Isobutane/butene alkylation on microporous and mesoporous solid acid catalysts: probing the pore transport effects with liquid and near critical reaction media
V. R.
Sarsani
ab and
Bala
Subramaniam
*ab
aCenter for Environmentally Beneficial Catalysis, Lawrence, KS 66045, USA
bDepartment of Chemical and Petroleum Engineering, University of Kansas, Lawrence, KS 66045, USA. E-mail: bsubramaniam@ku.edu; Fax: +1-785-864-6051; Tel: +1-785-864-2903
First published on 13th November 2008
Abstract
The alkylation of isobutane with 1-butene was investigated on microporous (β-zeolite) and mesoporous (silica supported heteropolyacids) catalysts in a slurry reactor. The reaction was investigated in the range of 25–100 bar and 15–95 °C in liquid phase and in near critical reaction media with either dense CO2 or dense ethane as diluent, partially replacing the excess isobutane. At 75 °C, the selectivity towards trimethylpentanes (TMP) in the liquid phase is 70%+ initially, but decreases with time on all the catalysts investigated. While near-critical reaction mixtures were employed in order to enhance pore diffusion rates, the conversion and selectivity profiles obtained with such mixtures are comparable to those obtained with liquid phase reaction mixtures in both microporous and mesoporous catalysts. This implies that pore diffusion effects play a limited role at higher temperatures (75–95 °C). In contrast, the liquid phase results at sub-ambient temperatures indicate that the catalyst is deactivated before the TMPs diffuse out of the pores, indicating that pore diffusion effects play an important role in the deactivation process at low temperatures. Our results suggest that novel approaches that enhance the pore-diffusion rates of the TMPs at lower temperatures must be pursued.
Introduction
The alkylation of isobutane with light olefins (C3–C5) is a refinery process used to produce alkylates, which constitute about 13% of the U.S. gasoline pool. Trimethylpentanes (TMPs) are the most desired compounds with high octane numbers within the alkylates. Within reformulated gasoline, the isobutane alkylates not only provide a high octane number with low vapor pressure but also the desired combination of low levels of sulfur, olefins and aromatics. Due to the consequences of the 1990 Clean Air Act and phasing out of MTBE, it is anticipated that the isobutane alkylate will play an even more significant role as refiners worldwide attempt to comply with environmental mandates such as reduction in volatile organic compounds, sulfur and nitrogen oxides.The most widely used catalysts in industry today for isobutane/olefin (I/O) alkylation are H2SO4 and HF. The use of environmentally benign solid acids as catalysts to replace these toxic mineral acids continues to be a major challenge of green chemistry and green engineering. To date, while there are claims of pilot scale plants1 and even a commercial scale plant,2 rapid catalyst deactivation by fouling hampers the commercial development utilizing solid acids. Detailed reviews on the subject can be found elsewhere.3–6 The current manuscript presents investigations aimed at better understanding the causative factors behind the rapid deactivation of solid acid catalysts by fouling. A better understanding of the pore size, temperature and solvent effects on catalyst deactivation is important for chemists and engineers to rationally develop practically viable solid acid catalysts. The work presented in this paper is thus relevant to green chemistry.
Researchers at Shell7 were one of the first groups to report a systematic study in a back-mixed slurry reactor to evaluate the activity of zeolites. Through a simple kinetic model, the authors reported that the I/O ratio is the most important parameter that determines alkylate selectivity and catalyst life time. Amongst various zeolites tested, the authors found that beta zeolite is the most promising catalyst for high activity and catalyst lifetime. Lercher's group has been active in systematically studying the mechanistic aspects of isobutane alkylation in a continuous slurry reactor on beta zeolites8 and more recently on Y9 and X zeolites.10 Froment's group has recently reported detailed mechanistic pathways of the reaction using studies in a plug flow reactor and by following the product distribution carefully at short time on stream (TOS). Single event kinetic modeling has been used to identify all the possible reactions.11 In these referenced reports, microporous acid catalysts were evaluated. The present study complements these studies by also including mesoporous acid catalysts for investigation.
Rapid catalyst deactivation by fouling is widely accepted as the major hindrance to commercial development. Use of supercritical (sc) reaction media is one way to reduce the deactivation process in situ by enhancing the transport properties. Early studies on the use of supercritical media for isobutane alkylation focused on operating above the critical point of the reaction mixture. However, the high temperatures (> 140 °C) needed to achieve the critical point lead to cracking products on strong solid acids, like sulfated zirconia,12Y zeolites,13heteropolyacids,14 adversely affecting the selectivity towards the desired products. Salinas et al.15 partly circumvent the high cracking activity at elevated temperatures by employing lower acid strength zeolites with high Si/Al ratios. Clark and Subramaniam16 reported on the use of CO2 as a diluent to yield supercritical mixtures at lower temperatures and demonstrated steady C8 alkylates production activity during isobutane/butene alkylation with zeolites and sulfated zirconia in a fixed bed reactor. However, the yield of alkylates was very low, less than 10%. All of the aforementioned studies in sc media are based on plug flow reactor studies and hence the I/O ratios in the reactor are lower compared to a well back-mixed system. The role of the media in the aforementioned studies was not fully understood.
Using mesoporous solid acid catalysts (SiO2-supported Nafion® or SAC-13 and heteropolyacids), it was shown that improved C8 yields can be obtained in a CSTR with CO2-based supercritical feed mixtures.17 By systematic pressure tuning studies, it was demonstrated that for the SAC-13 catalyst, the optimum conditions which maximize steady alkylates production are 95 °C, 80 bar, an olefin space velocity (OSV) of 0.05 h−1, and a molar I/O ratio of 5 : 1 with 70 mol% CO2 in the feed. Under these conditions, various heteropolyacid catalysts were evaluated and found that the maximum steady C8 alkylate selectivity was 25%, but still much lower compared to the current industrial processes (70%+).18
Corma and coworkers investigated zeolites and sulfated zirconia at temperatures of 0 °C and 50 °C and conclude that the optimum temperature to obtain desired alkylates depends on the acid strength, which determines the relative rates of cracking, alkylation and oligomerization.19 Olah and coworkers reported the effect of acid strength in their study with liquid acids (trifluoromethanesulfonic acid with trifluoroacetic acid/water) and report that H0 = −10.7 is the optimum acid strength.20
It is generally agreed that weak Brønsted acid sites catalyze only oligomerization.21 Some of the literature reports indicate that CO2 adsorbs strongly on the acid sites and that it can reduce the acid strength of the catalyst. Olah and coworkers report that liquid carbon dioxide acts as a competing weak base and decreases the acidity of the system, thereby affecting product quality. Their study was based on isobutane/isobutylene alkylation on liquid acids like HF, H2SO4 and CF3SO3H. However, the authors are uncertain about the extent of inhomogeneity in the reaction mixture with CO2.22 A recent report also confirms that CO2 can act as a Lewis base interacting with strong Lewis acids in solutions.23 Ruthven and coworkers report that CO2 adsorbs strongly compared to propane on a zeolitic catalyst.24
The main objective of the present work is to systematically investigate reaction media effects on conversion and TMP selectivity using diluents like dense CO2 and ethane on various solid acid catalysts. The extraction of heavy compounds by the near critical media is evaluated in a back-mixed slurry reactor with both microporous and mesoporous supported catalysts to clearly distinguish the effect of pore-diffusion limitations in these catalysts. Further, the low temperature (ambient/sub-ambient) region is explored to reduce the coke precursor formation rates and more effectively balance the coke removal rates. The results also shed light on the physico-chemical aspects of the reaction.
Experimental
Catalysts
Four different catalysts are used in this study. The zeolite catalysts were procured from GraceDavison. The beta zeolite (SMR 5-9858-01062) has a Si/Al ratio of 13 and USY zeolite (SMR 5-9858-01061) has Si/Al ratio of 3. The compositions, along with basic physical properties are listed in the Table 1. Zeolites were used in the powder form as received. The SEM images of these zeolites indicate that the particle size ranges from 1–10 μm. The supported heteropolyacid catalyst, 62% TPA/SiO2, was prepared as reported previously, but using n-butanol as solvent during impregnation.25 The SiO2-supported Nafion®catalysts (SAC-13) were supplied (in the form of pellets) by BASF Catalysts. The TPA/SiO2 and SAC-13 catalysts are size-reduced and sieved to 62–105 μm prior to use.The 62% TPA/SiO2 catalyst was pretreated in situ under flowing nitrogen at 175 °C for 1 h. The SAC-13 catalyst was pretreated in situ under flowing nitrogen at 150 °C for 3 h. The BET surface area (SA) and BJH pore volume (PV) measurements for these mesoporous catalysts were made using a Micromeretics Gemini instrument, employing nitrogen physisorption. Detailed pore size distribution on beta zeolites was experimentally measured by Micromeritics ASAP analyzer at DuPont.
Chemicals
The compressed gases used in the study, isobutane (Instrument purity grade, > 99.5%; Lot no. 102-51-02346-A2), 1-butene (Chemical purity grade, > 99.0%; Lot no. 102-51-02345-A1) and ethane (Chemical purity grade, > 99.0%), were procured from Matheson Tri-gas Inc. Liquified CO2 (Airgas) of 99.995% purity was used.Experimental setup
The details of the experimental setup are described elsewhere.17,26 Briefly, the catalyst is suspended as slurry in a continuous, back-mixed slurry reactor. Premixed I/O mixture and diluents (if any) are pumped separately using syringe pumps (500D from Teledyne Isco). Before admitting the feed mixture, the reactor is brought to the desired pressure and temperature with isobutane (when no diluent is used) or with isobutane and diluent in the same molar ratio as in the feed mixture. The products were analyzed online by gas chromatography (Agilent 6890 N), equipped with an FID, and a DB-Petro 100 m column (J & W Scientific), with a total analysis time of 90 min for each injection. The following definitions are used in discussing the results of this work.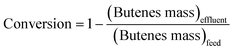
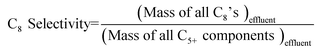
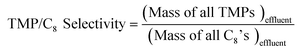
Results and discussion
Catalyst characterization
The total microporous pore volume (pore dia < 20 Å) is 0.222 cc/g, while the total pore volume for pore dia > 20 Å is 0.064 cc/g. Within pore sizes of 20–100 Å, the major fraction of pore volume is within 20–45 Å. The volume in inter-crystalline pores of large diameter (> 100 Å) is about 20% of the total pore volume (of pore sizes > 20 Å). The SEM images of zeolites shown in Fig. 1a (beta zeolite) and Fig. 1b (USY zeolite) depict that the crystallite sizes are on the order of 100 nm. The BET surface area and pore volume for mesoporous supported catalysts are discussed later.
Conversion and selectivity results
Liquid phase experiments
Initial experiments are conducted in the liquid phase with particular attention to the change in product distribution with TOS, which provides insights into the deactivation mechanism. By operating at a relatively long reactor residence time (ratio of volume of the reaction mixture to that of total volumetric flow rate at the exit) of ∼6.7 h, the initial product distribution is closely followed. At the beginning, the I/O ratios are very high since the reactor contains only isobutane. Model predictions suggest that high I/O ratios in the reactor favor C8 alkylate yields.26Fig. 2 gives the product distribution on beta zeolite in liquid phase at 75 °C, 40 bar, OSV = 0.11 h−1, I/O = 5.0. Several key observations are made from this experiment (see Figs. 2a–2c).
• As seen in Fig. 2a, almost complete butene conversion (99.5%+) is achieved up to 12 h, thereby providing a high I/O ratio in the reactor (CSTR) that favors the formation of the desired products, the trimethylpentanes. The formation of TMPs is high during this period (Fig. 2a), but decreases after 12 h, similar to that reported in the literature.7,9
• The decrease of ‘self alkylation’ activity (tracked by n-butane formation) is observed after 12 h (Fig. 2b), which indicates the decrease of hydride transfer activity and hence an important measure of catalyst lifetime, a correlation first noted by Lercher's group.9
• The formation of olefins within the C5–C7 fraction, C8 olefins and C2, C3 olefins is observed starting at about the same TOS of 12 h and increases with time (Fig. 2c). This indicates the onset of cracking reactions at about the same TOS when catalyst deactivation is observed.
• Within the C5–C7 fraction, the olefinic content reaches as high as 83%, with the fraction of C5olefin being dominant (74%). However, the fraction of C8 olefins among C8’s is only about 26% at longer TOS (see Fig. 2c).
• The selectivity towards heavier compounds (C9+) increases from 15% to about 50% until TOS of 23 h and then decreases (not shown in Fig. 2).
These observations show that the catalyst deactivation is concurrent with decrease in self-alkylation, build up of olefins from cracking and oligomerization reactions. Other insights from these results are discussed in the appropriate sections below.
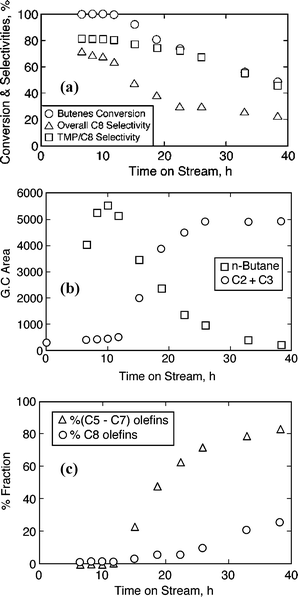 | ||
| Fig. 2 Temporal profiles on beta zeolite (75 °C, 40 bar, OSV = 0.11 h−1, I/O = 5.0): (a) conversion and selectivity profiles; (b) light ends; (c) olefins. | ||
Reaction media effects on zeolites: pressure tuning studies with ethane
Simulations based on the Peng–Robinson equation of state (PREOS) were performed to obtain the critical properties of the reaction mixtures containing isobutane, butene, 2,2,4 TMP, CO2 and ethane, as summarized in Table 2. The compositions in the table represent pure reactant, and reactants with partial/complete conversion of butenes to 2,2,4 TMP with either CO2 or ethane as diluent. Details may be found elsewhere.26 Based on the simulations, isothermal pressure tuning studies at 75 °C have been performed with sc-ethane as the solvent, with pressures ranging from 52–100 bars. At constant 1-butene space velocity and ethane flow rates, conversion and selectivity profiles were obtained at various fixed pressures at 75 °C. This pressure tuning is similar, in principle, to that reported for Fischer-Trøpsch synthesis with n-hexane28 and isobutane/butene alkylation with CO2 as diluents.17 In a typical experiment, the feed mixture contained about 74% (molar) ethane, the rest being made up of an I/O mixture at a molar ratio of 5.| Isobutane (mol%) | 1-Butene (mol%) | CO2 (mol%) | Ethane (mol%) | 2,2,4 TMP (mol%) | (Tc)mix (°C) | (Pc)mix (bar) |
|---|---|---|---|---|---|---|
| 25 | 5 | 70 | 0 | 0 | 67 | 74 |
| 21 | 1 | 70 | 0 | 4 | 78 | 89 |
| 20 | 0 | 70 | 0 | 5 | 81 | 92 |
| 25 | 5 | 0 | 70 | 0 | 78 | 55 |
| 21 | 1 | 0 | 70 | 4 | 90 | 63 |
| 20 | 0 | 0 | 70 | 5 | 93 | 65 |
| 20 | 4 | 0 | 76 | 0 | 70 | 55 |
| 16 | 0 | 0 | 76 | 4 | 82 | 64 |
| 15 | 3 | 0 | 82 | 0 | 62 | 55 |
| 12 | 0 | 0 | 82 | 3 | 71 | 62 |
| 10 | 2 | 0 | 88 | 0 | 53 | 53 |
| 8 | 0 | 0 | 88 | 2 | 59 | 59 |
The absence of any possible side effects of the diluent, ethane or its impurities is verified with the following experiment. After loading the pretreated catalyst, dense ethane is pumped to the reactor and maintained at 80 bar, 75 °C. After 2 h of exposure time at reaction conditions, isobutane is flowed into the reactor system until about 10 times the volume of the reactor to replace all the ethane with isobutane, after which the typical liquid phase experiment is started. The results show similar profiles to the liquid phase experiment without exposing the catalyst to ethane, confirming the absence of any adverse effects of ethane on catalyst activity and lifetime.
Figures 3a and 3b show the conversion and the TMPs/C8 selectivity profiles respectively during the pressure tuning studies on beta zeolite. In all cases, the results point to catalyst deactivation. The conversion stabilizes in the case of the mild pressure (52 bar) ethane experiment (Fig. 3a), while in the liquid phase (no ethane) run, the TMPs/C8 selectivity stabilizes at about 14% (Fig. 3b). Thus, the high initial TMP/C8 selectivity is not stabilized even with the use of dense ethane as the diluent. Note that in these experiments, the comparison of the ethane-based experiments are made with a liquid phase experiment such that the (I + diluent)/O ratio is 20 in both cases. The diluent is either additional isobutane or the supercritical fluid solvent (either CO2 or ethane). In this manner, the olefin residence times are unaffected in these experiments. However, the I/O ratios inside the reactor are different, which could affect the selectivity. To better understand the effect of feed I/O ratio on activity and catalyst life, the conversion and selectivity profiles of the experiments with feed I/O ratios of 5 and 20 (under otherwise identical conditions) are plotted with dimensionless time (defined as ratio of time on stream to that of reactor residence time) in Fig. 4. With identical olefin space velocity of 0.11 h−1, the reactor residence times for I/O ratios of 5 and 20 are calculated to be 6.7 h and 1.7 h respectively. Fig. 4 clearly shows that the I/O ratio does not affect the catalyst activity or selectivity significantly. It must be noted that at startup, the reactor contains isobutane only and that the results are compared across a duration of less than six reactor residence times which are insufficient to achieve a steady state even without catalyst deactivation.
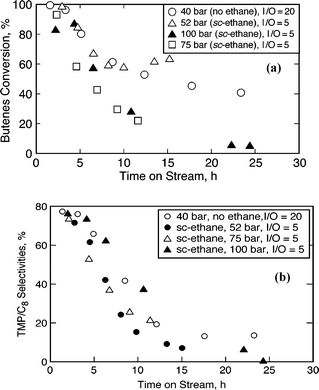 | ||
| Fig. 3 Temporal profiles on beta zeolite (75 °C, OSV = 0.11 h−1); (a) 1-butene conversion, (b) TMP/C8 selectivity. | ||
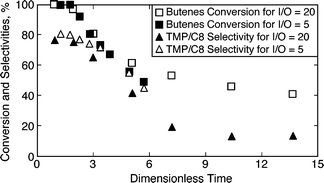 | ||
| Fig. 4 Comparison of conversion and selectivity profiles for molar feed I/O ratios of 5 and 20 (beta zeolite, 75 °C, 40 bar, OSV = 0.11 h−1, liquid phase). | ||
Activity at ambient/sub-ambient temperatures
Sub-ambient temperatures are used in both HF and H2SO4 based commercial processes. Hence, low temperature activity in the case of zeolites was explored since at sub-ambient temperatures, the coke precursor formation rates are expected to be much slower. Further, the TMPs are thermodynamically favored over DMHs at lower temperatures. Fig. 5a compares the performance of beta zeolite in liquid phase operation at 75 °C, 31 °C and 15 °C, under otherwise identical conditions. The conversion profiles of the low temperature experiments seem to be identical, but different from the profiles at 75 °C. As explained in the previous section, a steady state is possible only after a TOS of 40 h. Hence, the apparent steady activity until ∼12 h at 75 °C is only representative of the initial conversion.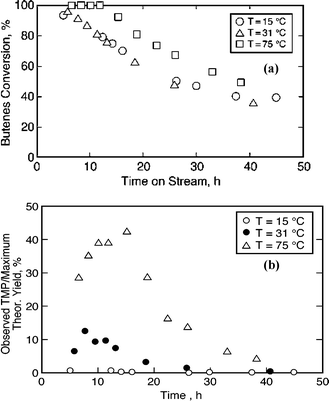 | ||
| Fig. 5 Comparison of temporal profiles at various temperatures on beta zeolite (40 bar, OSV = 0.11 h−1, molar feed I/O = 5.0); (a) 1-butene conversion; (b) TMP yields. | ||
To better understand the intrinsic activity, % TMP yields, expressed as a percentage of the maximum possible theoretical yields that can be achieved at steady state, are plotted in Fig. 5b. Because these are profiles from a CSTR at unsteady state, the plots also show that the deactivation started before 15 h. Note that while the TMP yields start to decrease beyond 15 h at 75 °C, they decrease much earlier (TOS of 8 h) at 31 °C. Negligible TMPs are observed at sub-ambient temperatures. These results indicate that the catalyst is deactivated before the TMPs diffuse out of the pores. Further, an experiment with dense ethane was conducted at 30 °C to see if the activity can be stabilized at low temperatures. The reaction in this case is expected to proceed in a gas-expanded liquid phase, as reviewed recently.27 It is clear that the activity declines continuously with time and the overall selectivity towards C8's is less than 10% after TOS of 20 h (see Fig. 6). It suggests that the enhanced diffusion rate in the expanded phase is not sufficient to ameliorate the deactivation process.
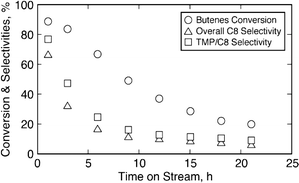 | ||
| Fig. 6 Temporal conversion and selectivity profiles on beta zeolite with dense ethane (73 mol% in feed) in liquid phase (30 °C, 40 bar, OSV = 0.11 h−1, molar feed I/O = 5.0). | ||
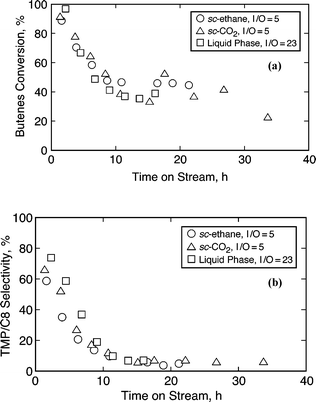 | ||
| Fig. 7 Comparison of temporal conversion profiles on 62%TPA/SiO2 (62–105 μm) with various media (95 °C, 40 bar, OSV = 0.11 h−1, molar fee I/O = 5.0); (a) 1-butene conversion; (b) TMP/C8 selectivity. | ||
Reaction media effects on mesoporous solid acids
Some reports have indicated that the media effects in microporous zeolitic catalysts are effective only on the processes occurring on the outer surface of these catalysts.16,29 Since the reaction media effects observed with ethane are not appreciable on zeolites, a new batch of heteropolyacids (62% HPA/SiO2) was also investigated with dense ethane, and the results compared with dense CO2 and liquid phase operation. Figs. 7a and 7b compare the conversion and selectivity profiles of these experiments. The conversion and the TMP/C8 selectivity stabilize at longer TOS of about 15–20 h in all the cases. These results indicate that the use of CO2vs.ethane as diluent does not alter the conversion/selectivity profiles and that the media effects are not appreciable for stabilizing the high initial TMP selectivity. This suggests that the desired reaction via intermolecular hydride transfer step is not diffusion controlled. Note that the higher temperature (95 °C) used with these catalysts is because of their lower acid strength compared to that of beta and USY zeolites.For comparison purposes, liquid phase experiments (with no additional diluents) were also conducted with USY, 62% HPA/SiO2 and SAC-13 catalysts with molar feed I/O ratio of 5, at 40 bar and olefin space velocity of 0.11 h−1. Such a comparison of initial activity for various catalysts is summarized in Table 3. It is interesting to note that despite lower than complete conversion (even at 95 °C) in case of HPA/SiO2 and SAC-13, the TMP/C8 selectivity is significant. The observed activity in zeolites is generally retained longer (TOS of 10 h) as compared to less than a few hours in the case of HPA/SiO2 and SAC-13. However, considering that the acid site density (ASD) of SAC-13 is an order of magnitude lower compared to zeolites and that HPA/SiO2 has an intermediate ASD between those of SAC-13 and zeolites, the results indicate that the net butene flow rate per acid site controls the observed duration of steady selectivity. This phenomenon was also pointed out recently by Lercher's group, based on their study on X zeolites.9 The net butene flow rate per acid site is greater in the case of SAC-13 and HPA/SiO2. The results on catalyst lifetime also suggest that deactivation of catalysts occurs site by site.
The physical properties of the spent mesoporous catalysts are characterized and compared to those of the fresh catalyst. The results are shown in Table 4. SAC-13 catalyst does not show any substantial loss of any surface area or pore volume but heteropolyacids lose greater than 98% surface area and pore volume in case of liquid phase experiments, but retain properties better when either dense CO2 or ethane is used as diluent. Despite these variations, all catalysts show deactivation with decreasing TMP/C8 selectivity with TOS, indicating that deactivation is not caused by pore blocking, at least on the SAC-13 catalyst. These results suggest that the deactivation on mesoporous catalysts should be studied in more detail.
| Catalyst | BET SA (m2/g) | Pore Volume (cc/g) | Avg. Pore Dia (Å) |
|---|---|---|---|
| SAC-13 N, Fresh | 129 | 0.51 | 162 |
| SAC-13 N, spent (liquid phase, 95 °C, 40 bar, I/O = 5) | 133 | 0.54 | 145 |
| SAC-13 N, spent (sc-ethane, 95 °C, 95 bar, I/O = 5) | 117 | 0.45 | 118 |
| 62%HPA/SiO2, Fresh | 220 | 0.29 | 48 |
| 62%HPA/SiO2, spent (liquid, 95 °C, 40 bar, I/O = 23) | 3.2 | 0.005 | 75 |
| 62%HPA/SiO2, spent (liquid, 95 °C, 40 bar, I/O = 5) | 4.2 | 0.008 | 83 |
| 62%HPA/SiO2, spent (sc-ethane, 95 °C, 95 bar, I/O = 5) | 38.3 | 0.09 | 72 |
| 62%HPA/SiO2, spent (sc-CO2, 95 °C, 95 bar, I/O = 5) | 25.1 | 0.06 | 72 |
Conclusions
This study represents a systematic evaluation of reaction media effects in highly active microporous and mesoporous solid acid catalysts. The liquid phase experiments in the back-mixed slurry reactor, with long reactor residence times provide a framework for comparing the initial activity of various solid acid catalysts. The zeolitic catalysts showed excellent selectivity towards TMP production, followed by HPA/SiO2 and SAC-13 catalysts.The use of either scethane or scCO2 as a diluent to replace the excess isobutane in the feed provides similar conversion and TMP selectivity profiles indicating that there are negligible adsorption effects due to the use of CO2 with the acid catalysts. Further, the conversion and selectivity profiles obtained with supercritical feed mixtures are comparable to those obtained in liquid phase experiments. These results demonstrate that reaction media effects are not significant during isobutane alkylation at higher temperatures (75–95 °C) implying that the desired alkylate forming reaction via the intermolecular hydride transfer step is not diffusion controlled at these temperatures.
The conversion and selectivity results at low temperatures show that the catalyst is deactivated even before the TMPs are transported out of the pores, thereby indicating that pore diffusion rates play an important role in the deactivation process at low temperatures. These results suggest that novel approaches that enhance the pore diffusion rates at lower temperatures need to be pursued.
The results presented here bridge the gap between alkylation reactions conducted in the liquid and supercritical phases, and have provided a better understanding of the physicochemical sequence of events occurring in solid acid catalyzed alkylations. An effort is currently underway in our laboratory to study the intrinsic adsorption, desorption and pore-transport rates of reactant and product molecules using a Tapered Element Oscillatory Microbalance (TEOM) on various solid acids under non-reactive conditions. The results of such TEOM investigations help discern if a chosen solid acid catalyst is a viable candidate for facile transport of the reactant and product molecules, a prerequisite for attaining stable catalyst activity.
Acknowledgements
We are thankful to GRACE for generously providing the samples of zeolitic catalysts, to Dr Keith W. Hutchenson of Dupont for providing the pore volume analysis of the beta zeolite, to Dr Yong Wang of the Pacific Northwest National Laboratory (PNNL) for providing supported heteropolyacid catalysts. Financial support from the National Science Foundation (EEC-0310689) is gratefully acknowledged.References
- J. A. Kocal, Green Chemistry Conference, Washington D.C. June, 2005 Search PubMed.
- A. Gaffney, Annual CREL meeting, Washington University, St. Louis, 2006 Search PubMed.
- A. Corma and A. Martinez, Catal. Rev.-Sci. Eng., 1993, 35, 483 CrossRef CAS.
- P. Rao and S. R. Vatcha, Oil Gas J., 1996, 94, 56 Search PubMed.
- J. Weitkamp and Y. Traa, Catal. Today, 1999, 49, 193 CrossRef CAS.
- R. Gläser, Chem, Eng. Techol., 2007, 30, 557 Search PubMed.
- K. P. deJong, C. M. A. M. Mesters, D. G. R. Peferoen, P. T. M. van Brugge and C. de Groot, Chem. Eng. Sci., 1996, 51, 2053 CrossRef CAS.
- G. S. Nivarthy, Y. He, K. Seshan and J. A. Lercher, J. Catal., 1998, 176, 192 CrossRef CAS.
- A. Feller, A. Guzman, I. Zuazo and J. A. Lercher, J. Catal., 2004, 224, 80 CrossRef CAS.
- C. Sievers, I. Zuazo, A. Guzman, R. Olindo, and J. A. Lercher, AIChE Annual Meeting, Salt Lake City, 2007 Search PubMed.
- G. Martinis and G. Froment, Ind. Eng. Chem. Res., 2006, 45, 940 CrossRef; G. Martinis and G. Froment, Ind. Eng. Chem. Res., 2006, 45, 954 CrossRef.
- G. Funamoto, S. Tamura, K. Segawa, K. Wan and M. Davis, Res. Chem. Intermed., 1998, 24, 449 CrossRef CAS.
- L. Fan, I. Nakamura, S. Ishida and K. Fujimoto, Ind. Eng. Chem. Res., 1997, 36, 1458 CrossRef.
- P. Y. Gayraud, N. Essayem and J. C. Vedrine, Stud. Surf. Sci. Catal., 2000, 130C, 2549 CAS.
- A. L. M. Salinas, D. Kong, Y. B. Taarit and N. Essayem, Ind. Eng. Chem. Res., 2004, 43, 6355 CrossRef CAS.
- M. Clark and B. Subramaniam, Ind. Eng. Chem. Res., 1998, 37, 1243 CrossRef CAS.
- C. J. Lyon, B. Subramaniam and C. Pereira, Stud. Surf. Sci. Catal., 2001, 139, 221 CAS.
- V. R. Sarsani, Y. Wang and B. Subramaniam, Ind. Eng. Chem. Res., 2005, 44, 6491 CrossRef CAS.
- A. Corma, Appl. Catal. A, 1994, 111, 175 CrossRef CAS.
- G. A. Olah, P. Batamack, D. Deffieux, B. Torok, Q. Wang, A. Molnar and G. K. S. Prakash, Appl. Catal. A, 1996, 146, 107 CrossRef CAS.
- A. Feller, A. Guzman, I. Zuazo and J. A. Lercher, Stud. Surf. Sci. Catal., 2003, 145, 67 CAS.
- G. A. Olah, E. Marinez, B. Torok and G. K. S. Prakash, Catal. Lett., 1999, 61, 105 CrossRef CAS.
- P. W. Bell, A. J. Thote, Y. Park, Ram B. Gupta and C. B. Roberts, Ind. Eng. Chem. Res., 2003, 42, 6280 CrossRef CAS.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339 CrossRef CAS.
- Y. Wang, A. Y. Kim, S. S. Li, L. Wang, C. H. F. Peden, and B. C. Bunker, in Shape-Selective Catalysis: Chemical Synthesis and Hydrocarbon Processing, ed. C. Song, C., M. M. Garces, and Y. Sugi, Am. Chem. Soc., Washington, DC, 1998 Search PubMed.
- V. R. Sarsani, Ph.D. Dissertation, University of Kansas, 2007.
- P. G. Jessop and B. Subramaniam, Chem. Rev., 2007, 107, 2615 CrossRef CAS.
- D. Bochniak and B. Subramaniam, AIChE J., 1998, 44, 1889 CrossRef CAS.
- R. Gläser and J. Weitkamp, Ind. Eng. Chem. Res., 2003, 42, 6294 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |