Field comparison of passive air samplers with reference monitors for ambient volatile organic compounds and nitrogen dioxide under week-long integrals
Shaibal
Mukerjee
*a,
Karen D.
Oliver
b,
Robert L.
Seila
a,
Henry H.
Jacumin Jr.
b,
Carry
Croghan
a,
E. Hunter
Daughtrey Jr.
b,
Lucas M.
Neas
c and
Luther A.
Smith
b
aNational Exposure Research Laboratory E205-02, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, USA
bAlion Science and Technology, Inc., Durham, NC 27713, USA
cNational Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, USA
First published on 8th October 2008
Abstract
This study evaluates performance of nitrogen dioxide (NO2) and volatile organic compound (VOC) passive samplers with corresponding reference monitors at two sites in the Detroit, Michigan area during the summer of 2005. Ogawa passive NO2 samplers and custom-made, re-useable Perkin-Elmer (PE) tubes with Carbopack X sorbent for VOCs were deployed under week-long sampling periods for six weeks. Precise results (5% relative standard deviation, RSD) were found for NO2 measurements from collocated Ogawa samplers. Reproducibility was also good for duplicate PE tubes for benzene, toluene, ethylbenzene, and xylene isomers (BTEX species, all ≤ 6% RSD). As seen in previous studies, comparison of Ogawa NO2 samplers with reference chemiluminescence measurements suggested good agreement. Generally good agreement was also found between the PE tubes and reference methods for BTEX species.
Introduction
Passive air samplers have been developed and used extensively for industrial hygiene and related occupational exposure purposes to measure gaseous air pollutants. Although a sampling integral (usually 24 h minimum) is required for passive samplers to measure air pollutants above detection limits, they are an attractive method for saturation and exposure monitoring owing to their small size, low cost, and ease of use in the field.1 Air quality studies have used diffusion samplers such as Palmes tubes and filter badges to passively monitor NO2.2,3–5 Other passive methods have been used to monitor volatile organic compounds (VOC) in ambient air, most notably organic vapor monitor badges and other diffusion samplers using an activated carbon sorbent.6,7Ogawa passive samplers have been used in ambient air monitoring networks to monitor urban ozone, assess ozone, nitrogen dioxide (NO2), and sulfur dioxide trends at national parks and forested areas, determine distance of nitrogen dioxide emissions from highway traffic, and monitor personal/indoor air.4,5,8–10Ogawa samplers can measure these varying gaseous species depending on the reactants used on the collection filter substrate.1,5 For this study, Ogawa passive samplers were used for the collection of NO2.
Limited evaluations of the Perkin-Elmer (PE) tubes packed with various sorbents for sampling of volatile organic compounds (VOCs) have been performed in other studies in controlled exposure chamber and field settings.11–15 In terms of field comparisons, diffusive sampling PE tubes packed with Carbopack X sorbent have been found to be comparable to pumped methods and canister monitoring, with apparent over- and under-estimation of concentrations. A cost advantage in using these samplers is that they are re-usable for field deployment after thermal desorption (see methods section).
During the summer of 2005, a study was conducted by the U.S. Environmental Protection Agency (EPA) in the Detroit, Michigan area to measure NO2 and VOCs at elementary schools as part of an overall study assessing respiratory effects in children due to traffic and related urban air pollution. As part of the study, concentrations obtained with passive samplers were compared to NO2 and VOC measurements obtained with continuous monitors at two compliance sites. A motivation for using these passive samplers was their evaluation and application in a concurrent EPA study called the Detroit Exposure and Aerosol Research Study (DEARS).14,16 Also, similar methods were used in other cities (El Paso17 and Dallas, Texas) for planned intercity comparisons. We hypothesized that if the passive samplers were comparable in performance to reference measurements in the field, then this could provide further validation of passive sampling networks for monitoring community exposures in other locations on a cost-effective basis.
Methods
Site locations and reference monitoring
Monitoring was conducted at two compliance sites operated by the State of Michigan Department of Environmental Quality (MDEQ). The sites were selected due to their influence from traffic and heavy industry. The MDEQ data used in this comparison was the final, quality assured version of data reported in the EPA Air Quality System (AQS; data available at http://www.epa.gov/ttn/airs/airsaqs/). The MDEQ reference measurements were continuous and reported as hourly averages.The first site was in a residential section of northeast Detroit called the East 7 Mile site (AQS Site 26-163-0019) at the corner of Linnhurst and Dresden Streets on the Bessy Playground and near Von Steuben Elementary School. The second site was in neighboring Dearborn, Michigan (AQS Site 26-163-0033) at 2842 Wyoming Avenue. This site was next to Paul Costea Park and Salina Elementary School, approximately 500 m from the River Rouge Industrial Complex and 200 m from an associated railroad yard.
As part of routine monitoring conducted by MDEQ, the East 7 Mile site measured NOx species (including NO2) on a continuous basis using a TECO Model 42C gas-phase chemiluminescent NOx analyser (Thermo Environmental Instruments, Franklin, MA); these data were compared to NO2 measurements from the passive samplers. The chemiluminescent NOx analysers used were Federal Reference Method (FRM) samplers (Designated Method RFNA-1289-074) in accordance with Title 40, Part 53 of the Code of Federal Regulations (40 CFR Part 53).18,19 All chemiluminescence data were reported in units of parts per billion by volume (ppbV) and above the method detection limit (MDL) of 5 ppbV.
Volatile organic compounds were continuously monitored by MDEQ at the East 7 Mile Site. The site is designated as an EPA Photochemical Assessment Monitoring Station (PAMS) to assess trends in ozone-related pollutants; an automated gas chromatograph (auto-GC) was established according to PAMS network monitoring protocol.19,20 VOCs were sampled hourly using a AutoSystem GC, and a Model ATD400 Automatic Thermal Desorption unit (Perkin-Elmer Life and Analytical Sciences, Shelton, CT) fitted with an air sampling accessory. This system cryogenically (−30 °C) pre-concentrated air on a proprietary dual bed sorbent trap at a flow rate of 15 ml min−1 for 40 min. The trap was heated to 320 °C to inject the cryogenically pre-concentrated sample onto a non-polar 50 m × 0.22 mm ID BP1 column (SGE, Austin, TX) which was connected through a Deans switch to a second column, a 50 m × 0.32 mm ID Al2O3/Na2SO4 (alumina) porous layer open tubular column (Varian, Palo Alto, CA). After passing through the BP1 column, light hydrocarbons (C2 to C4) were further separated on the alumina column and quantified by a flame ionization detector (FID). Activation of the Deans switch sent the heavy fraction (C5 to C12 hydrocarbons) directly to a second FID. The columns were temperature programmed from 46 °C for 15 min to 170 °C at 5 °C min−1, then to 200 °C at 15 °C min−1 held for 6 min. Total analysis time was 48 min. All auto-GC measurements were above the MDL (0.1 ppbC).
The MDEQ did not conduct VOC/NO2 sampling at Dearborn. Thus, VOCs were collected at this site in 32 L Summa-polished stainless steel canisters (BRC/Rasmussen, Hillsboro, OR) that were cleaned, evacuated, and subsequently sampled to one-half atmosphere at 1.5 ml min−1 using a Model 423 Series Precision Low Flow Controller (Veriflo Division, Parker Hannifin Corporation, Richmond, CA) over a one week period. Pumps were not used for canister sampling. Analyses of VOCs were performed using GC-FID. Volatile organic sampling and analysis was based on U.S. EPA Compendium Methods TO-14a.21 All VOC analyses with the canisters were above the MDL (0.1 ppbC). No reference NO2 measurements were performed at Dearborn.
Passive sampling and analysis
Volatile organic compounds were measured using Carbopack X sorbent (approximately 650 mg of 40/60 mesh, unwashed) (Supelco, Bellefonte, PA) packed in stainless steel thermal desorption tubes (Perkin-Elmer); the sampler will be referred to as the PE tube. The PE tubes were 6 mm outside diameter by 90 mm long and were the same as described by McClenny and others13,14 with the exception that the inside diameter of the tube body was lined with a thin layer of ceramic to avoid rusting and permit more reproducible results from sampler to sampler; this tube was a custom product from the manufacturer (Supelco) following discussions with EPA. This sorbent has been demonstrated as applicable for VOC collection, including 1,3-butadiene, with minimal ozone interference.13,15 The PE tubes were packed with 6 cm Carbopack X sorbent and conditioned by the manufacturer. The tubes are sealed with 0.25 inch brass Swagelok™ fittings with combined polytetrafluoroethylene ferrules and stored in glass culture tubes with Teflon-lined caps. Once the tubes were received from the manufacturer, they were conditioned at 350 °C for 3 hours using the Dynatherm (Model 60) and CDS Analytical (Model 9600, Oxford, PA) tube conditioners. A subset of the tubes was blanked by thermal desorption using a TurboMatrix ATD (Perkin-Elmer) followed by gas chromatography/mass spectrometry (GC/MS) using a Saturn 2000 GC/MS (Varian) to assess potential artifact values. Results indicated that the tube background was generally low and reproducible from tube to tube. The effective sampling rate averaged over one week, based on a similar approach discussed in Martin at al. (2005),22 was used to determine the exact active sampling duration. In this study, VOC results from benzene, toluene, ethylbenzene, o-xylene, m- and p-xylene (collectively referred as BTEX species) and 1,3-butadiene are reported.Analysis of the tubes was performed using a TurboMatrix ATD and Saturn 2000 GC/MS system. Further details on the analytical method and experiments conducted to evaluate response of the selected VOCs under varying temperature and humidity conditions are presented elsewhere.13 All VOC samples from the PE tubes were field blank-corrected and reported in units of parts per billion carbon (ppbC) to match the original measurement units of the reference methods. The tubes were ready for re-use after thermal desorption and were re-deployed in the field.
The Model 3300 Ogawa Passive Samplers (Ogawa & Company, Pompano Beach, FL) were used for collecting NO2. This two-sided sampler consists of a cylindrical polymeric body (2 cm in diameter and 3 cm long) with a diffusion barrier and two stainless steel screens on each side. The device holds a glass-fiber collection pad coated with triethanolamine (TEA) at each end for sampling. The TEA-coated pads were loaded in the lab just prior to deployment to minimize contamination and degradation. All components, except the collection pad, are re-useable. Samples were field blank-corrected and reported in units of ppbV to match the original measurement units of the chemiluminescence analyser reported in AQS. The NO2 content was determined by an ion chromatograph (IC, Model 500, Dionex, Sunnyvale, CA) equipped with an IonPac™ AS-14 anion-exchange analytical column with 1.2 mL min−1eluent flow, a 50 µL sample loop, an anion self-regenerating suppressor (ASRS-1), and NaHCO3/Na2CO3-eluting buffers to quantify nitrite ion as a measure of NO2 to analyse the water extracts of the pads. Additional details on the Ogawa sampler for NO2 are discussed elsewhere.4
Evaluation of week-long monitoring of PE tubes
The study design was based on week-long sampling time integrals. This was based on previous studies that have demonstrated the passive samplers can be deployed for week-long and longer sampling time integrals.4,17,22,23 Thus, the PE tubes were also evaluated for linearity of response under week-long integrals prior to field deployment. A subset of the tubes were exposed to 2 ppbV of a TO-14/1,3-butadiene mixture in an exposure chamber at 75% relative humidity for 1, 2, 3, 5, and 7 day periods. The exposure chamber used for evaluating the tubes is described in McClenny and others.13 Samples were subsequently analysed using automated thermal desorption/GC/MS. For the majority of the 25 compounds evaluated (all but cis-1,2-dichloroethene and 1,2-dichloroethane), linear response was observed over the 1 day to 1 week exposure time. The results for benzene are in Fig. 1 to exemplify this linearity of the PE tube response and, hence, its applicability for week-long exposures. A calibration curve was subsequently generated by tubes exposed in the chamber for one week to 90 pptV, 180 pptV, 2 ppbV, and 10 ppbV of the TO-14/1,3-butadiene mixture. | ||
| Fig. 1 Week-long passively loaded PE tube series for benzene using 2 ppbv TO-14 VOC mixture in exposure chamber. | ||
Field monitoring and data analysis
Monitoring occurred for six consecutive weeks, from July 19 to August 30, 2005. Passive samplers were sheltered in specially-designed 2.8 L (22 cm widest diameter, 10 cm height) stainless steel bowls to avoid potential of outgas effects with other shelter materials; dimensions of the shelter permitted free flow of air at sampling inlets while minimizing effects from precipitation. The shelters were designed for installation on chain-link fences or stand-alone posts. A female Swagelok adapter welded at the bowl base and a rod installed inside approximately 8 cm from the bowl base was used to secure the PE tubes; Ogawa samplers were hung from this rod. Week-long sampling periods commenced on Tuesday afternoons through the following Tuesday afternoons (average of 169 h). Duplicate samples were collected at both sites during each sampling period; separate shelters were used for the duplicate samplers to avoid potential interference. Shelters were placed approximately 1 m above ground level at East 7 Mile and approximately 3.5 m above ground at Dearborn in well-ventilated areas. Passive samplers could not be placed at the same height as the continuous monitors at East 7 Mile; the passive samplers were approximately 5 to 6 m away from the reference sampling inlets. The passive-reference sampling height in Dearborn was the same. Passive samplers were collected and replaced with new ones at the same time each week. A CapLok™ tool (Markes International Limited, Llantrisant, UK) was used to tighten and loosen the Swagelok™ brass fittings on the PE tubes. A diffusive sampling cap (part number L4070207, Perkin-Elmer) replaced one of the tube Swagelok fittings and was installed in the field during sampling. The tubes were attached to the shelter with the sampling caps facing down during sampling. Samplers were shipped to the analysis laboratory on a weekly basis and refrigerated at 4 °C upon receipt.To make the comparisons described here, hourly AQS data from the continuous samplers were averaged on the same time frames as the six passive sampling periods. Missing hourly values were not imputed; averages were calculated only from available data. Measurements from the continuous monitors are typically quality assured prior to being reported in AQS. The AQS data were required to exhibit a data completeness level of at least 75% (i.e., 75% of valid hourly data) within each of the sampling periods. The Dearborn canister sample on week 2 was not used owing to flow controller problems, thus yielding only five sampling periods for VOC comparison purposes at that site.
For both NO2 and VOC data collected, simple linear regressions of the passive sampling measurements on the averaged values from AQS data were utilized to assess the performance of the passive samplers at each site. Regression analysis was chosen because it is an efficient way to compare the two methods across the range of observed values. Given that duplicate sampling was part of the study design, precision error (as percent relative standard deviation) was also calculated. All statistical analyses were performed with SAS 9.1 software.24
Results and discussion
Ambient assessment
Table 1 shows summary statistics of the NO2 and select VOC concentrations from the passive samplers at each site. The MDEQ continuous measurements at East 7 Mile and the 32 L canister samplers at Dearborn are also summarized in Table 1. Nitrogen dioxide and BTEX species were above corresponding MDLs for the passive measurements. Nitrogen dioxide is commonly associated with transportation, power plant, industrial combustion sources, and secondary formation from reactions between primary emissions of NO and O325,26 while BTEX species are associated with motor vehicle tail pipe and gasoline emissions.27–29Ogawa NO2 measurements were consistently higher at Dearborn than East 7 Mile (Fig. 2). (Note that the chemiluminescence data are shown as gray area peaks for the hourly data and as dots to represent week-long means of the hourly data.) Industrial and railroad emissions located in the immediate vicinity of the Dearborn site were possible reasons for higher NO2 levels. The average and standard deviation for sum of BTEX species was similar at East 7 Mile (20 ± 4 ppbC) and Dearborn (18 ± 3 ppbC). Pearson correlation for the sum of BTEX species and NO2 was 0.9 at East 7 Mile, indicative of mobile source emissions for these pollutants. Comparison of median NO2 levels reported with week-long measurements using Ogawa samplers in El Paso17 suggest that ambient levels at the Detroit sites were a third to half lower. At Dearborn, Pearson correlation between sum of BTEX species and NO2 was negatively correlated (−0.01) indicating non-transportation sources (such as the River Rouge industry) may have been impacting either ambient NO2 or BTEX VOCs or both.| Compound | East 7 Mile passivesa (N = 12)b | Dearborn passives (N = 12) | MDLc | East 7 Mile referenced (N = 6 for NO2; N = 5 for VOCs) | Dearborn referencee (N = 5) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Median | Min | Max | %RSDf | Median | Min | Max | %RSD | Median | Min | Max | Median | Min | Max | ||
| a Samples were field blank-corrected with data summarized based on pollutants below detection limit assigned a value equal to ½ the detection limit. b Numbers in parentheses are the number of observations designated at the top of site column. c MDL: method detection limit for passive samplers. d Reference methods at E-7 Mile site were auto-GC for VOC and chemiluminescence analyser for NO2 operated by MDEQ and reported in the EPA Air Quality System; these methods were continuous and are summarized based on their average value matched to the same time integral as the co-located passive samplers. Co-located NO2 and VOC measurements from reference samplers in same units. e Reference methods at Dearborn were passive 32 L summa-polished canister samplers operated by EPA for this study; values measurements are matched to the same time integral as the co-located passive samplers. f %RSD: relative pooled standard deviation (standard deviation ÷ mean as percentage) for analysis of precision of duplicate samples. g NM: not measured. | |||||||||||||||
| NO2 | 14 | 11 | 17 | 3 | 21 | 20 | 28 | 5 | 0.7 | 13.7 | 12.1 | 17.8 | NM g | NM | NM |
| 1,3-Butadiene | 0.3 | 0.2 | 0.4 | 10 | 0.2 | 0.1 | 0.4 | 13 | 0.2 | NM | NM | NM | 0.2 | 0.1 | 0.3 |
| Benzene | 2.6 | 1.8 | 3.5 | 4 | 2.2 | 1.4 | 2.9 | 6 | 0.1 | 2.3 | 2.0 | 4.7 | 3.5 | 2.2 | 5.5 |
| Toluene | 9.3 | 7.6 | 14 | 2 | 7.6 | 5.4 | 9.2 | 2 | 0.1 | 8.1 | 7.0 | 10.9 | 8.7 | 6.0 | 10.3 |
| Ethylbenzene | 1.4 | 1.2 | 1.9 | 1 | 1.5 | 1.0 | 2.2 | 4 | 0.1 | 1.4 | 1.1 | 1.7 | 1.8 | 1.1 | 2.4 |
| m,p-Xylene | 4.5 | 4.1 | 6.5 | 2 | 5.2 | 3.2 | 7.4 | 4 | 0.3 | 4.8 | 4.1 | 5.9 | 5.4 | 3.3 | 6.4 |
| o-Xylene | 1.6 | 1.4 | 2.3 | 1 | 1.5 | 1.0 | 1.8 | 3 | 0.1 | 2.5 | 2.1 | 4.0 | 1.7 | 1.2 | 2.3 |
| Styrene | 0.2 | 0.1 | 0.2 | 5 | 0.2 | 0.2 | 0.3 | 9 | 0.2 | 0.6 | 0.5 | 0.8 | 1.2 | 0.5 | 3.0 |
| 1,3,5-Trimethylbenzene | 0.6 | 0.5 | 0.9 | 4 | 0.7 | 0.5 | 1.8 | 6 | 0.1 | 0.8 | 0.7 | 1.0 | 1.1 | 0.4 | 1.5 |
 | ||
| Fig. 2 Average concentrations of duplicate Ogawa passive samplers (as horizontal lines) during the study period. Hourly NO2 concentration from the chemiluminescence analyser (as gray area peaks) are also averaged per week (as dots in the middle of the sampling week). | ||
Precision
Table 1 lists the relative standard deviations (%RSD, [standard deviation ÷ mean as percent]) for the duplicate Ogawa NO2 and PE tube VOC samples. Precision error for the Ogawa samplers was determined to be ≤ 5%. Measurements for NO2 and VOCs at Dearborn were less precise (higher %RSD) compared to East 7 Mile. The %RSDs for PE tube samples were low (≤10% at East 7 Mile and ≤ 11% at Dearborn) indicating good reproducibility. The %RSD values for the BTEX measurements in this study were similar to studies using this sampler for 24 h sampling periods.14 Reproducibility for Ogawa NO2 measurements from this study was similar when compared to duplicate week-long sampling of NO2 using Ogawas in El Paso.17Comparison to reference methods
Fig. 3 presents comparisons between BTEX concentrations reported by the PE tubes and the weekly averaged auto-GC at East 7 Mile. Fig. 4 shows BTEX comparisons between PE tubes and 32 L canister samples at Dearborn. Regression results and tests on the parameter estimates are shown in the figures. | ||
| Fig. 3 Regression of PE tube vs. auto-GC at East 7 Mile: (a) benzene; (b) toluene; (c) ethylbenzene; (d) o-xylene; (e) m,p-xylene. ap < 0.05 for y intercept significantly different from zero and/or slope significantly different from 1. | ||
 | ||
| Fig. 4 Regression of PE tube vs. 32 L canister sample at Dearborn: (a) benzene; (b) toluene; (c) ethylbenzene; (d) o-xylene; (e) m,p-xylene. ap < 0.05 for y intercept significantly different from zero and/or slope significantly different from 1. | ||
Initial examination of Fig. 3 suggest that PE tubes tended to track the auto-GC for all BTEX species. While the regressions for benzene and o-xylene were statistically different from the 1:1 line, note that the regression lines for benzene (Fig. 3a) and o-xylene (Fig. 3d) were heavily influenced by a single outlier in each case. Review of the individual hourly data from the auto-GC revealed that the outliers for benzene and o-xylene were driven by extremely elevated readings over a particular five hour period. For this five hour period, benzene was elevated to a level approximately ten times higher than any other level seen outside this period. Similarly, o-xylene levels were at least five times higher than any other observation outside this five hour period. This illustrates the distortion of the mean value from the auto-GC as a representative concentration for the entire week when a relatively few elevated values are present. The levels for the other BTEX species were relatively elevated during this period but not nearly to the degree as for benzene and o-xylene. Note that without this outlier, the agreement with the PE tubes and auto-GC was very good for benzene (R2 = 0.82) and o-xylene (R2 = 0.98). However, review of quality assurance data for both the PE tubes and auto-GC did not indicate a problem with the observations and, therefore, all observations are reported in Fig. 3. The overall results from Fig. 3 indicate good agreement of PE tubes and auto-GC with the exception of weeks when extremely elevated concentrations occur during a few hours.
For BTEX measurements compared between PE tubes and 32 L canisters (Fig. 4), agreement as indicated by R2 was generally good except for benzene. Though the regression lines for benzene, ethlybenzene and o-xylene were statistically different from the 1:1 line and the R2 for benzene was only 0.32, note that levels for all species were reported to be very low by both methods. The largest discrepancy was observed for benzene and this difference was < 3 ppbC. Note that the median differences for the PE tubes and 32 L canisters at Dearborn did not exceed 1 ppbC (Table 2).
| Compound | Bias (%)a | Differenceb |
|---|---|---|
| a Bias ((passive–reference) ÷ reference × 100) reported as the median of all sampling period mean biases. b Median difference between passive method minus reference method; units of ppbV for NO2 and ppbC for VOCs. c NO2 comparison from Ogawavs. chemiluminescence analyser. d VOC comparisons from Carbopack X tubes vs. auto-GC at East 7 Mile. e VOC comparisons from Carbopack X tubes vs. 32 L canister samples at Dearborn. | ||
| East 7 Mile | ||
| NO2c | −2 | −0.2 |
| Benzene d | −7 | 0.4 |
| Toluene | 19 | 3 |
| Ethylbenzene | 5 | 0.2 |
| m,p-Xylene | 0.5 | 0.3 |
| o-Xylene | −40 | −0.8 |
| Styrene | −70 | −0.4 |
| 1,3,5-Trimethylbenzene | −24 | −0.2 |
| Dearborne | ||
| 1,3-Butadiene | 19 | 0.03 |
| Benzene | −38 | −0.8 |
| Toluene | −7 | −0.3 |
| Ethylbenzene | −12 | −0.2 |
| m,p-Xylene | 1 | −0.3 |
| o-Xylene | −18 | −0.3 |
| Styrene | −84 | −1 |
| 1,3,5-Trimethylbenzene | −17 | −0.5 |
Biases and absolute differences in Table 2 indicate that the PE tubes tended to under-predict VOC levels in comparison to 32 L canisters. For the PE tubes versus auto-GC, slight over-predictions and under-predictions were observed for VOC species (Table 2). Fig. 5a to c display the agreement between the PE tubes and reference methods.
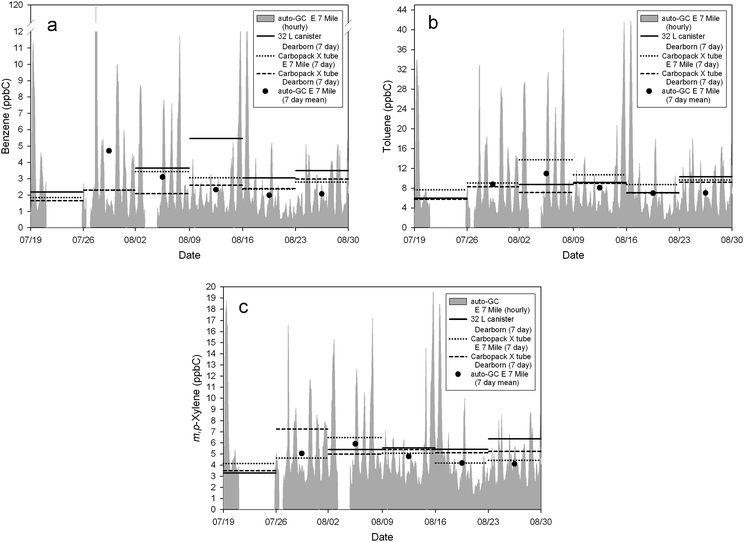 | ||
| Fig. 5 Average concentrations of duplicate Carbopack X tubes and canisters (as horizontal lines) during the study period with hourly average concentrations from the auto-GC. (a) Benzene; (b) toluene; (c) m,p-xylene. Hourly VOC concentration from auto-GC (as gray area peaks) are also averaged per week (as dots in the middle of the sampling week). | ||
An assessment of quality of the auto-GC VOC data was done by plotting total non-methane organic compound (TNMOC) concentration versus individual VOCs from the hourly averaged auto-GC data. In urban airsheds dominated by traffic, BTEX compounds have been found to be highly correlated with TNMOC.30 Scatterplots (not shown) suggested that BTEX compounds were properly identified in the chromatograms. Separate analysis of the AQS and passive lab data did not reveal any quality issues with the data used.
Nitrogen dioxide measurements reported from the Ogawa samplers and chemiluminescence analyser at the East 7 Mile site were comparable. Fig. 2 shows the good agreement between Ogawas and weekly mean chemiluminescence data at East 7 Mile. The NO2 regression results (Fig. 6) revealed a reasonable R2 (0.6) with the y intercept and slope (3.6 and 0.7, respectively) indicating that the Ogawa and reference method were close to the 1 : 1 line; p values indicated that the slope was not different from 1 and intercept not different from 0. The Ogawas at East 7 Mile tracked NO2 levels well with the chemiluminescence analyser (Fig. 2) and the bias was only 2% (Table 2).
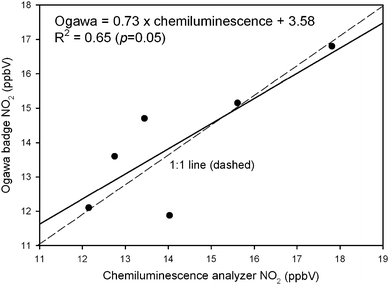 | ||
| Fig. 6 Regression of Ogawa sampler vs. chemiluminescence analyser at East 7 Mile. | ||
Finally, Fig. 2 and 5 also show respective data from the chemiluminescence analyser for NO2 and from the auto-GC for benzene, toluene and m,p-xylene superimposed on the week-long passive and week-long canister sample results; duplicate passive sampler data were averaged for presentation purposes. Potential episodic or short-term influences are smoothed out by the passive samplers and canisters relative to the hourly-averaged reference values. It is conceivable that these episodic ‘spikes’ may have affected the comparison results. Although week-long periods using the hourly data that had < 75% data capture were not used (such as week 1 for the auto-GC—see Fig. 5), it is possible that the reduced sample sizes affected the power of these regressions.
Conclusion
The overall conclusion from this limited study was that passive samplers were capable of reproducible measurements. The high level of precision of these samplers is important in spatial analysis studies in urban areas which have typically relied on passive samplers at multiple sites for modeling urban-gradients of air pollutants.31,32The samplers were generally comparable to reference methods given the low concentrations and small sample sizes. Absolute differences between passive and reference methods for VOCs were small. Results from this study indicate that the auto-GC and the PE tubes could show discrepancies for a week-long sampling period. However, these discrepancies were driven by a few hours of highly elevated concentrations. Thus, the passive samplers are useful for integrated samples while continuous monitors are necessary to assess short term temporal variations.
The primary caveat attached to these results is that this was a short-term study with collection of few samples, and only one measurement method available for the reference samplers. Comparisons of data were, thus, limited. Because of this, it is recommended that additional method evaluations be conducted with these passive samplers to more definitively establish their precision and accuracy. Accordingly, comparison of passive samplers with corresponding reference methods should be a routine quality assurance component where such samplers are deployed for air pollution measurement and modeling studies.
Acknowledgements
We thank Chris Fortune, Mike Wheeler, Dennis Williams and Mariko Porter, all from Alion Science and Technology, for their technical support. We also thank Ron Williams, William McClenny, Paul Killough and the EPA DEARS Team for input related to the passive methods and contacts with MDEQ. We also thank Mary Ann Heindorf, Ann Chevalier, Deborah Sherrod, Amy Robinson, and Craig Fitzner, all from MDEQ, for access to the MDEQ sites and their data and Alan Vette and Mark Sather of EPA for reviewing the manuscript. The U.S. EPA through its Office of Research and Development funded and managed the research described here under contract EP-D-05-065 to Alion Science and Technology, Inc. It has been subjected to Agency review and approved for publication. Mention of trade names or commercial products does not constitute an endorsement or recommendation for use.References
- S. V. Krupa and A. H. Legge, Environ. Pollut., 2000, 107, 31–45 CrossRef CAS.
- E. D. Palmes, Environ. Int., 1981, 5, 97–100 CrossRef CAS.
- Y. Yanagisawa and H. A. Nishimura, Environ. Int., 1982, 8, 235–242 CrossRef CAS.
- H. Y. Chang, M. T. Morandi and C. P. Weisel, J. Expos. Anal. Environ. Epidemiol., 2008, 18, 441–451 Search PubMed.
- R. M. Cox, Environ. Pollut., 2003, 126, 301–311 CrossRef CAS.
- C. W. Chung, M. T. Morandi, T. H. Stock and M. Afshar, Environ. Sci. Technol., 1999, 33, 3666–3671 CrossRef CAS.
- R. G. Lewis and S. M. Gordon, in Principles of Environmental Sampling, ed. L. H. Keith, American Chemical Society, Washington, DC, 1996, pp. 401–470 Search PubMed.
- J. L. Varns, J. D. Mulik, M. E. Sather, G. Glen, L. Smith and C. Stallings, Environ. Sci. Technol., 2001, 35, 845–855 CrossRef CAS.
- J. D. Ray, TheScientificWorld, 2001, 1, 483–497 Search PubMed.
- N. L. Gilbert, S. Woodhouse, D. M. Stieb and J. R. Brook, Sci. Total Environ., 2003, 312, 43–46 CrossRef CAS.
- R. H. Brown, J. Environ. Monit., 1999, 1, 115–116 RSC.
- U. Wideqvist, V. Vesely, C. Johansson, A. Potter, E. Brorström-Lundén, K. Sjöberg and T. Jonsson, Atmos. Environ., 2003, 37, 1963–1973 CrossRef CAS.
- W. A. McClenny, K. D. Oliver, H. H. Jacumin Jr, E. H. Daughtrey Jr. and D. A. Whitaker, J. Environ. Monit., 2005, 7, 248–256 RSC.
- W. A. McClenny, H. H. Jacumin Jr, K. D. Oliver, E. J. Daughtrey Jr and D. A. Whitaker, J. Environ. Monit., 2006, 8, 263–269 RSC.
- J. H. Lee, S. A. Batterman, C. Jia and S. Chernyak, J. Air Waste Manage. Assoc., 2006, 56, 1503–1517 CAS.
- R. Williams, EM, October 2005, 43 Search PubMed.
- S. Mukerjee, L. A. Smith, G. A. Norris, M. T. Morandi, M. Gonzales, C. A. Noble, L. M. Neas and H. A. Özkaynak, J. Air Waste Manage. Assoc., 2004, 54, 307–319 CAS.
- U.S. Environmental Protection Agency, Fed. Reg., 1989, 54, 50820 Search PubMed , http://www.epa.gov/ttn/amtic/criteria.html (accessed 2008).
- U.S. Environmental Protection Agency, Technical Assistance Document for Sampling and Analysis of Ozone Precursors, EPA/600-R-98/161, Research Triangle Park, NC, 1998, http://www.epa.gov/ttn/amtic/files/ambient/pams/newtad.pdf (accessed 2008) Search PubMed.
- U.S. Environmental Protection Agency, PAMSGRAM Vol. 18. Supplemental Information on the Operation of the Ozone Precursor System, Research Triangle Park, NC, 2000, http://www.epa.gov/ttn/amtic/files/ambient/pams/pamsgrm18.pdf (accessed 2008) Search PubMed.
- U.S. Environmental Protection Agency, Compendium Method TO-14A. Determination of volatile organic compounds (VOCs) in ambient air using specially prepared canisters with subsequent analysis by gas chromatography, EPA/625/R-96/010b, Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, Cincinnati, OH, 2nd edn, 1999, http://www.epa.gov/ttn/amtic/files/ambient/airtox/to-14ar.pdf (accessed 2008) Search PubMed.
- N. A. Martin, P. Duckworth, M. H. Henderson, N. R. W. Swann, S. T. Granshaw, R. P. Lipscombe and B. A. Goody, Atmos. Environ., 2005, 39, 1069–1077 CrossRef CAS.
- M. E. Sather, E. T. Slonecker, J. Mathew, H. Daughtrey and D. D. Williams, Environ. Monit. Assess., 2007, 124, 211–221 CrossRef CAS.
- SAS Institue, Inc., SAS/STAT 9.1 User's Guide, SAS, Cary, NC, 2004 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics, Wiley, New York, 1998 Search PubMed.
- P. Warneck. Chemistry of the Natural Atmosphere, Academic Press, San Diego, 1988 Search PubMed.
- E. M. Fujita, Sci. Total Environ., 2001, 276, 171–184 CrossRef CAS.
- G. F. Evans, T. A. Lumpkin, L. Smith and M. C. Somerville, J. Air Waste Manage. Assoc., 1992, 42, 1319–1323 CAS.
- T. L. Conner, W. A. Lonneman and R. L. Seila, J. Air Waste Manage. Assoc., 1995, 45, 383–394 CAS.
- C. W. Lewis, R. C. Henry and J. H. Shreffler, J. Air Waste Manage. Assoc., 1997, 48, 71–76.
- Z. Ross, P. B. English, R. Scalf, R. Gunier R, S. Smorodinsky, S. Wall and M. Jerrett, J. Exposure Sci. Environ. Epidemiol., 2006, 16, 106–114 Search PubMed.
- L. Smith, S. Mukerjee, M. Gonzales, C. Stallings, L. Neas, G. Norris and H. Özkaynak, Atmos. Environ., 2006, 40, 3773–3787 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |