Green catalysis for selective CO oxidation in hydrogen for fuel cell
Shengjun
Huang
,
Kenji
Hara
and
Atsushi
Fukuoka
*
Catalysis Research Center, Hokkaido University, Kita-21 Nishi-10, Kita-ku, Sapporo, 001-0021, Japan. E-mail: fukuoka@cat.hokudai.ac.jp; Fax: +81 11-706-9139; Tel: +81 11-706-9140
First published on 29th July 2009
Abstract
Preferential oxidation of carbon monoxide in excess hydrogen (PROX) is one of the key processes for the production of clean hydrogen fuel for the polymer electrolyte membrane fuel cell (PEFC). Development of highly active and selective catalysts is among the most challenging works for decades. Platinum-based catalysts are regarded as the most promising candidates for this process. However, the activities of Pt catalysts seriously suffer from the strong poisonous adsorption by CO substrate at low reaction temperature. This article addresses the present approaches to improve catalytic performance at low temperature. The focus of this article is a new reaction route of CO oxidation over Pt/mesoporous silica under PROX reaction conditions. The high activity and selectivity of this catalyst has been attributed to the surface silanols in mesoporous silica. The perspectives of Pt/mesoporous silica in the PROX reaction are also presented.
 Shengjun Huang Shengjun Huang | Shengjun Huang received his Bachelor's degree in 2000 from Gannan Normal College, Ganzhou, China. From 2000, he studied heterogeneous catalysis in Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences (CAS), where he received a Ph.D. in industrial catalysis in 2006. In 2007, he joined the research group of Prof. Fukuoka of Hokkaido University as a postdoctoral research follow. His background was synthesis and application of zeolites in hydrocarbon transformation processes. Currently, he works on the synthesis and catalytic application of mesoporous material. |
 Kenji Hara Kenji Hara | Kenji Hara received his Bachelor's and Master's degrees in 1998 and 2000 from the University of Tokyo, respectively. Then he worked as a JSPS Researcher in Graduate School of Science, the University of Tokyo. He moved to Graduate School of Science, Hokkaido University as Assistant Professor in 2001. He received his Ph.D. from Hokkaido University in 2006. He was promoted to Associate Professor at Catalysis Research Center, Hokkaido University in 2007. He received the Young-Researcher Lecture Award from Surface Science Society of Japan in 2008. His current research interests are in preparation of functionalized surfaces and its application including catalysis. |
 Atsushi Fukuoka Atsushi Fukuoka | Atsushi Fukuoka received his Bachelor's and Master's degrees in 1982 and 1984 from the University of Tokyo. Then he moved to Hokkaido University as Assistant Professor. He received his Ph.D. from the University of Tokyo in 1989. He worked for Tokyo University of Agriculture and Technology for 6 years, and then he moved to Catalysis Research Center, Hokkaido University, where he has been Professor since 2007. He received Young Scientist Award from The Catalysis Society of Japan in 1994. His current research interests are in the synthesis and catalysis of mesoporous materials and the catalytic conversion of biomass. |
Broader contextThe production of hydrogen fuel free of carbon monoxide (concentration less than 10 ppm) is an essential requirement for the application of polymer electrolyte membrane fuel cell (PEFC). Preferential oxidation of CO by oxygen in excess H2 is the most economical and cost-efficient resolution. The most challenging work is the development of highly active and selective catalysts at the operation temperature (∼353 K) of PEFC, in which the Pt-based anodes are extremely susceptible to the poisoning by CO (>10 ppm). The surface sites of Pt-based catalysts are easily covered with CO, leading to the blockage of the sites for O2activation. Generally, modification of Pt architecture and usage of reducible support are approaches to overcome this constraint effect. However, these methods are always accompanied by the competitive H2oxidation due to the difficulty in the precise control of the O2activation. Herein, we show another approach by using Pt/mesoporous silica catalyst. It is revealed that surface silanols can be an oxidant in the reaction, by which Pt/FSM-16 exhibits ca. 100% CO conversion and >95% selectivity from 298 to 423 K. This overcomes the obstacle of O2activation and provides a new route for the final resolutions. |
Introduction
Polymer electrolyte membrane fuel cell (PEFC) uses the chemical energy of hydrogen to efficiently produce electricity with water as a by-product. In terms of its high energy-efficiency and ultra-low emission of pollutants, the PEFC is regarded as one of the most promising candidates to replace the internal combustion engine in vehicles, and power supply in stationary and portable power applications.1 The hydrogen fuel can be produced from fossil fuels (such as natural gas and coal) and other renewable resource such as biomass, solar and wind energy. However, in the medium term, pure hydrogen generated from renewable or sustainable energy sources will not be extensively available. At present, steam reforming, partial oxidation and auto-reforming of alcohols and hydrocarbons are still the major routes in hydrogen production.1,2 The description for the whole process is shown in Fig. 1.3 A noticeable amount of CO, ca. 5–15 vol%, is formed together with the generation of H2. By subsequent water-gas-shift (WGS) processes at high temperature (623–823 K) and low temperature range (473–573 K), the CO concentration can be reduced to 0.5–1 vol%.4,5 Unfortunately, Pt-based anodes are still extremely susceptible to the poisoning by the CO (>10 ppm) at their operation temperature (∼353 K).1,6,7 Therefore, the delivery of clean H2 to Pt anodes becomes the essential and imminent requirement for the application of PEFC. However, the complete reduction of CO cannot be accomplished by the WGS process due to the limitation in thermodynamic equilibrium.1–5 To achieve the acceptable ppm level of CO concentration, methanation (Eq. (1)) and preferential oxidation of carbon monoxide (PROX) (Eq. (2)) are designed for the fine cleaning of hydrogen:4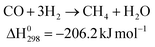 | (1) |
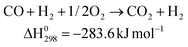 | (2) |
 | ||
| Fig. 1 The simplified description for the current production of hydrogen. | ||
However, the methanation of CO consumes rather higher amount of H2, which decreases the total efficiency of fuel cells.1 The PROX is regarded as the most effective and cost-efficient way to achieve the goal. The ideal solution is the direct integration of PROX process and PEFC as shown in Fig. 2. In order to approach this goal, the catalytic systems should match following key requirements:1,8
(1) Extremely high activity for CO oxidation. This deserves a priority due to the sensitivity of Pt anodes. In the stream containing 1 vol% CO, a conversion over 99.9% has to be achieved for the reduction to ppm level.
(2) High selectivity for CO oxidation. The catalysts should be inert for the reverse water-gas-shift reaction, which hampers the deep reduction of CO in the presence of abundant H2 and CO2. In order to minimize the hydrogen loss, the catalysts are expected to be inactive for the side oxidation of H2.
(3) Wide operation temperature window. Since the PROX is placed between the low temperature WGS reactor and PEFC, the catalysts should be applicable from 353 K to the outlet temperature of low temperature WGS reactor.
(4) Compatible with the operation parameters and conditions of upstream process. The catalysts should be resistant to the high concentration of CO2 and steam from the upstream WGS processes, and preserve their performances to the fluctuations in the space velocities and start-stop operation cycles.
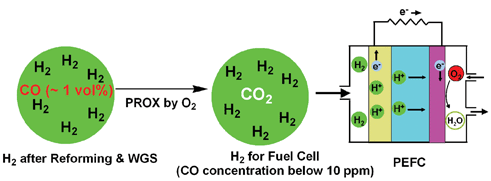 | ||
| Fig. 2 The diagram of ideal PROX process for the deep purification of H2 for PEFC. | ||
Various catalysts have been studied including precious metals (Ru9–11 Pd3,9,12,13 Rh14–16Au17–21 and Pt22–30), non-precious metals (Cu31–33 and Co31,34) and the combination of multiple metallic systems. The traditional Ru-based catalysts are operated at 413–473 K and always accompanied with the methanation of H2. Gold-based catalysts are rather active and selective at 353 K; however, the catalytic activities are still insufficient. Pt-based catalysts have been widely investigated and regarded as the promising candidates for this reaction. In fact, Pt/Al2O3catalysts were used to remove carbon monoxide from hydrogen prior to ammonia synthesis as early as in the 1960s.35–37 Unfortunately, these conventional catalysts are not directly applicable to the present system because they cannot match the requirement for the operation temperature, and hence cannot be directly integrated with the PEFC. In general, the operation temperature window for the complete reduction of CO is 443–473 K for conventional Pt-based catalysts in PROX reaction. The extension to the lower temperature range has become the target for decades. What is the most challenging point for Pt-based catalysts at low reaction temperature? It is well recognized that the surface of Pt catalyst will soon be fully covered by adsorbed CO under a steady-state flow of CO and O2, which prevents oxygen adsorption and hence suppresses the oxidation reaction.38 This problem can only be overcome with temperatures higher than 450 K, at which continuous desorption of adsorbed CO occurs and gaseous O2 may compete for these free adsorption sites.
Based on this understanding, the basic strategy is to promote the O2 adsorption at low temperature, i.e. to transform the competitive Langmuir–Hinshelwood reaction pathway to non-competitive dual-site one. So far, the efforts in this area can be divided into following aspects (Fig. 3):
(1) Incorporation of second metal or metal oxide as a promoter for the dissociation of O2 (route a);
(2) Usage of reducible support as the site for the activation of O2 (route b);
(3) Creation of new Pt structure to weaken the extremely strong interaction between metal and CO (route c);
(4) Pursuit of new catalyst support (mesoporous silica) (route d), which is basically inspired by our work.
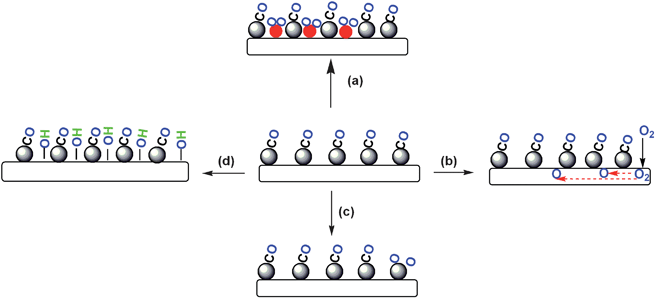 | ||
| Fig. 3 Reported strategies for the improvements of Pt-based catalysts. | ||
This paper gives examples of the ongoing research and advancement concerning the PROX reaction over Pt-based catalysts. The typical improvements in major catalyst systems have been referred. The results based on our recent work are also discussed, which reveals a newly proposed way for the oxidation of CO over the mesoporous silica. Furthermore, the perspectives of PROX reaction over mesoporous silica system are also presented.
1. Representative improvements for the catalyst systems
1.1 Incorporation of promoter for activation of O2
The direct way to realize the dual-site reaction pathway is to incorporate some metals or metal oxides, which are capable of O2 dissociation. The reported formulations include MnOx/Pt/Al2O3,39 Ce2O3/Pt/Al2O3,40 SnOx–Pt/Al2O341,42 and Fe–Pt/Al2O3(or TiO2).43–46 Compared with the supported monometallic Pt catalysts, the light-off temperatures are shifted to lower range over the promoted systems in usual CO oxidation reaction, which proves the contribution from improved O2activation. However, the complete CO reduction still cannot be achieved over these systems at low temperatures. For example, MnOx/Pt/Al2O3 (0.35 wt% Pt-15 wt% Mn) shows ca. 90% CO conversion at 413 K in the PROX reaction despite the high oxygen storage of MnOx species.39 SnO2–Pt/Al2O3 (1 wt% Pt-3 wt% Sn) can achieve ca. 100% CO conversion at 383 K with excess O2 conditions (O2/CO = 1.5).41 A series of Fe promoted Pt/zeolite catalysts have been reported to exhibit higher activity.23,26,27,47–49 The Fe–Pt bimetallic (4 wt% Pt-0.5∼2 wt% Fe) systems on mordenite zeolite show complete CO reduction between 373–403 K under the condition of excess O2/CO = 1. The reaction has been proposed to proceed through the “bifunctional mechanism” as shown in Fig. 4(b), where Pt acts as a CO adsorption site and the Fe site serves as an O2activation site, respectively.26 | ||
| Fig. 4 Incorporation of metal or metal oxide species for the dissociation of O2: (a) mono-metal without promoter, (b) neighbouring bimetallic system, (c) intimate bimetallic system. | ||
Depending on the pretreatment conditions, Fe species can be presented as a metal oxide strongly intimated with Pt or metallic form in metal-Pt alloy (Fig. 4c).26,42,44,48 It is reported that the bond strength between CO and Pt can be weakened due to the increase of Pt 5d-vacancy by alloying effect with metallic Fe.48 Besides improved O2activation capability, such intimate interaction can also suppress the CO adsorption on Pt as characterized by chemisorption and DRIFT-IR results.42,44,48 Furthermore, the effect of Fe promoter also depends on the type of support. The Fe promoted Pt/Al2O3catalysts do not exhibit such high activity and selectivity at low temperature. The cages of mordenite and ZSM-5 zeolites are speculated to play positive roles in reactions including the restricted space for fabrication of highly dispersed Pt nanoparticles and also “special reaction space” for CO oxidation. The effect of zeolites architecture was also investigated, and difference among various zeolites was observed as the following: Pt/A > Pt/mordenite > Pt/X. Moreover, dependence of oxidation activity on the pore diameter of A zeolite was also observed. The higher catalytic performance of Pt/3A was attributed to the “shape selectivity” by the dimension of 3 Å pore diameter, which may inhibit the admission of CO2 molecule and suppress the reverse water-gas-shift side reaction.3,23 Further efforts have been made to prepare bimetallic systems with uniform composition between Pt and promoters, which can improve the cooperative effects between CO adsorption and O2activation sites.50,51 By using organometallic precursors (Pt5Fe2(1,5-cyclooctadiene)(CO)12 or PtFe2(1,5-cyclooctadiene)(CO)8), the cluster-derived 1 wt% PtFe/SiO2catalysts show a relatively uniform Fe : Pt composition and particle-size distribution, which result in higher activity and selectivity than the 1 wt% Fe–Pt/SiO2 catalyst prepared by the incipient wetness impregnation method.50
1.2 Usage of reducible support
Besides the incorporation of promoters for Pt over inert Al2O3 and zeolite support, the usage of reducible–oxidizable support is an alternative route to provide the sites for O2activation. Ceria is typical and representative of such a type of support due to the well-known high oxygen storage capacity (OSC) of CeO2,52 which can improve the oxygen supply and subsequent oxidation reaction. As expected, Pt/CeO2 (0.54 wt% Pt) showed ca. 100% CO conversion at 373 K with a stoichiometric O2/CO ratio in the absence of H2.30 Although the CO conversion was improved in the presence of H2 at the low temperature range (<353 K), it decreased with further higher reaction temperature and far from the complete CO reduction.30 As noted above, the reaction follows the modified non-competitive Langmuir–Hinshelwood pathway, i.e. the O atoms are activated by the ceria support and react with adsorbed CO through spill-over or at the metal-support interface. The participation of surface oxygen from the support has already been proved from the observation of zero-order oxygen pressure dependence and also the oxygen-exchange measurements between C16O and 18O-predosed catalysts.53–55 The decreased CO conversion in PROX is attributed to the competitive side oxidation of H2, which occurs between the surface O atoms of ceria support and spill-over H atoms from Pt. The side reaction becomes more severe with higher OSC. The doping of ZrO2 to CeO2 improved the OSC and oxygen mobility of CeO2,8,56 and was able to oxidize CO even in the absence of O2.8 However, the CO conversions over the Pt/CeO2–ZrO2 (1.0 wt%) in PROX was much lower than those of Pt/CeO2. In contrast, the O2 conversions were always ca. 100%, indicative of more serious H2oxidation side reactions. This may suggest that it is very important to control the O2activation to avoid the competitive side reaction in PROX. Nevertheless, the controversy over the reaction mechanism still remains. It is proposed that the reaction may occur through the low temperature water-gas-shift route, i.e., the gas phase O2 is not only limited to the activation by the re-oxidation of reducible support, but also transformed into the surface water on the ceria surface that is reactive with CO on the Pt surface.57–59 Nonetheless, the redox properties of supports always lead to the serious competitive side oxidation of H2. Furthermore, the CeO2 support is not resistant to the CO2 due to the carbonation effect.30,58 These weaknesses manifest the intrinsic disadvantages of this type of material in the practical PROX reactions.1.3 New Pt structure with weakened CO adsorption
Bimetallic noble metal systems of Pt–Ru and Pt–Pd have been reported to show high CO-tolerance in eletrocatalysis.60,61 These combinations are naturally incorporated into the heterogenous catalysis application. In such systems, the catalysts work in the bifunctional way: the more oxophilic metal (with lower CO coverage, e.g.Ru) serves as an oxygen activator, which improves the oxidation of adsorbed CO on less oxophilic Pt center. The typical reported formulations include Pt–Ru/Mordenite,62 Pt–Ru/SiO263 and Pt–Ru/Al2O3.64 The metals can be presented as in monometallic particles and segregated intra-particles (alloy); the latter may weaken adsorption strength between CO and Pt. Another way is to directly weaken the interaction between Pt and CO by the addition of alkali base metal species.65–67 However, it is difficult to control the state and interaction of the components, while the improvement in catalytic performance is also limited.Very recently, a Ru–Pt core-shell (Ru@Pt) structure has been designed.64,68 Compared with the traditional Pt–Ru alloy and mixture of monometallic particles, the core-shell structure is characteristic of confined Ru core covered with an approximately thin monolayer-thick shell of Pt atoms. The full characterization results (XRD, TEM, XPS and FT-IR) validate only 1–2 Pt monolayer is exposed on the surface. For the 1000 ppm CO feeds, CO is reduced to ppm level from room temperature over the Ru@Pt/Al2O3 (1 wt% Pt loading, O2/CO = 5) catalysts, which is distinctly superior to the PtRu alloy and Ru + Pt monometallic mixture counterparts. Since the Ru is buried in the core, the traditional bifunctional mechanism is not operative. This means the electronic structure of Pt surface should be modified by the Ru core. The Ru substrate has a lateral compression on Pt monolayer and decreases the interaction between Pt and adsorbates.64,68 Moreover, the interaction between Pt-monolayer and Ru core downshifts the d-band center. Both effects contribute to the weakened interaction strength between Pt surface and adsorbates, and significantly destabilize CO on Pt. This leads to lower CO saturation coverage and provides much more CO-free sites for activation of other reactants. The density-function-theory (DFT) calculations show 2/3 monolayer of the pure Pt(111) surface is covered with CO at saturation state, whereas only 1/2 monolayer of Pt (Ru@Pt) is covered with CO. The CO-free sites decrease the H2 dissociation energy barrier and contributes to an O2 dissociation route spill-over hydrogen. These interpret the high CO oxidation activity of Ru@Pt architecture at low reaction temperatures.
2. Promotional effect of mesoporous silica for PROX reaction
The basic start point of current strategy is to improve the O2activation by weakening the CO adsorption on Pt or adding reducible promoter. However, the improved O2activation always results in the competition of H2oxidation, which suppresses the CO oxidation. So far, the architecture with precise controlled O2activation capability still requires more efforts. Among the reported results, the reduction of CO still requires temperatures around 373 K or high excess O2/CO ratio.49,68 Hence, there still remains a challenge of catalyst design for high activity and selectivity from the low temperature (<373 K). Instead of exploring new Pt architecture, we put our efforts into the pursuit of a support.Since the discovery of FSM-16,69,70MCM-4171 and SBA-1572 in the last decades, mesoporous silicas have been extensively investigated for applications in catalysis.73,74 The mesoporous silicas are characteristic of ordered pores (2–10 nm) with high surface area (ca. 500–1000 m2 g−1), which are attractive as catalysts and supports. However, understanding the role of mesoporous silica is generally limited to give higher dispersion of active sites than over the conventional silica. From the viewpoint of molecular level, the participation of mesoporous silica in the catalytic reactions is through the silanols (or hydroxyl groups) on the surface. If the surface silanols can participate in the CO oxidation, the limitation of O2activation on the activity can be decreased. Inspired by this point, we investigate the PROX reaction over the Pt/mesoporous silica.
2.1 Unique superior catalytic performance of Pt/FSM-16.
Pt nanoparticles and nanowires (5 wt% Pt loading) are synthesized in several types of mesoporous silica (FSM-16, MCM-41 and HMM-1) as previously reported.75 The surface area and dispersion of Pt nanoparticles over mesoporous silica are higher than those of conventional SiO2- and Al2O3-supported catalysts (Table 1).The Pt nanoparticles over FSM-16 (5 wt% Pt loading) show unprecedented high activity. As shown in Table 2, Pt(p)/FSM-16 provides remarkably higher activity of ca. 100% CO conversion at 353 K under the conditions of twice excess O2 (O2/CO = 1) over the stoichiometry (O2/CO = 1/2). In contrast, SiO2-, Al2O3- and HMM-1 supported catalysts show CO conversion less than 30% under the same reaction conditions, and require 423 K or even higher temperature to remove majority of CO.25 In particular, Pt(p)/FSM-16 gives ca. 100% conversion of CO from 313 K to 423 K,25 thus showing that this is one of the most active catalysts for PROX. The reported Pt catalysts always require excess O2 (O2/CO > 1/2) because the H2oxidation (H2 + 1/2 O2 → H2O) consumes the O2. As shown in Fig. 5a, in the stoichiometric O2/CO (1/2) gas flow mixture, Pt(p)/FSM-16 shows CO conversions over 95% at 298–423 K. This indicates that the CO selectivity is over 95% for Pt(p)/FSM-16.
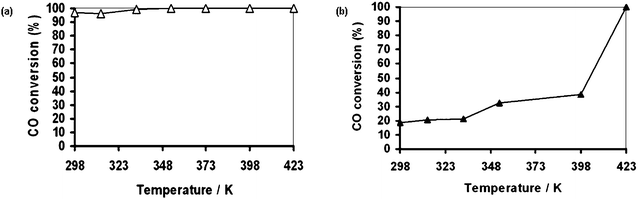 | ||
| Fig. 5 (a) PROX over Pt(p)/FSM-16 under stoichiometric conditions. Reaction conditions: CO 1%, O2 0.5%, N2 5%, H2 balance, SV 12000 ml g−1 h−1, 0.1 MPa. (b) Usual CO oxidation over Pt(p)/FSM-16. Reaction conditions: CO 1%, O2 1%, N2 5%, He balance, SV 12000 ml g−1 h−1, 0.1 MPa. | ||
For the mesoporous type silica, FSM-16 and MCM-41 show the similar catalytic performance, perhaps due to the similar structure. However, the performance of Pt(p)/organosilica HMM-1 is even lower than those of Pt/Al2O3 and Pt/SiO2 despite its higher Pt dispersion. This poses a question: what is the decisive factor for the activity?
For Pt(p)/FSM-16, 2.5 nm Pt particles inside the internal mesopore are observed in the TEM and STEM images. The Pt/Al2O3 and Pt/SiO2 have a broad particle-size distribution from 3 to 7 nm. However, the temperature-programmed CO desorption (CO-TPD) experiments show the interaction between CO and Pt is quite similar on different catalysts. Moreover, the CO conversion on Pt(p)/FSM-16 undergoes a sharp decrease if the reaction is carried out in the absence of H2 (Fig. 5b). This indicates the reaction mechanism of PROX over Pt(p)/FSM-16 is totally different from the usual CO oxidation, and the difference in the Pt morphology is not responsible for the different activities.
2.2 Mechanistic study of PROX by the isotope tracer method
IR spectra of CO adsorption (30 Torr) over Pt catalysts at 298 K were shown in Fig. 6a. All the typical three samples, active Pt(p)/FSM-16 and inactive Pt(p)/HMM-1 and Pt/SiO2, show a linear CO frequency on Pt at 2088 cm−1 (Fig. 6a). However, small gaseous CO2 peaks were observed at ca. 2350 cm−1 over Pt(p)/FSM-16 in the enlarged range of 2400–2200 cm−1 (Fig. 6b). Furthermore, in the adsorption of 13CO or a mixture of 12CO and 13CO, gaseous 13CO2 peak was also observed at 2280 cm−1. As a strong comparison, none of CO2 peak was observed in the CO adsorption over the inactive Pt(p)/HMM-1 or Pt/SiO2catalyst. It should be noted that no oxygen source was presented except the FSM-16 support in the adsorption of 12CO or 13CO. The most reasonable explanation is that the oxygen of silica in FSM-16 is incorporated into CO2 in the CO adsorption.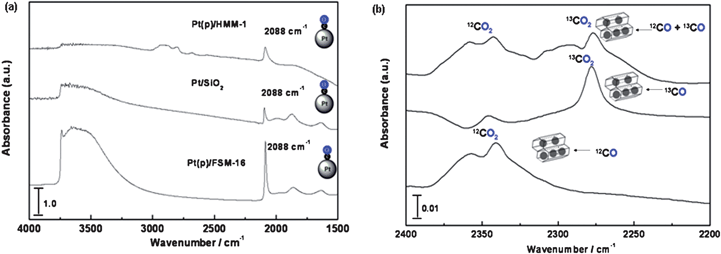 | ||
| Fig. 6 (a) IR spectra of 12CO adsorption over Pt(p)/FSM-16, Pt/SiO2 and Pt(p)/HMM-1. (b) Formation of gaseous 12CO2 or 13CO2 in the adsorption of 12CO and 13CO over the Pt(p)/FSM-16. | ||
In order to confirm this hypothesis, sequential addition of CO, 18O2 and D2 on Pt(p)/FSM-16 was further performed. After admission of CO (10 Torr) to the IR cell at 293 K, gaseous C16O2 peak appears at ca. 2350 cm−1 (Table 3). By further addition of 18O2 (10 Torr) to the cell, no peak of C16O18O was detected at 2331 cm−1. Even after introduction of D2 (60 Torr) for simulation of the PROX conditions (CO + 18O2 + D2), the peaks of 18O-labeled CO2 were not observed. Since no exchange was observed in the admission of C18O2 to Pt(p)/FSM-16, we can exclude the fast 16O–18O exchange between gaseous C18O2 product and siliceous FSM-16 (Si16O2). Thus, the direct participation of dissociated O from gas phase O2 can be denied. In the PROX reaction conditions, gas phase O2 may be involved through the H2O formation, i.e. WGS pathway between H2O and adsorbed CO. However, if we assume labeled water (D218O) generated from D2 and 18O2 attacked CO, C16O18O would be a main product of WGS. Based on these results, only the surface silanol groups should be responsible for the CO oxidation.
| Atmosphere | CO | CO2 | C16O18O (2331 cm−1) |
|---|---|---|---|
| a Sequential adsorption of CO (10 Torr), 18O2 (10 Torr), and D2 (60 Torr). | |||
| CO | 2088 cm−1 | 2350 cm−1 | Not observed |
| CO + 18O2 | 2088 cm−1 | 2350 cm−1 | Not observed |
| CO + 18O2 + D2 | 2084 cm−1 | 2350 cm−1 | Not observed |
Table 3 also shows the variation of Pt–CO peak position in the sequential adsorption of CO, 18O2 and D2 on Pt(p)/FSM-16. The Pt–CO peak at 2088 cm−1 was not shifted by the addition of 18O2 to the gas phase (Table 3). In contrast, by further addition of D2 to the gas phase, the Pt–CO peak was shifted to 2084 cm−1. This low frequency shift is attributable to the enhanced back-donation from Pt to CO by forming Pt–D bond in the dissociative adsorption of D2 to Pt. If 18O2 were adsorbed on Pt under the same conditions, the Pt–CO peak would be shifted to higher frequency by the decreased back-donation from Pt to CO. The shift of Pt–CO peak suggests the selective adsorption of H2 on Pt and O2 on FSM-16.
The unique property of silanols on FSM-16 can be further demonstrated by the H-D exchange experiments. When D2 was admitted to the IR cell after CO adsorption at 293 K, a broad band of OD was detected over Pt(p)/FSM-16 at ca. 2600 cm−1 (Fig. 7), while the OD band was not seen over Pt/SiO2, Pt(p)/HMM-1 or bare FSM-16 itself. Presumably, D2 is adsorbed on the Pt surface even in the presence of adsorbed CO, and the OD band reflects the spill-over process in the PROX atmosphere.
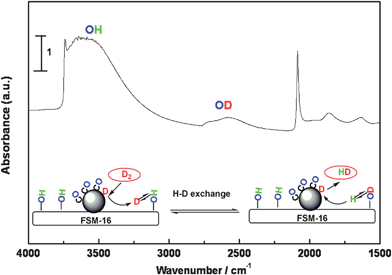 | ||
| Fig. 7 H-D exchange over Pt(p)/FSM-16 in the sequential adsorption of CO (10 Torr) and D2 (10 Torr). | ||
2.3 Overall mechanism for PROX reaction over Pt(p)/FSM-16
Combining the catalytic performances and the IR results, we propose that the surface OH on FSM-16 is highly reactive toward CO adsorbed on Pt nanoparticles at the interface of Pt and FSM-16. As depicted in Fig. 8, the OH groups attack CO on Pt to form CO2 and H2, and a vacant site is formed on FSM-16. O2 is adsorbed on FSM-16 and suggested to form the OH groups with the assistance of spill-over hydrogen from Pt.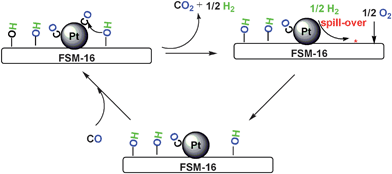 | ||
| Fig. 8 Proposed mechanism for PROX over Pt(p)/FSM-16. | ||
Perspectives and prospects
According to the present results, it can be concluded that the difference between mesoporous silica and conventional amorphous silica is not only in their physical parameters, but also in their reactivity in surface silanols. Since the high activity and selectivity of Pt/mesoporous silica is related with the surface silanols in FSM-16 and MCM-41, the following questions arise naturally:(1) Which type of surface silanols is closely related with the superior catalytic performances? In general, there are three types of surface silanols (free, geminal and hydrogen-bonded silanols) on the mesoporous silica. We have revealed the involvement of surface silanols in the reaction. However, we have no more information about the possible different activities among these silanol groups.
(2) What is the effect of pore size on the catalytic performance? The pore diameter may influence the encapsulation of Pt nanoparticles into the mesopore, which affects the proximity between metal sites and surface silanols. On the other hand, the distribution of silanols is different as the function of pore size,76,77 which can give more clarifications about the catalytic role of surface silanols.
(3) How about the catalytic performance of other mesoporous silicas? FSM-16 (MCM-41) is one of the representative materials in the mesoporous silica family. Can we observe such promotional effect on other typical mesoporous silica? This will improve the understanding of the catalytic role of mesoporous silica not only from the viewpoint of structure but also from their possible intrinsic different reactivity in surface silanols.
(4) How can we decrease the Pt loading to a practical level? It has been reported that the introduction of Al into mesoporous silica can improve the activity of Au catalysts in CO oxidation.78 Since the incorporation of Al will change the type and interaction of surface silanols, similar effect may also be observed on the Pt/mesoporous silica in PROX reaction.
(5) Why is there remarkable difference between siliceous FSM-16 and organic-inorganic HMM-1 silica?
We have just opened the application of mesoporous silica in PROX reaction. The ongoing and forthcoming investigations will promote the catalytic performance and improve understanding the intrinsic catalytic role of mesoporous silica. We hope it will be helpful not only for PROX reaction but also for the general understanding of these materials in wide applications.
Conclusions
In general, the PROX reaction and normal CO oxidation are regarded to follow the same competitive Langmuir–Hinshelwood reaction mechanism. As a result, one of the crucial problems for Pt-based catalysts in the PROX reaction is to create the CO-free sites for the selective and controlled activation of gas phase O2. The reported methods include the introduction of O2 activator, usage of reducible support and designed structure for weaker CO adsorption. Despite these efforts, further efforts are still required for the innovation of extremely high active, selective and stable catalyst systems. At the same time, the controversy and unresolved problems still remain in the basic research on this process, e.g. the reaction intermediate, variation of surface species plus their real roles in reaction, the origin of the deactivation and the possibly transformed states under different reaction condition (space velocity, O2/CO ratio and concentration, etc.). The application of mesoporous silica in PROX opens a new potential way for the resolution. The most valuable and interesting finding is that the surface silanols in mesoporous silica can play as the “oxidant” to initiate the CO reaction. This may release the confliction in the modification for the adsorption between CO and Pt, in which too much weakened adsorption will accelerate the side oxidation of H2. Compared with the system with O2 activator, it also decreases the side reaction at higher temperature. Joint efforts from above mentioned methods will help to approach the final solutions for PROX process.Acknowledgements
This work was supported by a Grant-in Aid for Scientific Research on Priority Areas (No. 18065001, “Chemistry of Concerto Catalysis”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.References
- R. J. Farrauto, Y. Liu, W. Ruettinger, O. Ilinich, L. Shore and T. Giroux, Catal. Rev. Sci. Eng., 2007, 49, 141–196 CrossRef CAS.
- D. L. Trimm, Appl. Catal., A, 2005, 296, 1–11 CrossRef CAS.
- I. Rosso, C. Galletti, G. Saracco, E. Garrone and V. Specchia, Appl. Catal., B, 2004, 48, 195–203 CrossRef CAS.
- A. F. Ghenciu, Curr. Opin. Solid State Mater. Sci., 2002, 6, 389–399 CrossRef.
- T. V. Choudhary and D. W. Goodman, Catal. Today, 2002, 77, 65–78 CrossRef CAS.
- R. A. Lemons, J. Power Sources, 1990, 290, 251–264 CrossRef.
- H. Igarashi, T. Fujino and M. Watanabe, J. Electroanal. Chem., 1995, 391, 119–123 CrossRef CAS.
- A. Wootsch, C. Descorme and D. Duprez, J. Catal., 2004, 225, 259–266 CrossRef CAS.
- S. H. Oh and R. M. Sinkevitch, J. Catal., 1993, 142, 254–262 CrossRef CAS.
- Y. F. Han, M. J. Kahlich, M. Kinne and R. J. Behm, Phys. Chem. Chem. Phys., 2002, 4, 389–397 RSC.
- M. Echigo and T. Tabat, Appl. Catal., A, 2003, 251, 157–166 CrossRef CAS.
- F. Marino, C. Descorme and D. Duprez, Appl. Catal., B, 2004, 54, 59–66 CAS.
- W. Li, F. J. Gracia and E. E. Wolf, Catal. Today, 2003, 81, 437–447 CrossRef CAS.
- H. Tanaka, S. Ito, S. Kameoka, K. Tomishige and K. Kunimori, Appl. Catal., A, 2003, 250, 255–263 CrossRef CAS.
- S. Ito, H. Tanaka, Y. Minemura, S. Kameoka, K. Tomishige and K. Kunimori, Appl. Catal., A, 2004, 273, 295–302 CrossRef CAS.
- S. Ito, T. Fujimori, K. Nagashima, K. Yuzaki and K. Kunimori, Catal. Today, 2000, 57, 247–254 CrossRef CAS.
- M. M. Schubert, V. Plzak, J. Garche and R. J. Behm, Catal. Lett., 2001, 76, 143–150 CrossRef CAS.
- B. Schumacher, Y. Denkwitz, V. Plzak, M. Kinne and R. J. Behm, J. Catal., 2004, 224, 449–462 CrossRef CAS.
- R. M. Torres Sanchez, A. Ueda, K. Tanaka and M. Haruta, J. Catal., 1997, 168, 125–127 CrossRef CAS.
- F. Arena, P. Famulari, G. Trunfio, G. Bonura, F. Frusteri and L. Spadaro, Appl. Catal., B, 2006, 66, 81–91 CrossRef CAS.
- M. M. Schubert, A. Venugopal, M. J. Kahlich, V. Plzak and R. J. Behm, J. Catal., 2004, 222, 32–40 CrossRef CAS.
- A. Manasilp and E. Gulari, Appl. Catal., B, 2002, 37, 17–25 CrossRef CAS.
- H. Igarashi, H. Uchida, M. Suzuki, Y. Sasaki and M. Watanabe, Appl. Catal., A, 1997, 159, 159–169 CrossRef CAS.
- O. Korotkikh and R. Farrauto, Catal. Today, 2000, 62, 249–254 CrossRef CAS.
- A. Fukuoka, J. Kimura, T. Oshio, Y. Sakamoto and M. Ichikawa, J. Am. Chem. Soc., 2007, 129, 10120–10125 CrossRef CAS.
- M. Kotobuki, A. Watanabe, H. Uchida, H. Yamashita and M. Watanabe, J. Catal., 2005, 236, 262–269 CrossRef CAS.
- M. Kotobuki, A. Watanabe, H. Uchida, H. Yamashita and M. Watanabe, Appl. Catal., A, 2006, 307, 275–283 CrossRef CAS.
- M. Shou and K. Tanaka, Catal. Lett., 2006, 111, 115–118 CrossRef CAS.
- S. H. Cho, J. S. Park, S. H. Choi and S. H. Kim, J. Power Sources, 2006, 156, 260–266 CrossRef CAS.
- J. L. Ayastuy, A. Gil-Rodriguez, M. P. Gonzalez-Marcos and M. A. Gutierrez-Ortiz, Int. J. Hydrogen Energy, 2006, 31, 2231–2242 CrossRef CAS.
- F. Marino, C. Descorme and D. Duprez, Appl. Catal., B, 2005, 58, 175–183 CrossRef CAS.
- P. Ratnasamy, D. Srinivas, C. V. V. Satyanarayana, P. Manikandan, R. S. Senthil Kumaran, M. Sachin and V. N. Shetti, J. Catal., 2004, 221, 455–465 CrossRef CAS.
- M. Tada, R. Bal, X. Mu, R. Coquet, S. Namba and Y. Iwasawa, Chem. Commun., 2007, 4689–4691 RSC.
- G. Marban, I. Lopez, T. Valdes-solis and A. B. Fuertes, Int. J. Hydrogen Energy, 2008, 33, 6687–6695 CrossRef CAS.
- M. Brown, and A. Green, US Pat., 3 088 919, 1963.
- J. Cohn, US Pat., 3 216 782, 1965.
- J. Cohn, US Pat., 3 216 783, 1965.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS.
- J. L. Ayastuy, M. P. Gonzalez-Macros, J. R. Gonzalez-Velasco and M. A. Gutierrez-Ortiz, Appl. Catal., B, 2007, 70, 532–541 CrossRef CAS.
- U. Oran and D. Uner, Appl. Catal., B, 2004, 54, 183–191 CrossRef CAS.
- G. Uysal, A. N. Akin, Z. I. Onsan and R. Yildirim, Catal. Lett., 2006, 111, 173–176 CrossRef CAS.
- M. M. Schubert, M. J. Kahlich, G. Feldmeyer, M. Huttner, S. Hackenberg, H. A. Gasteiger and R. J. Behm, Phys. Chem. Chem. Phys., 2001, 3, 1123–1131 RSC.
- O. Korotkikh and R. Farrauto, Catal. Today, 2000, 62, 249–254 CrossRef CAS.
- X. S. Liu, O. Korotkikh and R. Farrauto, Appl. Catal., A, 2002, 226, 293–303 CAS.
- K. Tanaka, Y. Moro-oka, K. Ishigure, T. Yajima, Y. Okabe, Y. Kato, H. Hamano, S. Sekiya, H. Tanaka, Y. Matsumoto, H. Koinuma, H. He, C. Zhang and Q. Feng, Catal. Lett., 2004, 92, 115–121 CrossRef CAS.
- G. W. Roberts, P. Chin, X. L. Sun and J. J. Spivey, Appl. Catal., B, 2003, 46, 601–611 CrossRef CAS.
- M. Kotobuki, A. Watanabe, H. Uchida, H. Yamashita and M. Watanabe, Chem. Lett., 2005, 34, 866–867 CrossRef CAS.
- M. Kotobuki, T. Shido, M. Tada, H. Uchida, H. Yamashita, Y. Iwasawa and M. Watanabe, Chem. Lett., 2005, 103, 263–268 CAS.
- N. Maeda, T. Matsushima, H. Uchida, H. Yamashita and M. Watanabe, Appl. Catal., A, 2008, 341, 93–97 CrossRef CAS.
- A. Siani, B. Captain, O. S. Alexeev, E. Stafyla, A. B. Hungria, P. A. Midgley, J. M. Thomas, R. D. Adams and M. D. Amiridis, Langmuir, 2006, 22, 5160–5167 CrossRef CAS.
- J. Yin, J. Wang, T. Zhang and X. Wang, Catal. Lett., 2008, 125, 76–82 CrossRef CAS.
- A. Trovarelli, Catal. Rev. Sci. Eng., 1996, 348, 439–520 CrossRef.
- E. Bekyarova, P. Fornasiero, J. Kaspar and M. Graziani, Catal. Today, 1998, 45, 179–183 CrossRef CAS.
- A. Holmgren, D. Duprez and B. Andersson, J. Catal., 1999, 182, 441–448 CrossRef CAS.
- C. Descorme, Y. Madier and D. Duprez, J. Catal., 2000, 196, 167–173 CrossRef CAS.
- H. S. Roh, H. S. Potdar, K. W. Jun, S. Y. Han and J. W. Kim, Catal. Lett., 2004, 93, 203–207 CrossRef CAS.
- O. P. Tellinger, D. Teschner, J. Krohnert, F. C. Jentoft, A. K. Gericke, R. Schlogl and A. Wootsch, J. Phys. Chem. C, 2007, 111, 5426–5431 CrossRef.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Krohnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. K. Gericke, Z. Paal and R. Schlogl, J. Catal., 2006, 237, 1–16 CrossRef CAS.
- D. Teschner, A. Wootsch, O. P. Tellinger, J. Krohnert, E. M. Vass, M. Havecker, S. Zafeiratos, P. Schnorch, P. C. Jentoft, A. K. Gericke and R. Schlogl, J. Catal., 2007, 249, 318–327 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 259–266 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 275–283 CrossRef CAS.
- H. Igarashi, H. Uchida and M. Watanabe, Chem. Lett., 2000, 29, 1262–1263 CrossRef.
- S. Y. Chin, O. S. Alexeev and M. D. Amiridis, J. Catal., 2006, 243, 329–339 CrossRef CAS.
- S. Alayoglu and B. Eichhorn, J. Am. Chem. Soc., 2008, 130, 17479–17486 CrossRef CAS.
- C. Pedrero, T. Waku and E. Iglesia, J. Catal., 2005, 233, 242–255 CrossRef CAS.
- Y. Minemura, S. Ito, T. Miyao, S. Naito, K. Tomishige and K. Kunimori, Chem. Commun., 2005, 1429–1431 RSC.
- M. Kuriyama, H. Tanaka, S. Ito, T. Kubota, T. Miyao, S. Naito, K. Tomishige and K. Kunimori, J. Catal., 2007, 252, 39–48 CrossRef CAS.
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333–338 CrossRef CAS.
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS.
- S. Inagaki, Y. Fukushima and K. Kuroda, Chem. Commun., 1993, 680–682 RSC.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2372–2419.
- A. Taguchi and F. Schuth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- A. Fukuoka, Y. Sakamoto, T. Higuchi, N. Shimomura and M. Ichikawa, J. Porous Mater., 2006, 13, 231–235 CrossRef CAS.
- M. Katoh, K. Sakamoto, M. Kamiyamane and T. Tomida, Phys. Chem. Chem. Phys., 2000, 2, 4471–4475 RSC.
- T. Ishikawa, M. Matsuda, A. Yasukawa, K. Kandori, S. Inagaki, T. Fukushima and S. Kondo, J. Chem. Soc., Faraday Trans., 1996, 92, 1985–1989 RSC.
- C.-W. Chiang, A. Wang, B.-Z. Wu and C.-Y. Mou, J. Phys. Chem. B, 2005, 109, 18042–18047 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |