed. Oxidative bromination of trans-stilbene (8) and trans-4-octene (23) were performed on a 5 mmol scale (5 mmol of alkene, 10 mmol of 48% aq. HBr, 70 μL of 3.6 M aq. NaNO2, 10 mL of CH3CN). After stirring the reaction mixture for either 16 h or 24 h at room temperature, the acetonitrile was distilled off and regenerated. In the case of the solid product 7, the crude reaction mixture was poured into water and the resultant white precipitate was filtered off, and after drying 95% of meso-1,2-dibromo-1,2-diphenylethane (7) was obtained. In the case of the liquid product 24, a minimum quantity of EtOAc was added, the resultant organic phase was dried over Na2SO4 and the solvent was evaporated. The result was anti-4,5-dibromooctane (24) in a 94% yield.
When comparing alternative protocols, the relevant environmental, health and safety aspects must also be taken into consideration. Regarding resource efficiency, the conventional procedure using molecular bromine16 (Scheme 6a) or hydrobromic acid and hydrogen peroxide8a (Scheme 6b) compare favourably to the twenty-four procedures examined.5b These procedures had been chosen to be juxtaposed to the larger-scale procedure for aerobic oxidative dibromination of trans-stilbene (8) with HBr/air/NaNO2(cat.) system (Scheme 6c).
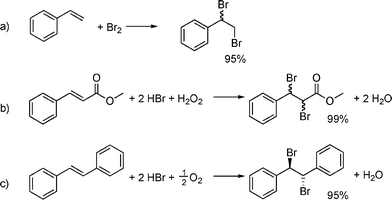 | ||
| Scheme 6 Bromination methods using (a) molecular bromine,16 (b) hydrobromic acid and hydrogen peroxide,8a and (c) hydrobromic acid and oxygen. | ||
The protocols are from the laboratory stage. Against this background, the environmental factors E = ∑ Waste [kg]/Product [kg] given in Fig. 1 demonstrate that there is no significant difference regarding waste production and solvent demand between this method and other bromination methods5b with much higher E factors. Protocol (a) has the advantage that no water is used so waste water treatment is unnecessary.
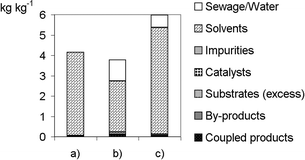 | ||
| Fig. 1 Environmental factor E of different bromination methods shown in Scheme 6 using the software EATOS.17 For details see Table 2. Work-up procedures have not been described in detail8a,16 and, thus, were not considered here: (a) recrystallization; (b) washing with water, brine and drying over sodium sulfate; (c) pouring into 10 mL of water. | ||
Contrary to the quantitative results, the nature of the reagents used distinguishes themselves notably. Both, molecular bromine and an aqueous solution of hydrobromic acid have corrosive properties. Additionally, hydrogen peroxide is not only corrosive but can decompose in a runaway scenario.18 However, bromine is much more volatile and has a higher risk potential than hydrobromic acid. Another important issue is the use of toxic and environmentally problematic substances such as chloroform (solvent of protocol (a)) or tetrachloromethane (solvent of protocol (b)). Evidence, although limited, suggests that CCl4 is a carcinogen. Moreover it can have long-term adverse effects on the aquatic environment, is ozone depleting and a greenhouse gas. The authors of protocol (b) suggest dioxane as an alternative solvent, but dioxane is an irritant, highly flammable, can form explosive peroxides and potentially carcinogenic. Obviously, the advantage of substituting bromine with hydrobromic acid in protocol (b) becomes revoked because technology equipment has to avert the potential risk associated with the use of halogenated solvent or dioxane. In this regard, the application of acetonitrile, albeit highly flammable but merely harmful instead of toxic in protocol (c) respresents a significant progress and, furthermore, air is used instead of hydrogen peroxide.
The actual mechanism of aerobic oxidative bromination using hydrobromic acid and sodium nitrite as a catalyst is yet to be investigated. However, bromine was proposed as a reactive brominating species for the bromination of aromatic compounds and aryl ketones.10c Alkenes are convenient substrates to study the mechanism of halogenation and the nature of halogenating reagents. We performed a study on the effect of a brominating reagent's structure and the mode of its addition on the course of bromination of styrene (1) and observed some interesting results. Classical bromination of 1 with bromine in acetonitrile follows a different pathway to that of an HBr/air/NaNO2(cat.) system; only 60% of 1,2-dibromo-1-phenylethane (2) was formed, with the remainder being acetamide 27 and oxazole 28 formed by the incorporation of acetonitrile into the carbocation intermediate (Table 3, entry 2). An analogous reaction with an additional 10 equivalents of water, as is present when using aqueous HBr in aerobic oxidative bromination, yielded a similar amount of dibromide 2 (entry 3). Also, significant differences were noticeable when molecular bromine was added slowly to a solution of styrene (1) in acetonitrile by its diffusion via the gaseous phase. In this case similar results to aerobic oxidative bromination were obtained, with the selective formation of the dibrominated product 2 (entry 4), thus pointing to the importance of the slow formation of the brominating species during the course of the oxidative bromination. A similar result was observed when HBr3 was formed as a brominating agent prior to the addition of 1; less bromohydrine 3 was formed despite aqueous HBr being used (entry 5). When using BrOH/NaBr for bromination (entry 6) and when the brominating agent is formed in situviahaloperoxidase-like oxidation of HBr with hydrogen peroxide (entry 7), the formation of bromohydrin 3 is more expressed. It appears that the slow generation of bromine could be the key for the selective formation of vic-dibromides with the possibility of forming HBr3 due to an excess of HBr at the early stages of the reaction.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time (h) | Distribution (%)a | ||||||
| 2 | : | 3 | : | 27 | : | 28 | |||
| a Determined by 1H NMR spectroscopy. b Styrene (1 mmol) was added to the solution of Br2 (1 mmol) (and H2O (10 mmol)) in CH3CN (10 mL). c A similar reactivity was observed to that reported in ref. 19. d Bromine vapours were added by diffusion from the gaseous phase20 to the solution of styrene (1 mmol) in CH3CN (10 mL). e Br2 (1 mmol) and HBr (1 mmol) were mixed together in CH3CN (10 mL) followed by the addition of styrene (1 mmol). f Br2 (1 mmol) and NaOH (1 mmol) were mixed together and then the solution of styrene (1 mmol) in CH3CN (10 mL) and H2O (10 mmol) was added. g 30% aq. H2O2 (1 mmol) and 48% aq. HBr (2 mmol) were added to the solution of styrene (1 mmol) in CH3CN (10 mL). | |||||||||
| 1 | HBr (2eq.), NaNO2 (0.05 eq.), air | 5 | 98 | : | 2 | : | / | : | / |
| 2 | Br2 (1eq.)b,c | 3 | 60 | : | / | : | 20 | : | 20 |
| 3 | Br2 (1eq.), H2O (10 eq.)b | 3 | 61 | : | 23 | : | 11 | : | 5 |
| 4 | Br2 (1eq.)d | 24 | 96 | : | 4 | : | / | : | / |
| 5 | HBr3 (1eq.)e | 3 | 98 | : | 2 | : | / | : | / |
| 6 | BrOH/NaBr (1eq.), H2O (10 eq.)f | 3 | 84 | : | 14 | : | 1 | : | 1 |
| 7 | H2O2 (1eq.), HBr (2 eq.)g | 24 | 90 | : | 10 | : | / | : | / |
Also, what is noteworthy is the selective formation of trans-addition products in aerobic bromination. This result is different to bromination with bromine in chlorinated solvents, where cis-addition was also observed in the bromination of acenaphthylene (11, trans : cis 70 : 30),21trans-stilbene (8, trans : cis 84 : 16)22 and diphenylethyne (25, trans : cis 60 : 40).23 Alternatively, when H2O2/HBr was used for bromination in a biphasic system with CCl4 or an ionic liquid only trans-bromination takes place in the bromination of methyl trans-cinnamate.8a,24 A similar effect was observed in bromination with pyridinium tribromide, where a reaction with trans-stilbene (8) in CH2Cl2 produces an 86 : 14 mixture of trans : cis-adducts, while the reaction in a water suspension yields only the trans-adduct.22
Nitrogen oxides were suggested to be the catalyst in this reaction due to a well known decomposition of nitrite under acidic conditions into a mixture of nitrogen oxides. By mixing NaNO2 with a strong acid (HClO4) in MeCN, colorless bubbles immediately start to evolve and solution remained colorless even after a prolonged period of time in air. When HBr is used as an acid, dark yellow bubbles appear and the solution gradually becomes yellow-orange which, together with the fact that a solution of bromine in MeCN is red-brown, suggests the formation of a different decomposition product of NaNO2 with HBr. A review of available literature suggests that the formation of a nitrosyl bromide could explain the yellow-orange colour of the solution (Scheme 7A). NOBr reportedly decomposes at room temperature into nitric oxide and bromine (Scheme 7B).25a We turned to UV-vis spectroscopy to determine the presence of NOBr as a product of the decomposition of NaNO2 with HBr in acetonitrile. In the spectra obtained for a solution of Br2 in MeCN, two peaks at 269 (s) and 390 (br) nm are observed, whereas for a degassed suspension of NaNO2 in MeCN treated with HBr, a spectra is obtained with peaks at 269 (br) and 330 (br) nm. According to the literature data, NOBr has a peak at 225 nm that is in our case overlapped by the strong signal of Br2,25b,c while a broad band at 330 nm corresponds with the literature one.25b However, after 30 min only absorption signals for Br2 are observed. Our conclusion is that during NaNO2 catalyzed aerobic dibromination of alkenes, nitrite is first transformed into NOBr, which then decomposes to bromine and nitric oxide (Scheme 7). Thus, a small amount of bromine is already formed in this pre-catalytic phase and is rapidly consumed by the alkene. This shifts the equilibrium towards complete decomposition of NOBr.
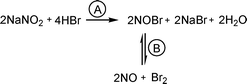 | ||
| Scheme 7 | ||
At which point, nitric oxide enters into the catalytic cycle of aerobic bromination, where it is oxidized into NO2 by air. This in turn oxidizes HBr into Br2 (Scheme 8). It appears that the slow generation of bromine is crucial for selective trans-dibromination of alkenes, while the presence of unreacted HBr in the reaction mixture makes the formation of HBr3 possible and its role in selective dibromination can not be precluded (Table 3, entries 4, 5).
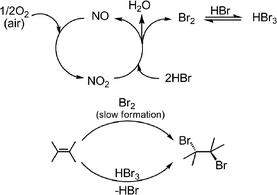 | ||
| Scheme 8 | ||
Conclusion
Reported herein is a mild and simple method for the efficient and selective bromination of alkenes, in which aqueous HBr is used as a source of bromine, air at near ambient pressure as the oxidant and NaNO2 as a low-cost catalyst. This procedure performed in acetonitrile provides a practical alternative to other procedures by avoiding the use of volatile bromine and problematic halogenated solvents. Furthermore, the isolation procedure is straightforward requiring only the separation of the organic product from the inorganic salts. It was established that NOBr is first formed from the catalyst and HBr and its decomposition leads to the generation of NO that acts as the true catalyst in aerobic oxidative bromination. In the bromination step, the formation of different active brominating agents is possible, however it seems that the selectivity of bromination is governed by the slow formation of bromine through aerobic oxidation, while its conversion into HBr3 with residual HBr could also play an important role.Both, environmental and health and safety aspects were also considered and compared with alternative protocols and aerobic dibromination. The obtained E-factors show that this aerobic protocol is similar to bromination using Br2 and an H2O2/HBr system in chlorinated solvents. However, aerobic bromination with HBr/NaNO2/air replaces Br2 with the less volatile aqueous HBr and hydrogen peroxide with air, while chlorinated solvents are eliminated altogether. These and the good selectivity and yield makes this method a promising alternative to other dibromination protocols.
Experimental
General
All chemicals were obtained from commercial sources and were used without further purification. Acetonitrile was distilled before use. Column and thin layer chromatography were carried out using silica gel 60 (0.063–0.200 mm) and silica 60F-245 plates, respectively. 1H and 13C NMR spectra were recorded in CDCl3 using a Varian Inova 300 MHz spectrometer. The chemical shifts (δ) are reported in ppm units relative to TMS as an internal standard for 1H NMR and CDCl3 for 13C NMR spectra. Melting points were determined using a Büchi 535 melting point apparatus. Mass spectra were obtained using an Autospec Q mass spectrometer with electron impact ionization (EI, 70 eV).Typical reaction procedure for aerobic oxidative dibromination of alkenes and 1,2-diphenylethyne with HBr/air/NaNO2(cat.) system
In a typical experiment 1.0 mmol of substrate was dissolved in 10 mL of freshly distilled acetonitrile, then 2.0 mmol of 48% aqueous solution of HBr (226 μL) and 20 μL of 2.5 M aqueous solution of NaNO2 (0.05 mol of NaNO2) were added. The flask was immediately covered with an air filled balloon (1 L) and the solution stirred at room temperature for 5–24 h. The progress of the reaction was monitored by TLC. At the end of the reaction, 5 mL of ethylacetate was added. The organic phase was discoloured by adding a few drops of 37% aq. NaHSO3, neutralized by NaHCO3 and finally dried over anhydrous Na2SO4. After the removal of solvent, the isolated reaction mixture was analyzed by 1H NMR spectroscopy. Finally, products were isolated by flash or column chromatography (SiO2, hexane–EtOAc) or purified by crystallization and their structures determined by comparison with the literature data.Larger-scale procedure for aerobic oxidative dibromination of trans-stilbene (8) with HBr/air/NaNO2(cat.) system
5.0 mmol (901 mg) of 8 was mixed with 10 mL of freshly distilled acetonitrile, then 10.0 mmol of a 48% aqueous solution of HBr (1.13 mL) and 70 μL of a 3.6 M aqueous solution of NaNO2 (0.25 mol of NaNO2) were added. The flask was immediately covered with an air filled balloon (1 L) and solution stirred at room temperature for 12 hours. The progress of the reaction was monitored by TLC. At the end of the reaction MeCN was evaporated under reduced pressure, crude reaction mixture was poured into 10 mL of water and white precipitate was filtered off. After drying, 1.615 g (4.75 mmol) of 7 was obtained as white crystaline product.Larger-scale procedure for aerobic oxidative dibromination of trans-4-octene (23) with HBr/air/NaNO2(cat.) system
5.0 mmol (561 mg) of 23 was mixed with 10 mL of freshly distilled acetonitrile, then 10.0 mmol of a 48% aqueous solution of HBr (1.13 mL) and 70 μL of a 3.6 M aqueous solution of NaNO2 (0.25 mol of NaNO2) were added. The flask was immediately covered with an air filled balloon (1 L) and solution stirred at room temperature for 24 hours. The progress of the reaction was monitored by TLC. At the end of the reaction MeCN was evaporated under reduced pressure. Crude reaction mixture was diluted with 6 mL of EtOAc, dried with 0.25 g of Na2SO4 and inorganic material filtered off. After evaporating the solvent 1.278 g (4.70 mmol) of 24 was obtained as oily product.Acknowledgements
This research has been supported by the Ministry of Higher Education, Science and Technology of the Republic of Slovenia and the Young Researcher Program (A.P.) of the Republic of Slovenia. The authors are grateful to the staff of the National NMR Centre at the National Institute of Chemistry in Ljubljana and the staff of the Mass Spectroscopy Centre at the JSI. We are grateful to Prof. Dieter Lenoir for helpful discussions.Notes and references
- The Chemistry of Functional Groups: Supplement D2, The Chemistry of Halides, Pseudo-Halides and Azides, Part 1 & 2, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1995 Search PubMed.
- M. J. Dagani, H. J. Barda, T. J. Benya and D. C. Sanders, Bromine Compounds, in Ullmann's Encyclopedia of Industrial Chemistry, electronic edition, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) K. Tanaka, R. Shiraishi and F. Toda, J. Chem. Soc., Perkin Trans. 1, 1999, 3069–3070 RSC; (b) J. Salazar and R. Dorta, Synlett, 2004, 1318–1320 CrossRef CAS; (c) G. Bellucci, R. Bianchini, R. Ambrosetti and G. Ingrosso, J. Org. Chem., 1985, 50, 3313–3318 CrossRef CAS; (d) R. Bianchini and C. Chiappe, J. Org. Chem., 1992, 57, 6474–6478 CrossRef CAS; (e) V. Kavala, S. Naik and B. K. Patel, J. Org. Chem., 2005, 70, 4267–4271 CrossRef CAS.
- L.-X. Shao and M. Shi, Synlett, 2006, 8, 1269–1271 CrossRef.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press Inc., New York, 1998 Search PubMed; (b) M. Eissen and D. Lenoir, Chem.–Eur. J., 2008, 14, 9830–984 CrossRef.
- (a) F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef; (b) D. G. Fujimori and C. T. Walsh, Curr. Opin. Chem. Biol., 2007, 11, 553–560 CrossRef CAS; (c) K. H. van Pee, C. J. Dong, S. Flecks, J. Naismith, E. P. Patallo and T. Wage, Adv. Appl. Microbiol., 2006, 59, 127–157 Search PubMed.
- (a) W. J. Jones, Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed; (b) D. Lenoir, Angew. Chem., Int. Ed., 2006, 45, 3206–3210 CrossRef CAS.
- (a) N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. V. Bedekar, Tetrahedron, 1999, 55, 11127–11142 CrossRef CAS; (b) A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Green Chem., 2007, 9, 1212–1218 RSC; (c) A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Tetrahedron Lett., 2006, 47, 7245–7247 CrossRef CAS; (d) J. Iskra, S. Stavber and M. Zupan, Synthesis, 2004, 1869–1873 CrossRef CAS; (e) J. Barluenga, M. Marco-Arias, F. Gonzalez-Bobes, A. Ballesteros and J. M. Gonzalez, Chem.–Eur. J., 2004, 10, 1677–1682 CrossRef CAS; (f) R. Ben Daniel, S. P. de Visser, S. Shaik and R. Neumann, J. Am. Chem. Soc., 2003, 125, 12116–12117 CrossRef CAS.
- (a) B. Sels, D. De Vos, M. Buntinx, F. Pierard, A. Kirsch-De Mesmaeker and P. Jacobs, Nature, 1999, 400, 855–857 CrossRef CAS; (b) B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350–8359 CrossRef CAS; (c) U. Bora, G. Bose, M. K. Chaudhuri, S. K. Dhar, R. Gopinath, A. T. Khan and B. K. Patel, Org. Lett., 2000, 2, 247–249 CrossRef CAS; (d) V. Conte, F. DiFuria and S. Moro, Tetrahedron Lett., 1996, 37, 8609–8612 CrossRef CAS; (e) V. Conte, B. Floris, P. Galloni and A. Silvagni, Adv. Synth. Catal., 2005, 347, 1341–1344 CrossRef CAS.
- (a) O. V. Branytska and R. Neumann, J. Org. Chem., 2003, 68, 9510–9512 CrossRef CAS; (b) L. Menini and E. V. Gusevskaya, Chem. Commun., 2006, 209–211 RSC; (c) G. F. Zhang, R. H. Liu, Q. Xu, L. X. Ma and X. M. Liang, Adv. Synth. Catal., 2006, 348, 862–866 CrossRef CAS; (d) J. Iskra, S. Stavber and M. Zupan, Tetrahedron Lett., 2008, 49, 893–895 CrossRef CAS; (e) F. Radner, J. Org. Chem., 1988, 53, 3548–3553 CrossRef; (f) R. Raja and P. Ratnasamy, J. Catal., 1997, 170, 244–253 CrossRef CAS; (g) R. Neumann and I. Assael, J. Chem. Soc., Chem. Commun., 1988, 1285–1287 RSC.
- D. Lenoir and C. Chiappe, Chem.–Eur. J., 2003, 9, 1037–1044.
- (a) G. Rothenberg and J. H. Clark, Green Chem., 2000, 2, 248–251 RSC; (b) W. Adam, C. Mock-Knoblauch, C. R. Saha-Moller and M. Herderich, J. Am. Chem. Soc., 2000, 122, 9685–9691 CrossRef CAS; (c) V. Conte, F. DiFuria and S. Moro, Tetrahedron Lett., 1994, 35, 7429–7432 CrossRef CAS; (d) V. Conte, F. DiFuria, S. Moro and S. Rabbolini, J. Mol. Catal. A: Chem., 1996, 113, 175–184 CrossRef CAS; (e) M. Andersson, V. Conte, F. DiFuria and S. Moro, Tetrahedron Lett., 1995, 36, 2675–2678 CrossRef CAS.
- T. Moriuchi, M. Yamaguchi, K. Kikushima and T. Hirao, Tetrahedron Lett., 2007, 48, 2667–2670 CrossRef CAS.
- T. K. Ying, W. L. Bao and Y. M. Zhang, J. Chem. Res., 2004, 806–807 Search PubMed.
- C. Limberg and J. H. Teles, Adv. Synth. Catal., 2001, 343, 447–449 CrossRef CAS.
- H. G. O. Becker in Organikum: Organisch-chemisches Grundpraktikum, Wiley-VCH, Weinheim, 21st edn, 2001, p. 299 Search PubMed, (see ref. 5b for a translation).
- M. Eissen and J. O. Metzger, Chem.–Eur. J., 2002, 8, 3580–3585 CrossRef CAS.
- M. Eissen, A. Zogg and K. Hungerbühler, J. Loss Prev. Process Ind., 2003, 16, 289–296 CrossRef.
- G. Bellucci, R. Bianchini and C. Chiappe, J. Org. Chem., 1991, 56, 3067–3073 CrossRef.
- A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Eur. J. Org. Chem., 2006, 483–488 CrossRef CAS.
- G. Bellucci, C. Chiappe, R. Bianchini, P. Lemmen and D. Lenoir, Tetrahedron, 1997, 53, 785–790 CrossRef CAS.
- K. Tanaka, R. Shiraishi and F. Toda, J. Chem. Soc., Perkin Trans. 1, 1999, 3069–3070 RSC.
- R. Bianchini, C. Chiappe, G. Lo Moro, D. Lenoir, P. Lemmen and N. Goldberg, Chem.–Eur. J., 1999, 5, 1570–1580 CrossRef CAS.
- T. Ying, W. Bao and Y. Zhang, J. Chem. Res., 2004, 806–807 Search PubMed.
- (a) C. T. Retcliffe and J. M. Shreeve, Nitrosyl Halides, in Inorganic Syntheses, ed. W. L. Jolly, McGraw-Hill Book Company, Inc., 1968, vol. XI, pp 194-200 Search PubMed; (b) H.-P. Loock and C. X. W. Qian, J. Chem. Phys., 1998, 108, 3178–3186 CrossRef CAS; (c) G. Maier, H. P. Reisenauer and M. De Marco, Chem.–Eur. J., 2000, 6, 800–808 CrossRef CAS.
- C. F. Ye and J. M. Shreeve, J. Org. Chem., 2004, 69, 8561–8563 CrossRef CAS.
- S. Lin and W. H. Saunders, J. Am. Chem. Soc., 1994, 116, 6107–6110 CrossRef CAS.
- R. Maidan, Z. Goren, J. Y. Becker and I. Willner, J. Am. Chem. Soc., 1984, 106, 6217–6222 CrossRef CAS.
- K. Yates and R. S. Mcdonald, J. Org. Chem., 1973, 38, 2465–2478 CrossRef CAS.
- G. W. Kabalka, K. Yang, N. K. Reddy and C. Narayana, Synth. Commun., 1998, 28, 925–929 CAS.
- S. Sternhel and P. W. Westerma, J. Org. Chem., 1974, 39, 3794–3796 CrossRef.
- V. F. Anikin and T. I. Levandovskaya, Zh. Org. Khim., 1988, 24, 1064–1070 CAS.
- A. Annunziata, C. Galli, P. Gentili, A. Guarnieri, M. Beit-Yannai and Z. Rappoport, Eur. J. Org. Chem., 2002, 2136–2143 CrossRef CAS.
- W. M. Jones and R. Damico, J. Am. Chem. Soc., 1963, 85, 2273–2278 CrossRef CAS.
- D. Lexa, J. M. Saveant, H. J. Schafer, K. B. Su, B. Vering and D. L. Wang, J. Am. Chem. Soc., 1990, 112, 6162–6177 CrossRef CAS.
- J. H. Espenson, Z. L. Zhu and T. H. Zauche, J. Org. Chem., 1999, 64, 1191–1196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of (2,3-dibromo-1,1,1-trifluoropropan-2-yl)benzene 20, Environmental factor E of different bromination methods, UV-vis spectra of Br2 in CH3CN and UV-vis spectra of NOBr. See DOI: 10.1039/b814989e |
| This journal is © The Royal Society of Chemistry 2009 |