Improved performance of porphyrin-based dye sensitised solar cells by phosphinic acid surface treatment
Alessandra
Allegrucci
a,
Naomi A.
Lewcenko
a,
Attila J.
Mozer
b,
Lynn
Dennany
b,
Pawel
Wagner
b,
David L.
Officer
*b,
Kenji
Sunahara
c,
Shogo
Mori
c and
Leone
Spiccia
*a
aSchool of Chemistry, Monash University, Victoria 3800, Australia. E-mail: leone.spiccia@sci.monash.edu.au; Fax: +61 3 9905 4597; Tel: +61 3 9905 4526
bIntelligent Polymer Research Institute, ARC Centre for Excellence for Electromaterials Science, University of Wollongong, Wollongong, 2522, Australia. E-mail: davido@uow.edu.au; Fax: +61 2 4221 3114; Tel: +61 2 4221 469
cDepartment of Fine Materials Engineering, Shinshu University, Nagano, 386-8567, Japan
First published on 22nd June 2009
Abstract
Chemical surface treatment of porphyrin-sensitised titania films using bis-(4-methoxyphenyl)phosphinic acid after dye adsorption, results in large improvements in DSSC efficiencies which originate primarily from higher short circuit currents. The result was attributed to a positive shift in the TiO2 quasi-Fermi level with simultaneous retardation of charge recombination. High device performances have been achieved even using simplified electrolyte matrices devoid of the common additives, LiI and t-butylpyridine.
Broader contextRecombination losses are one major factor that can limit the efficiencies of dye sensitised solar cells (DSSCs). Zinc-porphyrins, for example, are promising alternatives to Ru-sensitisers which exhibit recombination losses. Various strategies are being employed to minimise these losses, many of them aiming to block exposed sites on the nanostructured titania surface. One successful strategy has involved the co-adsorption of organic acids, such as decylphosphonic acid or chenodeoxycholic acid, and the dye. We demonstrate that chemical surface treatment of porphyrin-sensitised titania films using a diarylphosphinic acid, post dye adsorption, results in large improvements in DSSC efficiencies that directly parallel increases in the short circuit currents. Notably, the most striking improvements were found for electrolytes without the additives, i.e., no LiI and/or t-butylpyridine, commonly used to enhance DSSC performance. Controlled functionalisation of the semiconductor surface with carefully selected phosphinic acids, as a tool for improving DSSC efficiencies, is an exciting concept that can be applied for sensitisers where recombination reactions are currently limiting and opens up new possibilities for designing simplified electrolyte systems. |
Introduction
As international concerns about energy and climate change increase, there is an intense global research effort to reduce the cost and improve the performance of solar powered devices. Dye sensitised solar cells (DSSCs) are one such technology that has been intensively investigated since reported by O'Regan and Grätzel in 1991.1 In a DSSC, the dye, following excitation by light (D + hν → D*), injects an electron into the TiO2 conduction band (D* → D+ + CB), which then moves through the external circuit converting I3− → I− (typical redox couple) at the platinised counter electrode. Iodide then reduces the oxidised dye (3I− + 2D+ → 2D + I3−). Due to the nanostructured nature of these devices, molecular interactions involving the titania film and the adsorbed dye molecules,2,3 electrolyte components,4,5 or treatment agents6,7 can alter the conduction band potential of the semiconductor causing changes in the open circuit voltage (Voc) and/or the short circuit current (Jsc) leading to changes in the overall device efficiency (η) {η = Voc × Jsc × FF/Pin, where Pin is the power input and FF is the fill factor}. While Voc is dictated by the difference between the quasi-Fermi level of the TiO2 and the potential of the redox couple, Jsc arises from the efficiency of injection from the dye into the TiO2 and the efficiency of charge collection. Therefore, both values are limited by the recombination processes2,8,9 that occur when the photoinjected electron recombines with the oxidised dye molecule (R1) or reacts with the oxidised form of the redox couple in the electrolyte (R2). Process R2 has been well studied10–14 and has been deemed to have a greater impact on device performance than R1.15Various strategies are being employed to overcome this intrinsic limitation, many of them aiming to protect exposed sites on the titania surface. Two simple approaches involve protection of the unfunctionalised titania surface by either molecular engineering of the sensitising dye molecule to contain, e.g., long alkyl chains16 or blocking vacant titania sites by reaction with an organic acid. Co-adsorption of decylphosphonic acid7 or chenodeoxycholic acid (CDCA)6 with the dye leads to increased Voc and Jsc, while CDCA also has a beneficial effect when added to the electrolyte. Phosphinic acids also form strong interactions with titania17 but, in contrast to carboxylic or phosphonic acids, they have two organic substituents that can potentially better insulate the titania surface on attachment. Herein, we show that bis-(4-methoxyphenyl)phosphinic acid (BMPPA), which contains two bulky aryl substituents (Fig. 1), is a good candidate as a chemical surface treatment agent.
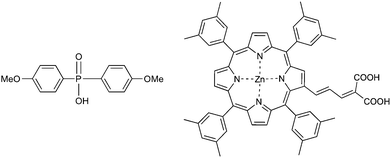
To date, literature reports have predominantly focussed on co-adsorption techniques, where the treatment agent is added to the dye solution. Herein we present a new approach involving the controlled exposure of the titania surface to the surface treatment agent for various times after dye-adsorption. The dye is a zinc-porphyrin (GD2, Fig. 1) previously used for high efficiency solar cells.18 Despite the great potential of zinc-porphyrins as alternatives to Ru-dyes in DSSCs, these compounds exhibit considerable recombination losses, as reported recently by several groups.19,20 Thus, this class of dyes is ideal for studying improvements in DSSC efficiency by surface treatment. For comparison, we have also undertaken measurements with the commonly used ruthenium dye, N719.
Results and discussion
Four electrolyte matrices were used during the course of this investigation (see Materials and methods for composition). Electrolyte A is the standard electrolyte for high-performing porphyrin-based DSSCs18 and contains an iodide source, iodine, LiI and TBP in an acetonitrile : valeronitrile (75 : 25) solvent (AN : VN). Electrolyte B is essentially identical to Electrolyte A without LiI. Electrolyte C contains only the iodide source and iodine in AN : VN, while Electrolyte D is the same as Electrolyte C but uses the less volatile 3-methoxypropionitrile (MPN) as the solvent. These electrolytes were chosen to gain insight into the device performance in the presence and absence of the various additives commonly used in the construction of DSSCs.Interestingly, the data for GD2 (Table 1) clearly shows that for all electrolyte systems, regardless of the presence or absence of performance enhancing additives, the films treated with BMPPA resulted in higher device efficiencies due to improved current densities. That is, the efficiency of devices constructed after film treatment, mirrors the improvement in current.
| Min | V oc/mV | J sc/mA cm−2 | FF | % η | |
|---|---|---|---|---|---|
| GD2 Electrolyte A | 0 | 728 (±2) | 8.6 (±0.4) | 0.71 (±0.01) | 4.1 (±0.2) |
| 5 | 718 (±9) | 8.8 (±0.3) | 0.72 (±0.01) | 4.3 (±0.1) | |
| 30 | 698 (±4) | 9.4 (±0.4) | 0.73 (±0.02) | 4.8 (±0.1) | |
| 60 | 706 (±2) | 9.9 (±0.1) | 0.73 (±0.01) | 4.9 (±0.1) | |
| GD2 Electrolyte B | 0 | 714 (±2) | 1.9 (±0.1) | 0.75 (±0.01) | 1.0 (±0.1) |
| 5 | 721 (±8) | 2.3 (±0.1) | 0.75 (±0.01) | 1.3 (±0.1) | |
| 30 | 690 (±10) | 5.1 (±0.2) | 0.76 (±0.04) | 2.7 (±0.2) | |
| 60 | 702 (±5) | 4.9 (±0.1) | 0.77 (±0.04) | 2.7 (±0.1) | |
| GD2 Electrolyte C | 0 | 669 (±2) | 5.2 (±0.1) | 0.76 (±0.06) | 2.9 (±0.5) |
| 5 | 647 (±7) | 5.9 (±0.6) | 0.74 (±0.03) | 3.2 (±0.4) | |
| 30 | 647 (±9) | 7.9 (±0.4) | 0.76 (±0.02) | 3.9 (±0.1) | |
| 60 | 659 (±10) | 8.9 (±0.5) | 0.75 (±0.01) | 4.4 (±0.3) | |
| GD2 Electrolyte D | 0 | 643 (±4) | 4.8 (±0.3) | 0.68 (±0.06) | 2.1 (±0.5) |
| 5 | 624 (±15) | 5.5 (±0.1) | 0.72 (±0.01) | 2.5 (±0.1) | |
| 30 | 626 (±17) | 6.6 (±0.5) | 0.69 (±0.04) | 2.9 (±0.4) | |
| 60 | 623 (±11) | 6.7 (±0.4) | 0.72 (±0.04) | 3.0 (±0.4) | |
| N719 Electrolyte E | 0 | 821 (± 15) | 13.7 (± 0.5) | 0.68 (± 0.01) | 7.7 (± 0.3) |
| 30 | 814 (± 7) | 13.6 (± 0.3) | 0.69 (± 0.02) | 7.6 (± 0.4) | |
| 60 | 805 (± 21) | 13.1 (± 0.01) | 0.70 (± 0.01) | 7.8 (± 0.3) | |
| 120 | 798 (± 12) | 13.4 (± 0.3) | 0.68 (± 0.01) | 7.2 (± 0.1) |
Cells constructed using Electrolyte A (both LiI and TBP) showed the best performance after 60 min of BMPPA treatment. Current increases of 15% (8.6 mA cm−2 to 9.9 mA cm−2) were accompanied by a 20% efficiency increase (4.1% to 4.9%).
DSSCs constructed using elecrolyte B (no LiI), showed the poorest overall performances. These devices exhibited similar open circuit voltages to those constructed using Electrolyte A but with poorer currents and efficiencies due to the inclusion of the TBP without the ameliorative effects of Li cations. Although device performance with this electrolyte was unsatisfactory, the currents improved by over 2.5 times from 1.9 to 4.9 mA cm−2 with increasing BMPPA treatment times while the efficiency increased from 1.0% (untreated) to 2.7% (60 min BMPPA treatment).
For Electrolyte C, which contained neitherLiInorTBP, striking improvements in performance were found for cells constructed with the treated films. The efficiency of devices made with films treated with BMPPA for 60 min was almost as high as those obtained using the standard Electrolyte (A): Jsc increased 1.6 fold and efficiencies improved from 2.9% (untreated) to 4.4% (treated) making these cells as efficient as those assembled using the untreated films and the high performing electrolyte. The IV profiles for Electrolyte C and different treatment times clearly demonstrate the link between treatment time and cell current, viz., the improvement in current densities with increasing treatment times shown in Fig. 2.
 | ||
| Fig. 2 IV profiles measured at 1 sun for DSSCs prepared with untreated and BMPPA treated titania films using Electrolyte C. | ||
Despite the poorer overall performances demonstrated by Electrolyte D, containing the less volatile 3-methoxypropionitrile (MPN), similar improvements in efficiency were observed which again originated from increases in Jsc.
The correlation between device efficiency and short circuit current is most evident when plotting these two parameters as a function of treatment time on the same graph, as is shown in Fig. 3. Here, the most striking improvements, of up to 300%, were observed following surface treatment for the electrolytes with no LiI (B and C).
 | ||
| Fig. 3 Plot of efficiency (η—full lines) and short circuit current (Jsc—dotted lines) versusBMPAA treatment time for devices constructed using the four electrolytes; composition of electrolytes A–D is given in footnotes to Table 1. | ||
Interestingly, surface treatment with BMPPA did not improve the performance of DSSCs constructed with N719 in conjunction with the standard electrolyte (which contains GuNCS instead of LiI). This data is summarised under N719 in Table 1. This result indicates that the BMPPA is not beneficial for DSSCs constructed with this commonly applied ruthenium dye.
To further investigate the origin of the improved cell current, IPCE (incident photon-to-current conversion efficiency) measurements were made on the cells prepared using Electrolyte A (Fig. 4). The treated films clearly provide improved IPCE profiles. The IV and IPCE data highlight the enhanced performance of devices constructed using titania films that had been treated with the surface protection agent after dyeing, which results from large increases in short circuit current. The observed improvements could result from decreased recombination due to surface protection, but could also arise from increased driving forces for electron injection due to decreased conduction band potential. Tuning of the conduction band potential has been proposed to be the origin of the beneficial effects observed when LiI is added to the electrolyte.21
 | ||
| Fig. 4 IPCE curves for cells constructed using Electrolyte A. | ||
To further elucidate how the treatment agent was influencing DSSC performance, electron lifetimes and diffusion coefficients were measured. In these experiments, devices were constructed using titania films that had been treated with BMPPA for 0, 5 and 30 min after dye adsorption and Electrolyte A. A plot of the electron diffusion coefficient as a function of short circuit current (Fig. 5a) suggests that longer treatment times result in smaller diffusion coefficients, however, the plot of the electron lifetime vs. short circuit current (Fig. 5b) clearly shows an improvement in electron lifetime after surface treatment.
 | ||
| Fig. 5 (a) Electron diffusion coefficient as a function of short circuit current. (b) Electron lifetime as a function of short circuit current. (c) Electron lifetime as a function of electron density. (d) Voc as a function of electron density. | ||
Fig. 5d suggests that there is a positive shift of up to 50 mV in the conduction band potential with increased treatment time. These results are consistent with the observed increase in Jsc and slight decrease (10–20 mV) in Voc after surface treatment. Interestingly, the increase of IPCE was prominent in the long wavelength region, suggesting that the excited electrons are injected without internal relaxation as is observed with Ru complex dyes.22 At open circuit, the 50 mV conduction band shift is mostly compensated by an increased electron density due to better injection and reduced charge recombination which results in overall changes to the Voc of less than that predicted.
The positive conduction band shift after the surface treatment is a major difference compared to the effect of CDCA which indicates that the origin of the improvement in cell performance must also be different. In the case of CDCA, Jsc is increased due to the suppression of aggregation of dye molecules on the surface of the titania. As such, it is only effective with dyes that suffer reduced performance as a result of surface aggregation. For our porphyrin dyes, the addition of CDCA leads to only slightly increased Jsc values, indicating that for these porphyrin systems, aggregation is not a major problem.23 With respect to charge recombination, CDCA seems to retard this process, but large effects are only seen when a sizeable portion of adsorbed dye is replaced by CDCA. Thus, in order to have a significant effect, a decrease in Jsc may be inevitable.24,25 For the case of BMPPA, the increase of Jsc is partly due to the positive shift of the quasi-Fermi level and, thus, the effect is expected for all dyes having relatively low LUMO levels. The drawback of the positive shift is the decreased Voc arising from a decreased difference between the conduction band edge and I−/I3−redox potential. However, BMPPA also retards charge recombination as evidenced by the increased electron lifetimes for treated devices which partially compensates this drawback. The mechanism of retardation is also likely to differ between CDCA and BMPPA. It has been pointed out that some dyes increase the local concentration of electron acceptor species.19,26 Therefore, replacement of surface-attached dye molecules with CDCA may simply reduce the degree of attraction to the acceptors. On the other hand, BMPPA can occupy vacant titania sites between the adsorbed dye molecules, preventing the approach of the acceptor species to the TiO2 surface. Although the mode of action of BMPPA requires further study, this concept provides the potential to apply surface treatment to manipulate both conduction band potentials and recombination rates simultaneously, which is an exciting development.
To confirm chemical attachment and/or adsorption of BMPPA to the titania surface, ATR FTIR spectra were recorded for 6 µm films that had been treated with 0.2 mM BMPPA in acetonitrile for 5–60 min. Bands attributable to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches in the 1500–1600 cm−1 region and OP–O stretches in the 1000–1200 cm−1 region clearly signified the presence of attached/adsorbed phosphinate on the titania surface (see Fig. 6).
C stretches in the 1500–1600 cm−1 region and OP–O stretches in the 1000–1200 cm−1 region clearly signified the presence of attached/adsorbed phosphinate on the titania surface (see Fig. 6).
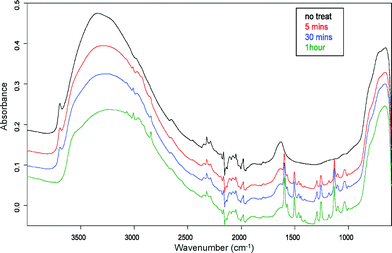 | ||
| Fig. 6 ATR FTIR profile of titania films treated with 0.2 mM BMPPA in acetonitrile for various times. | ||
Conclusion
Surface treatment of porphyrin-sensitised titania films with bis-(4-methoxyphenyl)phosphinic acid improved the efficiency of DSSC devices. These effects were witnessed with all electrolyte systems, although the most striking improvements were found for an electrolyte with no additional additives, i.e., no LiI and/or TBP. This discovery opens up new possibilities for designing simplified electrolyte systems using a variety of phosphinic acid treatment agents. Furthermore, such a post-treatment could be applicable in not only porphyrin-sensitised solar cells, but also in non-ruthenium dyes where recombination reactions are currently limiting.26 Controlled surface functionalisation as a tool for DSSC behaviour modification is an exciting concept worthy of further investigation.Materials and methods
Solvents and reagents were sourced from commercial suppliers and used as received. GD2 was prepared by a literature procedure.18Titania paste (18 nm) was provided by JGC Catalysts and Chemicals Ltd. (Kitakyushu-Shi, Japan). FTO glass was purchased from Nippon Sheet Glass (13 Ω □−1) and Surlyn (DuPont) from Solaronix (Aubonne, Switzerland).General cell construction protocols were adapted from literature procedures.27 The deposition of the titania working electrodes involved the application of a dense layer of titania to clean FTO glass, by spray pyrolysis of titanium diisopropoxide bis(acetylacetonate) 75% in isopropanol, followed by screen printing of one 6 µm layer of 18 nm titania nanoparticles. The working electrodes were sintered and stored in dust-free conditions. Prior to device construction, they were re-sintered at 450 °C for 30 min, cooled to 60 °C before being dipped in 0.2 mM ethanol solutions of GD2 in the dark for 2 h. The dyed films were removed in low light, washed with ethanol and dried before some were treated with 0.2 mM BMPPA in acetonitrile in the dark for 5, 30 or 60 min. After treatment, films were washed with acetonitrile, dried and used to construct DSSC devices as follows. Pre-drilled and cleaned counter electrodes were platinised using 10 mM hexachloroplatinic acid in isopropanol and sintered at 400 °C for 15 min. The working and counter electrodes were sandwiched with 25 µm Surlyn and sealed using a pneumatic finger and resistive heater (150 °C) for 20 s. The electrolyte was introduced into the cavity through pre-drilled holes in the counter electrode by vacuum backfilling. The filling port was sealed using 25 µm Surlyn and a microscope coverslip. Electrode contact was achieved using Cerasol CS186 solder at 220 °C with an oscillation frequency of 60 kHz prior to attaching copper wires using standard 60–40 tin–lead solder.
For GD2, four electrolytes were used: (1) Electrolyte A, standard electrolyte for porphyrin solar cells;18 0.6 M N-propyl-N′-methylimidazolium iodide (PMII), 0.03 M I2, 0.1 M LiI and 0.5 M tert-butylpyridine (TBP) in 75 : 25 acetonitrile:valeronitrile (AN : VN); (2) Electrolyte B, as Electrolyte A but with no LiI; (3) Electrolyte C, as Electrolyte A but with no LiIorTBP; and (4) Electrolyte D, the same as Electrolyte C but using the less volatile 3-methoxypropionitrile (MPN) as solvent. For N719, the electrolyte composition was 0.6 M PMII, 0.03 M I2, 0.5 M TBP and 0.1 M GuNCS in 85 : 15 AN to VN (Electrolyte E).
DSSC devices were tested using simulated sunlight (AM1.5 1000 W m−2) provided by an Oriel solar simulator with an AM1.5 filter. Current–voltage characteristics were measured using a Keithley 2400 source meter. Cells were biased from +800 to −300 mV with 10 mV steps and a 40 ms settling time (delay between application of the potential and current measurement) Short circuit current and open circuit voltage decays were determined by illuminating devices with a 635 nm diode laser, reducing the intensity and measuring the voltage or current response with a fast multimeter.19 For the IPCE measurements, cells constructed with dyed titania films that had been treated with BMPPA for various times (0, 5, 30, 60 min) were held under short circuit conditions and illuminated with monochromatic light in 10 nm steps using a Newport lamp. Electron densities (ED, cm−3) at the same illumination intensities were determined by a charge extraction method where the applied light bias on the device was removed, accompanied by a simultaneous switch from open circuit to short circuit. The resulting current was integrated and the electron density was calculated from the amount of charge extracted.
Acknowledgements
The authors thank the Australian Research Council and the Australian Centre of Excellence in Electromaterials Science for funding and JGC Catalysts and Chemicals Ltd. Kitakyushu-Shi, Japan for providing the titania paste. AA thanks EES and UNSW for the best poster award received at the 17th International Conference on Photochemical Conversion and Storage of Solar Energy held in Sydney in July 2008.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- S. Nakade, T. Kanzaki, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2005, 109, 3480–3487 CrossRef CAS.
- J.-W. Lee, K.-J. Hwang, D.-W. Park, K.-H. Park, W.-G. Shim and S.-C. Kim, J. Nanosci. Nanotechnol., 2007, 7, 3717–3721 CrossRef CAS.
- S. Nakade, T. Kanzaki, S. Kambe, Y. Wada and S. Yanagida, Langmuir, 2005, 21, 11414–11417 CrossRef CAS.
- H. Paulsson, L. Kloo, A. Hagfeldt and G. Boschloo, J. Electroanal. Chem., 2006, 586, 56–61 CrossRef CAS.
- N. R. Neale, N. Kopidakis, J. van de Lagemaat, M. Grätzel and A. J. Frank, J. Phys. Chem. B, 2005, 109, 23183–23189 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, R. Humphry-baker, J. E. Moser and M. Grätzel, Adv. Mater., 2003, 15, 2101–2104 CrossRef CAS.
- S. A. Haque, E. Palomares, B. M. Cho, A. N. M. Green, N. Hirata, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2005, 127, 3456–3462 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538–547 CrossRef CAS.
- A. J. Frank, N. Kopidakis and J. van de Lagemaat, Coord. Chem. Rev., 2004, 248, 1165–1179 CrossRef CAS.
- T. Kanzaki, S. Nakade, Y. Wada and S. Yanagida, Photochem. Photobiol. Sci., 2006, 5, 389–394 RSC.
- Q. Wang, S. Ito, M. Grätzel, F. Fabregat-Santiago, I. Mora-Sero, J. Bisquert, T. Bessho and H. Imai, J. Phys. Chem. B, 2006, 110, 25210–25221 CrossRef CAS.
- F. Fabregat-Santiago, J. Garcia-Canadas, E. Palomares, J. N. Clifford, S. A. Haque, J. R. Durrant, G. Garcia-Belmonte and J. Bisquert, J. Appl. Phys., 2004, 96, 6903–6907 CrossRef CAS.
- J. R. Durrant, S. A. Haque and E. Palomares, Coord. Chem. Rev., 2004, 248, 1247–1257 CrossRef.
- D. Kuciauskas, M. S. Freund, H. B. Gray, J. R. Winkler and N. S. Lewis, J. Phys. Chem. B, 2001, 105, 392–403 CrossRef CAS.
- N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS.
- A. J. Mozer, P. Wagner, D. L. Officer, G. G. Wallace, W. M. Campbell, M. Miyashita, K. Sunahara and S. Mori, Chem. Commun., 2008, 4741–4743 RSC.
- A. Forneli, M. Planells, M. A. Sarmentero, E. Martinez-Ferrero, B. C. O'Regan, P. Ballester and E. Palomares, J. Mater. Chem., 2008, 18, 1652–1658 RSC.
- D. F. Watson and G. J. Meyer, Coord. Chem. Rev., 2004, 248, 1391–1406 CrossRef CAS.
- R. Katoh, C. R. Chim., 2006, 9, 639 CrossRef CAS.
- Q. Wang, W. M. Campbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397–15409 CrossRef CAS.
- J.-H. Yum, S.-R. Jang, R. Humphry-Baker, M. Grätzel, J.-J. Cid, T. Torres and M. K. Nazeeruddin, Langmuir, 2008, 24, 5636–5640 CrossRef CAS.
- X.-F. Wang, O. Kitao, H. Zhou, H. Tamiaki and S. Sasaki, J. Phys. Chem. C, 2009, 113, 7954–7961 CrossRef CAS.
- M. Miyashita, K. Sunuhara, T. Nishikawa, N. Koumura, K. Hara, A. Mori, T. Abe, E. Suzuki and S. Mori, J. Am. Chem. Soc., 2008, 130, 17874–17881 CrossRef CAS.
- D. Kuang, S. Ito, B. Wenger, C. Klein, J. E. Moser, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4146–4154 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |