Design of solid catalysts for the conversion of biomass
Roberto
Rinaldi
and
Ferdi
Schüth
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470, Mülheim an der Ruhr, Germany. E-mail: schueth@mpi-muelheim.mpg.de
First published on 1st April 2009
Abstract
The discovery and investigation of novel and efficient pathways for the conversion of biomass into fuels and chemicals are among the big challenges facing heterogeneous catalysis nowadays. However, not all experience gained in the transformation of hydrocarbons over the last 100 years can directly be transferred to biomass conversion. In this article, we will discuss how the specific properties of biomass pose new requirements on the processes and on the solids that are used as catalysts for their conversion. Due to the importance of lignocellulosic materials, which constitute ca. 95% of the total plant biomass, we will focus mostly on the desired properties of solid catalysts for the conversion of these polymeric biomolecules. Research in this field is very intense presently and novel transformations and catalysts are being discovered at a high rate. This paper thus focuses on the concepts that govern biomass transformation instead of giving a complete and comprehensive survey of the literature, which would be outdated within a short time.
 Roberto Rinaldi Roberto Rinaldi | Dr Roberto Rinaldi studied Chemistry at the State University of Campinas, São Paulo, Brazil, where he received a PhD in Chemistry in 2006 under supervision of Prof. Dr Ulf Schuchardt. For his PhD work, he received the Evonik-Degussa Award conferred by the Brazilian Catalysis Society. In 2007 he joined the research group of Prof. Dr Ferdi Schüth as Post-Doc. Nowadays, he works on the transformation of cellulose in ionic liquids and on the development of novel catalysts for biomass utilization. |
 Ferdi Schüth Ferdi Schüth | Prof. Dr Ferdi Schüth studied Chemistry and Law at Münster, where he received a PhD in Chemistry in 1988 and the State Examination in Law in 1989. In 2001 he received the Award of the Stifterverband für die Deutsche Wissenschaft and in 2003 the Leibniz-Award of the German Science Foundation. He is vice president of the German Science Foundation (DFG) and serves on the editorial boards of several international journals. His research interests include catalysis, porous solids, hydrogen storage, high-throughput-experimentation in catalysis and materials science, and solids formation from solution. |
Broader contextBiomass is a very valuable feedstock, both for the production of chemicals and of novel fuels, which in the future could replace crude oil and gas as the current major raw materials. The most interesting—since the most abundant—fraction of biomass is lignocellulose, which, however, is highly protected and rather difficult to activate for further process steps. Solid catalysts are in principle very suitable for the processing of biomass. However, the requirements for biomass conversion are rather different, compared to the processing of hydrocarbon feedstocks, which form the backbone of our current energy and raw materials supply. The specific demands, novel process options and perspectives for the utilization of biomass by heterogeneously catalyzed pathways are highlighted in this article and contrasted to conventional value chains used nowadays in the petrochemical and chemical industries. |
Introduction
Forests and crops capture around 1% of the incoming solar radiation into biomass.1 This energy is stored in complex molecules, such as carbohydrates, lignins, proteins, glycerides, terpenes, and others.2 Unfortunately, tapping this resource directly to obtain fuels and chemicals is not possible, but instead new catalytic processes are required to facilitate and control the required conversions. Over the 20th century, the petrochemical and the chemical industry developed numerous processes to transform hydrocarbon-like compounds into almost everything that surrounds us in our modern world. However, most of these processes are not suitable for converting biomass. Looking at the general formula of monosaccharides—Cx(H2O)y—where x and y are equal in most of the cases, we see that each carbon atom is connected to an oxygen atom. In biorefineries, processing thus starts from highly oxygenated raw materials, and controlled de-functionalization is necessary, instead of functionalization used nowadays in the chemical industry. An efficient transformation of biomass should thus reduce its oxygen content—through efficient catalytic processes—providing alternative pathways for the production of fuels and chemicals.The major components of plant biomass—cellulose, hemicelluloses and lignins—possess much higher oxygen contents than gas, coal and crude oil (Fig. 1). The transformation of solid glucose into liquid fuels, such as ethanol or butanol, is an efficient strategy to decrease the O/C molar ratio. It is worth mentioning that the conversion of biomass to fuels does not aim at the total removal of the oxygen content from the raw materials—this would result in fuels with inferior anti-knocking properties. For instance, gasoline usually requires additives to improve its burning properties, since its O/C molar ratio is close to zero. Typically, oxygenated molecules (MTBE, ETBE, methanol, ethanol and others) are used as gasoline additives.3 These additives improve the anti-knocking properties of gasoline, which are related to the octane rating, enhancing the performance of internal combustion engines.
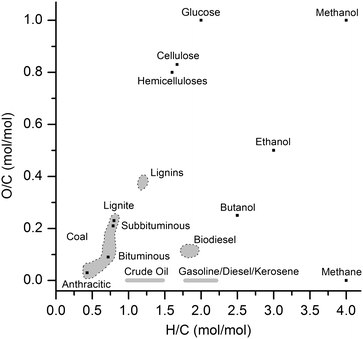 | ||
| Fig. 1 The O/C and H/C molar ratios of fossil and biomass raw materials and of fuels derived from them. | ||
Regarding the thermochemical data of combustion of organic compounds,4,5 the amount of energy liberated by the combustion of oxygenated compounds decreases with the O/C molar ratio (Table 1). For instance, the combustion of methanol and ethanol releases much less energy than burning of methane and ethane, respectively (Table 1, entries 1–4). Therefore, the emission of CO2 per amount of energy released (EE) is increased for compounds with higher O/C molar ratios (Table 1, entries 1–4). On the other hand, the combustion of organic compounds with high H/C molar ratio liberates much more energy, as can be seen in Table 1, entries 4–6. Consequently, the emission of CO2 per energy released is lower for higher H/C molar ratios. Methane therefore emits the lowest amount of CO2 per energy released in its combustion (Table 1, entry 2). One has to keep in mind though, that amounts roughly equivalent to the additional CO2 released in the combustion of oxygenates are produced during the de-functionalization of biomass to reduce the O/C ratio. Several processes of biomass de-functionalization require, in most of cases, external hydrogen supply. This CO2 released in the production of hydrogen should also be taken into account when calculating the amount of CO2 emitted in the combustion of transportation fuels made from biomass.126
| Entry | Reaction | O/C | H/C | −ΔcHa/kJ mol−1 | EE/mmol CO2kJ−1 |
|---|---|---|---|---|---|
| mol/mol | |||||
| a Values taken from ref. 4, 5. | |||||
| 1 | CH3OH(l) + O2(g) = CO2(g) + 2H2O(v) | 1 | 4 | 727 | 1.38 |
| 2 | CH4(g) + 2O2(g) = CO2(g) + 2H2O(v) | 0 | 4 | 891 | 1.12 |
| 3 | H3C–CH2OH(l) + 3O2(g) = 2CO2(g) + 3H2O(v) | 0.5 | 3 | 1367 | 1.46 |
| 4 | H3C–CH3 (g) + 3.5O2(g) = 2CO2(g) + 3H2O(v) | 0 | 3 | 1560 | 1.28 |
| 5 | H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2(g) + 3O2(g) = 2CO2(g) + 2H2O(v) CH2(g) + 3O2(g) = 2CO2(g) + 2H2O(v) |
0 | 2 | 1411 | 1.42 |
| 6 | HC≡CH(g) + 2.5O2(g) = 2CO2(g) + 3H2O(v) | 0 | 1 | 1300 | 1.54 |
The decisive factor for the success of our oil-based chemical industry was the association of the production of fuels with the production of chemicals. Nowadays, fuels correspond to 70.6% of the demand for crude oil.6 This market turns around US$ 385 billion a year only in the United States. On the other hand, petrochemicals and specialties based on these create revenues that correspond roughly to the same amount of money as the fuel market, but for their production only 3.4% of the crude oil is consumed (Fig. 2).6 The efficient relationship between the process chains for fuels and for chemicals from crude oil was weaved along with the development of the corresponding industries during the 20th century, ensuring the high profitability of these businesses.
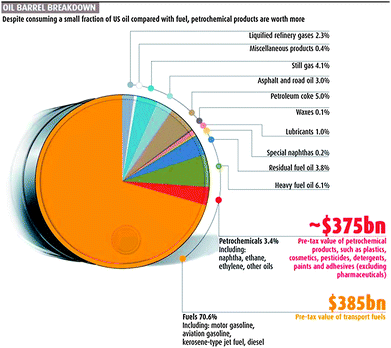
Also for the efficient utilization of biomass, it will be crucial to create a similar comprehensive network of process chains. However, the dynamics of this process are dictated rather by the market prices and political decisions than by purely chemical reasoning. For instance, the glycerol market faces downward pricing pressure due to the low demand growth and the abundant supply created by the massive increase of biodiesel production worldwide over the last five years. New outlets for glycerol are thus mandatory to improve the profitability of biodiesel market and the competitiveness of this sector.7,8
Catalysts are the philosopher's stones of the chemical industry. Indeed, most chemical processes utilize catalysts. The worldwide sales value of solid catalysts amount to ca. US$ 15 billion a year, creating an added value that is about 100 to 1000 times higher than their own price.9 In principle, one can use solids, molecules, or enzymes as catalysts. However, solid catalysts are the preferred option for most processes and dominate the chemical industry (it is estimated that 80–85% of the processes use solid catalysts).9 Whether and how this scenario would change, if the feedstock basis is changed to biomass, is difficult to predict, since research and development in biomass utilization are still in their infancy, with notable exceptions, like in the field of oleochemistry. However, it can be expected that an increasing importance of “biorefineries”, which would not only be restricted to the production of biodiesel or bioethanol (from sugarcane, maize or beet), would lead to an increase in the share of homogeneous and enzymatic catalysis, mainly for two reasons. Firstly, homogeneous catalysts and enzymes are dissolved in the reaction medium, facilitating the interaction with the dispersed solid biomass, e.g. cellulose, lignin, wood and others. Secondly, these catalysts usually require milder reaction conditions and are very selective. Recent reviews are dedicated to the promising role of homogeneous,10,11 enzymatic and biocatalysis12,13 in forthcoming biorefinery processes.
In this paper, we will focus on the challenges facing heterogeneous catalysis in biomass processing. We do not intend to cover comprehensively recent developments of new processes, but rather to provide a practical view on the challenges met in the development of solid catalysts for production of fuels and chemicals from biomass. Those readers who wish to obtain a more detailed discussion of emerging technologies in biomass transformation are referred to some thorough recent reviews on this topic.14–22
Why solid catalysts?
The main reason to consider heterogeneous catalysis for industrial processes is the ease of catalyst separation after the reaction. Separation processes represent more than half of the total investment in equipment for the chemical and fuel industries.23 In fact, when the picture of a refinery comes to our minds, the first view that we imagine is a landscape dominated by huge distillation towers, although the “invisible” cracking units are the heart of the fuel industry (Fig. 3). It is not an overstatement that the separation costs are a decisive factor in the final analysis of a new process. The ease of separation of solid catalysts can then be a crucial advantage. However, these catalysts should also be highly selective to guarantee the cost-effectiveness of the process. | ||
| Fig. 3 The distillation towers dominate the panorama and the costs of fuel industry. Courtesy of Petrobras. | ||
Homogeneous catalysts and enzymes have as a main advantage a high selectivity. In the case of molecular catalysts, which are usually metal complexes, the correct choice of ligands and metal center guarantee high chemoselectivity, regioselectivity, and/or enantioselectivity in the catalyzed reactions.24 On the other hand, enzymes are already very selective catalysts designed by nature, but they may be highly substrate specific. Despite the high selectivity achieved by molecular and enzymatic catalysts, many catalytic systems have not been commercialized because of the difficulties encountered when trying to separate molecular catalysts from the reaction media.24 A number of strategies have been developed to circumvent the problem of catalyst separation.24 The immobilization of these catalysts on solids through covalent or ionic bonding is an interesting possibility to heterogenize metal complexes25 or enzymes.26 Furthermore, biphasic systems are also alternatives to keep the catalyst separated from the products. In this approach, the reaction takes place in a liquid phase that is immiscible with the phase into which the product is continuously extracted.24
Some environmental issues of catalytic processing of biomass
Biomass as a starting point appears to lead easily to sustainable process chains. However, this is not necessarily the case, if one analyzes the processes used nowadays closer. The efficiency of processes regarding waste generation can be expressed by the “E-factor”, which is given by the ratio of mass of waste formed per mass of product.27–29 Although this concept does not capture the full impact of a process, since it does not take into account the quality of the waste, inspection of the E-factor can be illustrative in the analysis of a specific route.The dilute acid hydrolysis of wood followed by the fermentation of hexoses is able to convert about 11% of the energetic content of wood into ethanol.30 Most of the energy requirement for production of ethanol from wood can be supplied by burning the lignin fraction of wood. Fig. 4 shows the amounts of waste, byproducts and ethanol produced during this process.30
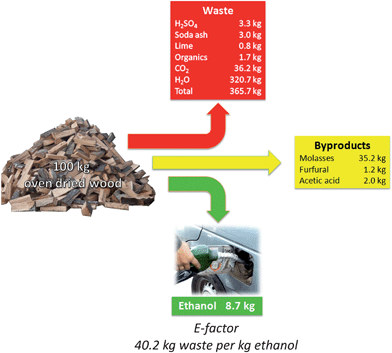 | ||
| Fig. 4 Ethanol, by-products and waste formed in the two-stage dilute acid hydrolysis of cellulose followed by fermentation of glucose to ethanol.30 | ||
The production of ethanol from wood generates 40.2 kg waste per kg of ethanol (Fig. 4), placing this process at the level of waste generation as found in the production of fine chemicals, according to R. A. Sheldon's classification.27 For sake of comparison, the manufacture of bulk-chemicals has much lower E-factors, typically ranging from <1 to 5.27 Most of the waste generated in the ethanol production from wood is industrial wastewater. Regarding a large-plant of cellulosic ethanol with a capacity to process 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of lignocellulosic material (wood, straw and other), producing 870 tons of ethanol a day, the amount of wastewater generated would reach 32070000 L. This amount is enough to supply a town in an industrialized area with ca. 300000 inhabitants daily. Although water is the most environmentally friendly solvent known, this statement is only valid when water is returned clean to the environment. In the case of the dilute acid process, several dehydration products, such as hydroxymethylfurfural, furfural, formic acid and even some phenols are formed (Fig. 5).31,36 The concentration of some of these impurities has to go down to the ppm level for the proper reuse of water. In other words, high-cost advanced oxidation processes for the industrial wastewater cleaning are mandatory. Among several other reasons, this also makes cellulosic ethanol still much less cost-competitive to ethanol from sugarcane or even from maize.
000 tons of lignocellulosic material (wood, straw and other), producing 870 tons of ethanol a day, the amount of wastewater generated would reach 32070000 L. This amount is enough to supply a town in an industrialized area with ca. 300000 inhabitants daily. Although water is the most environmentally friendly solvent known, this statement is only valid when water is returned clean to the environment. In the case of the dilute acid process, several dehydration products, such as hydroxymethylfurfural, furfural, formic acid and even some phenols are formed (Fig. 5).31,36 The concentration of some of these impurities has to go down to the ppm level for the proper reuse of water. In other words, high-cost advanced oxidation processes for the industrial wastewater cleaning are mandatory. Among several other reasons, this also makes cellulosic ethanol still much less cost-competitive to ethanol from sugarcane or even from maize.
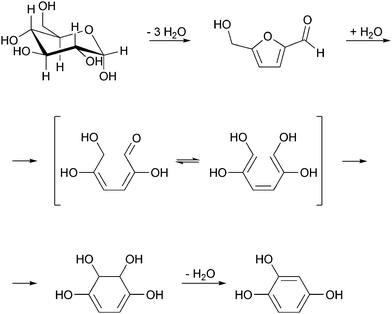 | ||
| Fig. 5 Formation of 1,2,4-benzenetriol from biomass processing.31 | ||
The biomass residues, such as sugar cane bagasse, can be used to supply energy for the biorefinery processes, reducing thus the emission of greenhouse gases (GHG). A fair estimation shows that ethanol produced from sugar cane emits 91% less CO2 than gasoline, while this value is only 18% for ethanol from maize.32,33 In Brazil, most of the energy required for the production of ethanol from sugar cane is supplied by burning the sugar cane bagasse, producing heat and electricity. This makes ethanol from sugar cane almost neutral in CO2 emissions. On the other hand, the production of ethanol from maize requires a massive supply of external energy, which in the US comes mostly from combustion of coal.32
Biomass composition
Biomass is the result of life on Earth, in which simple molecules, such as water, CO2, O2, N2 and several others, are activated by several chains of biochemical processes, e.g.photosynthesis, giving a myriad of complex substances that amount to ca. 170 × 109 tons per year.34 Carbohydrates or saccharides (cellulose, hemicellulose, starch and saccharose) correspond to around 75% of the plant biomass.2 Cellulose is the major constituent of the carbohydrate fraction of biomass, amounting to more than 40% of the total biomass composition.34 Hemicelluloses comprise ca. 25% of the biomass composition.36Lignin is the second most abundant “non-carbohydrate” component of biomass; its share in the biomass composition is estimated at ca. 20%.2 However, some authors also report values as high as 30%.35 Other natural compounds, such as oils, fats, proteins, and other substances, correspond to ca. 5% of the biomass on the Earth's crust.36 Chitin—the polymer that forms the exoskeletons of crustaceans—is also highly available in aquatic environments. The amount of chitin produced is estimated at around 10 × 109 tons a year.37 Those readers interested in a comprehensive view on the transformation of chitin into high value-added fine chemicals are referred to some reviews on this topic.37,38 Before addressing the role of heterogeneous catalysis in biomass conversion, it is worth to briefly address some important structural features of the major biomass components, since this has repercussions on the process options.Carbohydrates
The structural diversity of carbohydrates and the presence of a high number of asymmetric centers in their structures may frighten beginners in this field at first sight. However, it is important to keep in mind that most of the reactivity of these molecules is linked to the anomeric center and to the hydroxyl group (Fig. 6). The reversible aldol cyclization happens at the anomeric center (Fig. 6), making the carbonyl group of glucose and other sugars available for a number of reactions, e.g. C–C bond formation through aldol-condensations.39,40 The hydroxyl groups at positions other than the anomeric carbon are also reactive towards several reactions, such as dehydration to produce 5-hydroxymethylfurfural and furfural.39,40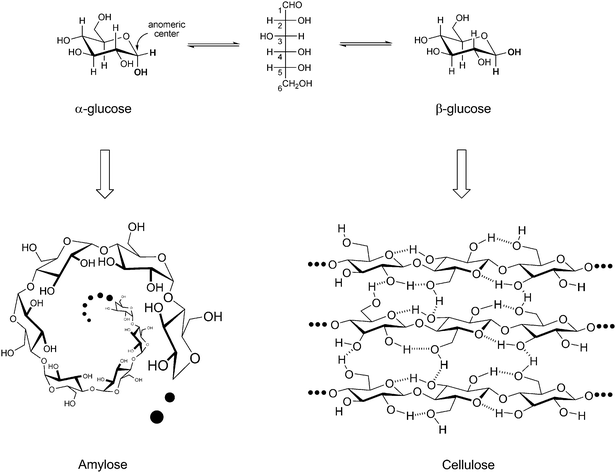
Glucose (C6H12O6) is the most abundant hexose in nature. Its acyclic structure represents only 0.02% of the glucose species in equilibrium at 31 °C.41 In aqueous systems glucose suffers aldol cyclization, involving the hydroxyl group at C-5 and the reducing-end (–CHO). This leads to the six-membered anomers α- and β-glucose (Fig. 6). The α : β ratio is 38 : 62 at 31 °C.41 The evolution took advantage of the structural differences between these anomers to build-up natural polymers with very specialized functions.
Starch is composed of a number of polymers, e.g.amylose and amylopectins, which are made of α-glucose. In contrast, cellulose is composed of β-glucose. Both amylose and cellulose share the formula (C6H10O5)n, where n is the number of repeating units or degree of polymerization (DP). The degree of polymerization of amylose is between 250–3000 whereas for cellulose values up to 10000 are reported.36 In both polymers, the glucose units are bonded via glycosidic linkages at the positions 1 and 4. The structural difference between the anomers plays a fundamental role in the tertiary structure of these polymers (Fig. 6).
In amylose, α-1,4-glycosidic linkages lead to an arrangement in which the pyranose rings are located beneath each other, resulting in a helical structure (Fig. 6), which is easily accessible by chemicals and by enzymes.36 Actually, this structural feature suggests why evolution developed amylose as an energy storage system in plant cells. The α-glycosidic linkage can easily be hydrolyzed, enabling then the mobilization of starch during the metabolic processes.36 On the other hand, in cellulose the β-1,4-glycosidic linkage leads to a side-by-side arrangement of the pyranose rings in a chain-like fashion, enabling an intense intramolecular hydrogen bonding between the groups nearby the glycosidic bond (Fig. 6).42 This feature locks the structure of cellulose as a “planar sheet”, which can be efficiently packed through intermolecular hydrogen bonding or van der Waals forces (Fig. 6).43,44 In this arrangement, most chemical functionalities of cellulose are not accessible to solvents, reagents and enzymes.
In addition to cellulose, a number of polysaccharides called hemicelluloses are also present in plants. These polymers share only little structural similarity with cellulose. Firstly, hemicelluloses are composed of several other sugars besides glucose (Fig. 7). Moreover, hemicelluloses are branched polymers with lower DP than cellulose (Fig. 8). In these materials, the chemical functionalities are not as well protected as by the crystalline structure in cellulose. This makes hemicelluloses easily hydrolysable by acids, bases or enzymes.36
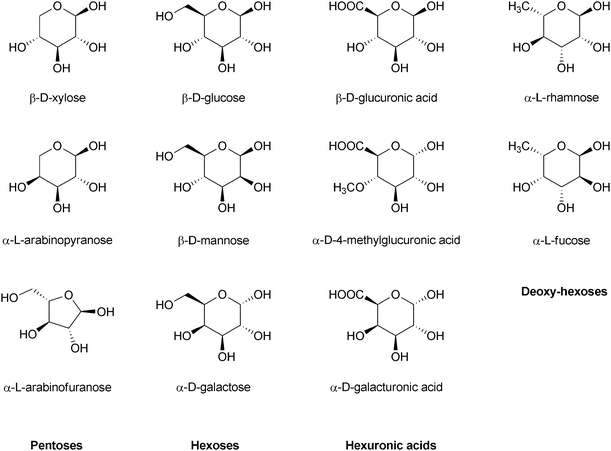 | ||
| Fig. 7 Structural building blocks of hemicelluloses.36 | ||
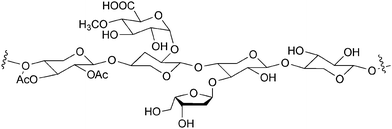 | ||
| Fig. 8 Structural model of hemicelluloses.36 | ||
Sucrose or table sugar is a very important biocommodity and a key component in the current biofuel scenario. Around 20 billion L of ethanol are being produced a year from fermentation of sucrose obtained from sugar cane and sugar beet.32 This disaccharide is formed by a glycosidic bond involving the anomeric centers of the glucose and fructose units (Fig. 9). In this manner, further polymerization of sucrose is not possible. Furthermore, sucrose shows no reducing properties, in contrast to its parent units, due to the absence of hydroxyl group in the anomeric carbon atoms. In diluted aqueous solution, sucrose lacks the intramolecular hydrogen bonding. However, at high concentrations glucose and fructose units twist around the glycosidic linkage, establishing intramolecular hydrogen bonds that lead to the sucrose structure found in solid state.45
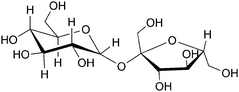 | ||
| Fig. 9 Sucrose structure. | ||
Lignins
The primary building units of lignin are p-coumaryl-, coniferyl- and synapyl alcohols (Fig. 10).46 These p-hydroxy-cinnamyl alcohols polymerize by random coupling reactions, forming a very complex structure (Fig. 10), which is still a matter of models.47 The structure and composition of lignins depend strongly on the type of wood and on the part of the plant.36Table 2 shows the typical substructures of lignin and their occurrence in lignins extracted from spruce (softwood) and beech (hardwood).46,47 The reactivity of lignins is dominated by the substructures containing aryl ethers, biaryls, phenols, benzyl and aliphatic alcohols. Furthermore, lignin is a very powerful radical scavenger because of its phenolic nature.48 These materials can easily suffer cross-linking under acid conditions or in the presence of radicals.| Sub-structure | Common name | Spruce lignin | Beech lignin |
|---|---|---|---|
| a Per 100 C9 units. | |||
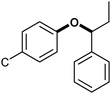
|
α-Aryl ether α–O–4 | 11–16 | 28–32 |
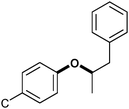
|
β-Aryl ether β–O–4 | 39–48 | 32–37 |
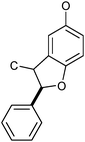
|
Benzofuran-like structure β–5 | 6–10 | 8 |
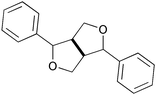
|
β–β in a resinol-like structure | 7–10 | 6.4 |
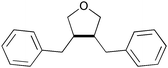
|
β–β in a THF-like structure | ||
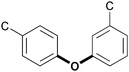
|
Diaryl ether 4–O–5 | 6–7 | 2 |
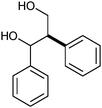
|
β–1 | 2 | 16 |
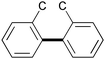
|
Biphenyl structure 5–5 | 7–9 | 2 |
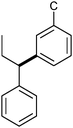
|
α–5 | 7 | — |
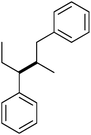
|
α–β | — | 4 |
| Aliphatic OHa | 92–98 | 88 | |
| Benzylic OH a | 18 | 4 | |
| Phenolic OH a | 29 | 16 | |
| Ketone groups a | 14 | 16 | |
| Aldehyde groups a | 3 | 4 | |
| Methoxy groups a | 92 | 136 |
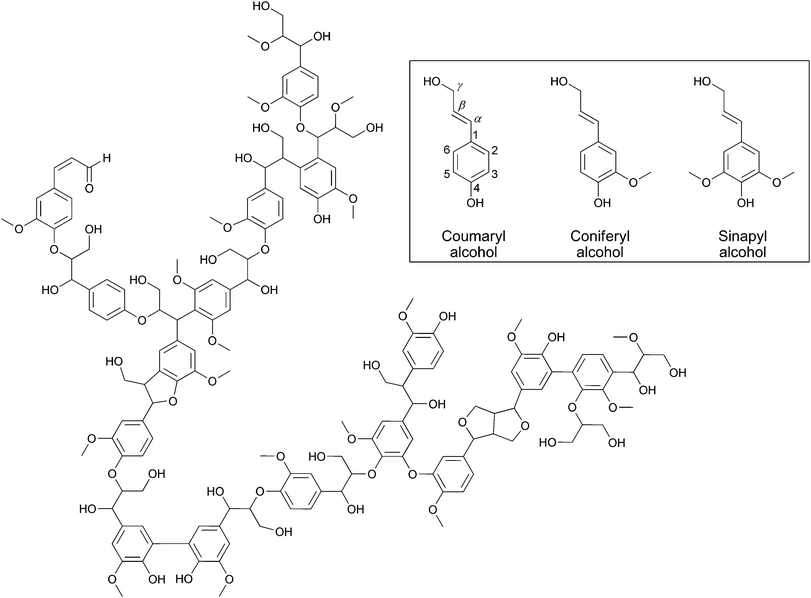 | ||
| Fig. 10 Proposed structure of lignin from spruce. The inset shows the structure of lignin building blocks.36 | ||
Fats and vegetable oils
Fats and vegetable oils are mainly composed of triesters of fatty acids and glycerol (Fig. 11). However, mono- and diglicerides are also found in these feedstocks. Usually, seeds of plants and animal fat are the main sources of triglycerides. Free fatty acids and phospholipids are important “impurities” of these raw materials that can poison solid catalysts.68Fig. 11 shows a typical triglyceride molecule and its reactive sites. Although biodiesel has attracted increasing attention, fats and vegetable oils can be transformed into a number of other products just by exploiting the reactivity of these sites.49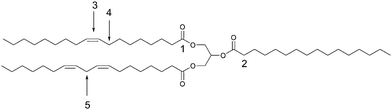 | ||
| Fig. 11 Reactive sites of triglycerides: (1) ester, (2) α–C, (3) carbon–carbon double bond, (4) allylic carbon, (5) diallylic carbon. | ||
Mineral components
The inorganic components of biomass can play an important role in the lifetime of solid catalysts, since several components (Cl, F, P, S, Na, Mg, etc.) can be potent poisons for the catalytic sites.68 Moreover, in any process based on biomass involving cyclic operation, one has to prevent the build-up of inorganic components in the cycle. The amount of inorganic components fluctuates widely, depending on the nature of the biomass.36 Some typical mineral components and their concentration ranges in wood are shown in Table 3.| Concentration (ppm) | Elements |
|---|---|
| 1000–100 | Fe, Mg, P, Zr, K, Ca, Mn |
| 100–10 | Ba, F, Ni, Si, Sn, Sr, Ti, Y, Zn |
| 10–1 | Cd, Ce, Cr, Ga, Ge, Hg, I, Nb, Nd, Pd, Pr, Pt, Rb, Ru, Se, Te, Tm |
| 1–0.1 | Ag, Al, As, Br, Co, Cs, Cu, Er, Hf, Ho, La, Os, Rh, Sb, Ta, Tb, W, Yb |
| 0.1–0.01 | Eu, In, Re, Sc, Sm, V |
| 0.01–0.001 | Au, Dy, Ir, Lu |
Challenges in the design of solid catalysts for biomass conversion
As highlighted above, biomass has a number of properties that places special requirements on solid catalysts used for their conversion. In the following sections we will address what we think are key aspects, when a heterogeneously catalyzed process for biomass conversion is considered. Since there is a wide variety of biomass types and process options, this section is not intended as a list of strict “do's” and “do not's” in the design of solid catalysts, but more as a guideline for the selection of some of the important issues that should be considered.Novel reaction media
The nature of biomass is essentially polymeric. A number of mechanisms protects these polymers against chemical and biological transformation. Due to these protective mechanisms, lignocellulosic materials also lack solubility in most of the conventional polar solvents. Although solvent-free operation is the best option for “green” processes, most of the biomass materials are solids, thus requiring at least a dispersant as reaction medium. The sluggish solubility of biomass components often makes the reactions heterogeneous concerning the substrate itself. In this case, processes on the substrate surface will affect reaction rates.50A recent breakthrough in lignocellulose chemistry was the finding of Rogers et al. that alkylmethylimidazolium ionic liquids can dissolve cellulose51 or even wood.52 The dissolution process disrupts the fibers of cellulose, leaving the hydroxyl groups and β-glycosidic linkages accessible and thus improving their reactivity to other reagents.53 For instance, cellulose54 and lignocellulosic materials55 dissolved in ionic liquids can be readily hydrolyzed using mineral acids as homogeneous catalysts at 100 °C.
We have shown that solid acids catalyze the depolymerization of cellulose dissolved in ionic liquids at 100 °C.56 This reaction proceeds efficiently using acid resins (Amberlyst 15DRY) as solid acid catalyst, yielding cellulose with low degree of polymerization in a controlled fashion.56 Although the homogeneous contribution of Brønsted acid species discharged into solution from the solid acid is unavoidable in this system,57 the use of this solid catalyst still has several advantages over that of molecular acids, such as mineral acids54 and p-toluenesulfonic acid.56
Remarkably, the breakdown of cellulose by Amberlyst 15DRY in 1-butylmethylimidazolium chloride (BMIMCl) produces selectively cellulose oligomers that are easily separated by precipitation through the addition of water.56 This is the crucial advantage of this process. The work-up for the extraction of sugar and dehydration products is normally very difficult due to the high solubility of these products in ionic liquids.58 This prohibits the practical use of the system employing homogeneously dissolved acids,54,55 since the reaction proceeds towards the formation of sugars and dehydration products at large amounts.
Ionic liquids are promising solvents not only for processing of lignocellulosic materials. Several papers reported that ionic liquid are interesting solvents for biodiesel production, enhancing the separation of glycerol.59–61 In spite of the window of possibilities opened by ionic liquid in biomass chemistry, the recovery and the reuse of ionic liquids—in a continuous process—is a big challenge to industrialization of new technologies.62 Due to the high costs of ionic liquids, this is mandatory for the development of commercially viable processes. Furthermore, the ionic liquids currently available cannot dissolve more than 10–15 wt % of cellulose. At these concentrations very viscous solutions or even “sticky”-gels are obtained.63 The future of R&D for biomass processing using ionic liquid depends on the development of new environmentally friendly ionic liquids that allow high concentrations of dissolved cellulose and still have good rheological properties.63 Additionally, it is fundamental that the products can be readily isolated from the ionic liquid, allowing then efficient solvent recycling.63 Moreover, also impurities from the biomass have to be efficiently separated, because their concentration would otherwise build up during the process, which may negatively affect the performance of the systems.
Regarding the solid catalysts, the effect of the ionic liquid species on the catalytic activity is still an open question, due to the lack of studies in this field. However, it is known from other systems that halide ions are potent modifiers of the acidity and catalytic activity.64–66 Both BF4− and PF6− can hydrolyze, forming HF, under certain conditions,67 which is surely a very potent poison for silica and alumina-based materials. Additionally, the organic ions can also interact with supported metal catalyst changing the catalytic activity, selectivity or even promoting the leaching of metal species.68,69 But, as stated above, little is known so far, and thus more comprehensive studies are urgently needed.
Subcritical water (T < 374 °C and at sufficient pressure to keep it liquid)70 and supercritical water (T > 374 °C and p > 22.05 MPa)70 are also interesting reaction media for the transformation of lignocellulose biomass.71,72Catalytic gasification of wood in sub- and supercritical water has attracted increasing attention in the last years, because this transformation requires a lower energy input than the catalytic steam gasification.71,73 In subcritical water, the hydrolysis of lignocellulosic materials requires a catalyst. Nickel, ruthenium, palladium, platinum and rhodium based-catalysts are typical examples.71 Under supercritical conditions, the gasification usually proceeds faster, and low-cost activated carbon and alkalis can be used as catalysts.71 However, the use of supercritical water requires more energy. Moreover, supercritical water is a highly corrosive reaction medium, limiting its application on a large scale.70
Liquid water superheated between 200 and 374 °C does not share the same physical and chemical properties that we are used to seeing at temperatures below 100 °C. Under subcritical conditions, water shows a decrease in the polarity and density due to the partial disruption of the H-bonding network.70 Surprisingly, toluene, for instance, becomes miscible with water at 308 °C and 22 MPa.74Water under subcritical conditions can also lead to detrimental changes in solid catalysts.72 For instance, metal oxides can suffer partial dissolution under sub- and supercritical conditions,75 which can induce significant changes in the structural and textural properties of these materials.76 The development of strategies for the stabilization of the structural and textural properties of solid catalyst in solvents under sub- and supercritical conditions is an important research topic for the application of solid catalysts in biorefinery processes.
Catalyst composition
As stated in the preceding section, biomass conversion is expected to be carried out in different solvents than those nowadays used. Considering that biomass components are typically relatively polar molecules, polar solvents or dispersants should be required for their chemical processing, at least in the initial stages of biomass conversion when the O/C ratio is still high. This brings new requirements with respect to catalyst stability, as alluded to at the end of the last section. Solid catalysts are nowadays selected to be highly temperature-stable, because conversions proceed often in the gas phase at high temperature, and to be resistant against relatively non-polar compounds, since the basis of the chemical industry are hydrocarbons. These characteristics immediately lead to the choice of inorganic oxide materials, which meet both of these requirements. Polymeric catalysts and catalyst supports are otherwise much less suitable for the oil-based chemical industry, because the temperature range for their use is limited and there are substantial swelling and stability problems in organic solvents.Most biomass conversion processes, with the exception of the important route viapyrolysis/gasification of biomass, typically take place at moderate temperatures, and in the liquid phase, mostly in polar solvents.22 This places new requirements on the properties of solid catalysts. They need to be stable against dissolution and leaching in aqueous or highly polar media under different pH conditions. In addition, many of the biomass components possess strongly chelating groups, which can facilitate leaching and poisoning of the catalytically active phases. Very careful choice of catalyst components is therefore mandatory, and the diversity of biomass will make this problem even more severe. Furthermore, the resistance of the solid catalysts against polar solvents is another significant issue, since the chemical processing of biomass should mainly be carried out in liquid phase because of the polymeric nature of most biomass components.
Having these points in mind, one may expect a shift of the solid materials used in biomass conversion towards polymeric organic solids, although inorganic solids will definitely continue to have major importance. Nevertheless, organic polymers or organic/inorganic composites could have quite interesting properties in biomass conversion processes, since they allow wide variation in surface functionality and can be tuned between hydrophobic and hydrophilic behavior (vide infra). However, the use of such materials as solid catalysts and catalyst supports is much less developed than that of the more classical catalytic materials, such as main group oxides and transition metal oxides.98 For instance, although porous polymers can be produced,77,78 this field is in a less mature state than the field of inorganic porous solids, and fine tuning of porosity for a wide variety of different polymers is still a difficult task. Nevertheless, this field appears to be an interesting direction for further research, which is expected to increase substantially in importance in the years to come. For instance, as we already showed, macroreticulated acid resins revealed to be interesting materials for the catalytic depolymerization of cellulose in ionic liquids.56
Porosity of the catalyst
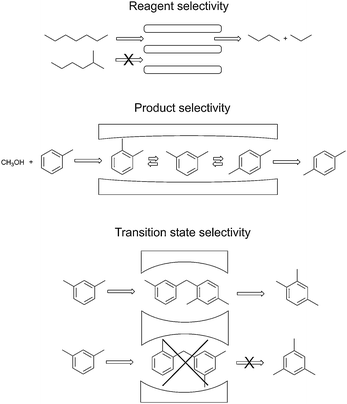
Porosity is one of the most important properties of solid materials, crucial for many practical applications, such as sorption and catalysis. These pore systems are often necessary to create sufficiently high surface areas needed for high activity. According to the IUPAC definition, porous materials are classified in three groups—microporous (pore size < 2 nm), mesoporous (2–50 nm), and macroporous (>50 nm) materials.79 Rationally designed porosity has been extensively exploited in many catalytic processes in the chemical industry.108 For instance, zeolites play an important role in cracking of crude oil and subsequent transformation of the fractions. These mixtures have very complex composition, but their selective transformation to important intermediates is possible, taking advantage of the well-defined micropore structure of zeolites and other molecular sieves.103Generally, in porous materials most of the catalytic sites are inside the pore system. In this manner, the molecular dimensions of reagents, products or transition-states (Fig. 12) can control the reaction selectivity.80 For instance, reagent selectivity occurs when only one type of reagent molecule can be transported into the pores. If several products with dimensions close to the pore sizes can be formed, only the molecules with proper dimensions to diffuse out will appear as observed products (product selectivity). Furthermore, the restricted space inside the pore system can prevent the formation of certain transition states that require more space than available, preventing the formation of several other products (transition-state selectivity).80
The most abundant components of biomass have high molecular weight (103 to 107 g mol−1)2 and are not soluble in conventional solvents. Both characteristics make the transport of biomass molecules into pores very cumbersome. The molecular dimensions of individual sugar molecules are rather small, for instance, β-glucose has a length of 0.58 nm along the axis C-1 and C-4 (Fig. 13).81 However, expanding this dimension to a single chain of cellulose containing 1000 anhydroglucose units, a length of 580 nm is found. Of course, in solution, such macromolecules are never fully extended, and diffusion of chain-like molecules in pore systems is possible by a reptation process. Nevertheless, for the processing of polymeric biomass severe mass transfer limitations have to be expected, requiring careful selection of the materials and tailoring of the pore sizes. Moreover, if the lignocellulosic materials are insoluble in the reaction medium, such as water, a dispersion of two solids (substrate and catalyst) is observed. In these cases, even if the individual cellulose chains would be able to enter the pore system of the catalyst, almost no interactions between substrate and catalyst are expected, since the solid particles of the substrate, consisting of polymeric chains, would not be able to enter the pore system.
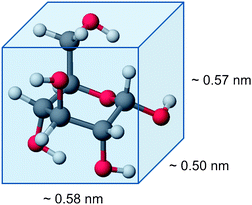 | ||
| Fig. 13 Dimensions of β-glucose. | ||
Several reports,82–84 however, show that cellulose or lignocellulosic slurries in water can suffer catalytic cracking to a certain extent, resulting in the formation of smaller molecules. We believe that two-step mechanisms are responsible in such systems. Firstly, a partial hydrolysis of biomass proceeds, involving Brønsted acidic species either released by the solid material or formed in the reaction medium,83,84 as we will discuss in the next section. Subsequently, the reactions involving the solid catalyst take place when the oligomers are small enough to access the pore system. Thus, the porosity of the solid catalyst can play a fundamental role in the catalytic activity and selectivity, because it determines to what extent the initial hydrolysis needs to proceed in homogeneous phase before starting the heterogeneously catalyzed reactions. There is also the possibility that the reaction starts on the external active sites of the catalysts. However, due to the fact that a dispersion system with two solid phases exists, such contributions are probably small.
Mesoporous and macroporous materials are the first choice for catalytic processes involving large molecules.85 However, if size selective transformations are desired, pores should have a defined architecture and be narrow enough to allow reagent, product and/or transition-state selectivity (Fig. 12). Although macroporous materials can be very active catalysts due to the high exposure of active sites to very bulky substrates, their pores are often too large to offer the steric restrictions that would direct the reaction pathway to selective transformations. Several metal oxides74,85–88 as well as carbonaceous materials89–91,94 can be assembled as ordered mesoporous materials. These materials have well-defined pore architecture, pore sizes from 2 to 30 nm and narrow pore size distribution. The control of these properties has been subject of intensive research and been reviewed many times in the literature.85–91
The stability of ordered mesoporous materials in liquid phase catalysis is an important issue.68 Metal oxides can suffer textural, morphological and structural changes in liquid phase. In aqueous reaction media several dissolution–precipitation equilibria can be established, which might reduce the lifetime of solid catalysts. Aluminum oxides are important supports for metals and oxide catalysts in chemical industry.92 However, water can affect the tailored properties of these materials. For instance, sol–gel derived mesoporous alumina can suffer deterioration of their properties by simple exposure of these materials to humidity.93 In a number of cases, the loss of metal from catalysts happens primarily due to support leaching by dissolution in the reaction medium.68 It is important to keep in mind that several acidic and chelating compounds are formed during the processing of biomass, which can make the reaction medium very aggressive to various solid catalysts. Above it was stated that ionic liquids can be interesting solvents for the processing of biomass. However, not much is known about the effects that different ionic liquids have on different solids used as catalysts and catalyst supports.67
Carbon supports are highly resistant to acidic and chelating media. They provide high surface area and are typically low cost materials and would thus appear to be a good base material for the production of solid catalysts for biomass conversion. However, the pore system of activated carbons obtained from nutshells, wood and coal almost exclusively consists of micropores. Consequently, these carbon supports do not appear as ideal choice for polymeric biomass conversion. Mesoporous carbons (ordered or disordered), on the other hand, have properties that could be interesting for the catalytic conversion of biomass. These materials can be obtained from the thermal treatment of low-molecular-weight phenolic resin precursors, affording mesoporous carbons with high surface areas (typically around 1000 m2 g−1, but also higher), large pore volumes (typically around 1 cm3 g−1, but also higher), and pore sizes adjustable almost over the entire mesopore range in a narrow and unimodal pore size distribution.94,95 However, up to now such systems seem to have been little exploited in biomass conversion.
Presently it is still a speculation that needs to be supported by solid evidence, but ordered mesoporous materials have the potential to achieve higher selectivity in cellulose and in lignin chemistry. In these materials, oligomers are size-allowed to diffuse inside the pore system; furthermore, the formation of polymerization byproducts, such as caramelan and tar, might be inhibited by mechanisms of product-selectivity and/or by transition-state selectivity (Fig. 12).
As in many other fields of catalysis, computational approaches could support the design of novel porous solid catalysts. Computational chemistry is nowadays an essential tool in the screening of novel drugs in the pharma industry, where exploring the guest–receptor interactions led to the design of several new drugsvia computational pre-screening. Similar approaches may be useful for catalyst screening and design.96,97 This route should certainly be more explored in the future. Of course, not all variables can be taken into account in the computational design; however, starting from transition states and molecular dimensions it may become possible to save experimental by simulation of the optimal architecture of the host void spaces that would be required to reach high selectivity. Computational design of solid catalysts is an exciting field still waiting to be fully explored in biomass chemistry.
Active sites
The productivity of the oil industry and the quality of liquid fuels could only be improved during the 20th century due to the massive R&D in the field of solid acids (Fig. 14).97–100 The typical acid-catalyzed reactions in chemical industry are shown in Fig. 15 (in some of these conversions, catalysts are bifunctional, and more than the acid functionality is needed).98 Solid bases, however, received much less attention, mainly because hydrocarbon conversions in the petrochemical industry, such as cracking, isomerization or alkylation, are governed by carbocation chemistry, which requires acid catalytic sites.101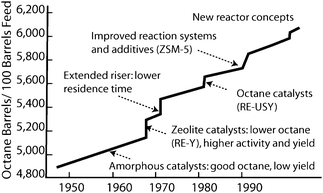 | ||
| Fig. 14 The effect of development of solid acid catalysts for catalytic cracking on the number of “octane” barrels that can be produced from oil feedstocks. Reproduced with permission of ACS.97 | ||
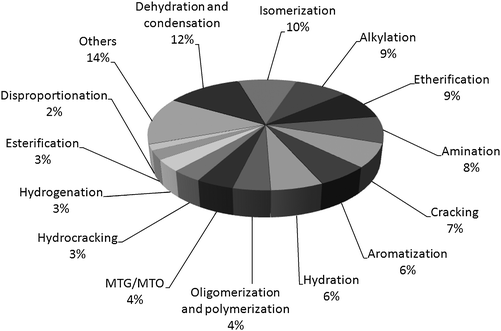
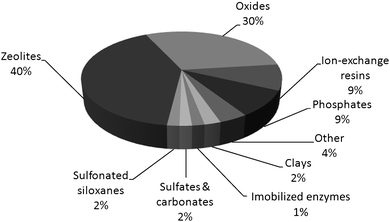
Reactions catalyzed by zeolites and oxides represent, respectively, ca. 40 and 30% of all acid–base catalyzed processes (Fig. 16).98 Despite the importance of zeolites as acid catalysts for the current chemical processes, the catalytic processing of biomass faces the transformation of much larger and bulkier substrates that cannot access the zeolite micropores. This suggests that this class of materials might not have the same central importance for the conversion of biomass as they had and still have in the petroleum industry. However, as soon as the polymeric biomass is broken down to a sufficient extent, the catalytic transformation of platform chemicals obtained from biomass, e.g.ethanol, furfural, glycerol and levulinic acid,102 can take advantage of the unique properties of zeolites as catalysts for organic reactions.103
The main contrast between fossil feedstocks (oil, coal and gas) and biomass is indeed the high O/C ratio, i.e. the oxygen content (Fig. 1). The transformation of biomass to liquid fuels thus means a reduction of the O/C ratio and an increase of the H/C ratio as much as possible. An example is the transformation of fructose into 2,5-dimethylfuran, a potential alternative biofuel. Dumesic et al.104 reported that in each step of this process the oxygen content decreases through the elimination of watervia dehydration and hydrogenolysis (Fig. 17). As a result, substances with lower boiling point and higher research octane number (RON) are obtained. In other words, fructose is systematically transformed to compounds with better burning properties.
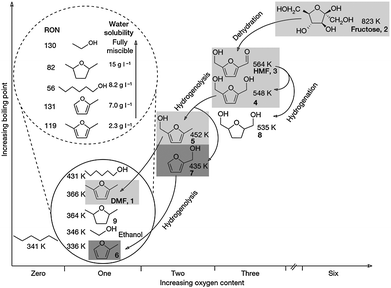 | ||
| Fig. 17 Production of 2,5-dimethylfuran starting from fructose. The intermediates are ranked by the boiling point and the oxygen content. Compounds: 2,5-dimethylfuran (DMF, 1); D-fructose (2), 5-hydroxymethylfurfural (HMF, 3); 2,5-dihydroxymethylfuran (4); 2-methyl,5-hydroxymethylfuran (5); 2-methylfuran (6); furfural alcohol (7); 2,5-dihydroxymethyltetrahydrofuran (8); and 2,5-dimethyltetrahydrofuran (9). The inset shows water solubility and research octane number (RON) of monooxygenated C6-compounds and (6) compared to ethanol. Reproduced with permission of Nature.104 | ||
The decrease in the oxygen content of biomass raw materials is possible by three types of reactions—dehydration, hydrogenolysis and hydrogenation. From these transformations, only hydrogenation is not an acid-catalyzed reaction, underlining the importance of acid catalysis also in biomass-based processes. The primary step, the hydrolysis of lignocellulosic materials, which is also acid catalyzed, does not change significantly the O/C and H/C ratios. However, this initial reaction is the prerequisite of the subsequent defunctionalization reactions, since the depolymerization provides the small molecules that are the entry point for subsequent transformations, which decrease the O/C ratio.
In the oil and petrochemical industries, solid catalysts are typically used in gas-phase or in liquid-phase transformations of hydrocarbons, where the reaction medium is typically non-polar.99,100 In these cases, the chemistry and the “intrinsic” strength of the sites are directly determined by the nature the of the acidic or basic sites exposed on the external or internal surfaces. Usually, the acid sites on the surfaces are highly polarized hydroxyl groups, acting as H+-donor (Brønsted acid), or coordinatively unsaturated cationic sites, which leave M+ exposed to interact with guest molecules as an acceptor of an electron-pair (Lewis acid). In the case of basic sites, loosely bonded-hydroxyls (OH−), or oxide (M–[O2−]–M and M–O−) groups, are typically responsible for the basicity of the materials.101 A number of synthetic parameters can tune the strength and amount of acidic and basic sites.101,118
The acid–base nature of the surface is strongly affected when the solid surfaces are immersed in polar liquids. In this case, several complex equilibria involving the surface groups and the polar solvent molecules take place.105 This much more complex state of the surface is expected in the transformation biomass due to their high polarity and to the requirement of a polar solvent or dispersant. The picture of the solid surface, usually thought to possess vacancies and defined sites in gas-phase systems, changes in a polar liquid-phase system. The molecules of the polar solvent, e.g.water, butanol, ketones or others, occupy the vacancies or coordinatively unsaturated sites (Lewis acid sites). The resulting surface groups and the already existing surface hydroxyl groups may then ionize as Brønsted acids or bases, leading to altered reactivities of the surface sites towards the substrate.105 Consequently, several charged groups cover the surface. These groups establish several equilibria with the polar solvent. H+-species can be taken up from the solvent or released into the reaction medium. In this manner, reactions can take place both on the catalyst surface and in the reaction medium.105 This may explain the relatively high yields of glucose and water-soluble byproducts observed in the hydrolysis of cellulose catalyzed by zeolite β and ZSM-5 in water, compared to materials with larger pores, such as sulfated zirconia, sulfonated active carbon (AC-SO3H) and Amberlyst 15 (Fig. 18).106 Cellulose or even glucose (Fig. 14) would have problems to access the microchannels of these medium-pore zeolites, which are 10-ring systems with an ellipsoidal tubular diameter of 0.55 × 0.56 nm.103
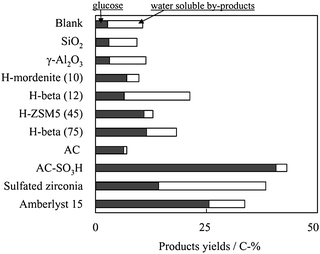 | ||
| Fig. 18 Hydrolysis of cellulose over various solid acid catalysts at 423 K. Reaction conditions: milled cellulose 45 mg, catalyst 50 mg, distilled water 5.0 mL, 24 h. Reproduced with permission of RSC.106 | ||
Since cellulose, hemicelluloses and lignins are large polymeric molecules, the access of these molecules even to mesopores is difficult. An interesting option to circumvent this problem is to use materials that can reversibly release H+-species into the medium, catalyzing thus the depolymerization of the solid substrate. In a second step, other catalytic sites on the catalyst surface can facilitate the transformation of the soluble oligomers, producing smaller molecules in a one-pot reaction. Fukuoka and Dhepe83 showed that cellulose is converted into sugar alcohols, allowing yields of ca. 30%, over supported Pt-catalysts. The authors proposed that the in situ generated acidic sites catalyze the hydrolysis of cellulose to glucose, and that the CO group in glucose is readily reduced by Pt or Ru with H2 to form sorbitol.83 In fact, only ca. 3% glucose yield is achieved in blank experiments using only the catalyst supports, indicating that the acid sites for the hydrolysis of cellulose should mostly be generated in situ from H2.83 It is thus very probable that hydrolysis of cellulose takes place in the reaction medium, since even zeolites-based catalysts showed activity in this transformation.107 Additionally, the reaction is carried out in a cellulose slurry, in which no transport of cellulose into the pores is expected.
Ruthenium supported on activated carbon also shows catalytic activity in the hydrogenolysis of cellulose to sugar alcohols and glycols, as reported by Liu and et al.84 In this work, Ru/C promotes the reversible formation of H+-species in hot water and the hydrogenolysis of glucose.84 The pioneering work of Fukuoka and Dhepe83 and Liu et al.84 showed that reversible formation of H+-species is possible. This feature can be combined with a rational design of the porosity of the solid catalyst to improve the catalyst selectivity in a combined homogeneous-heterogeneously catalyzed reaction.108
Hydrolysis, dehydration and hydrogenolysis are the most powerful pathways to convert biomass to liquid fuels. Acids catalyze these reactions. However, this does not mean that basic solid catalysts would play no or only a minor role in biorefinery processes. On the contrary, several important transformations of oxygenated molecules proceed more efficiently under basic conditions.109 Such a reaction is, for instance, aldol-condensation, which is a key intermediate reaction applied to form large organic molecules using carbohydrate-derived carbonyl compounds.40,110 Moreover, although much effort has been invested in the discovery of efficient solid catalysts for transesterification of vegetable oils, most industrial plants for the production of biodiesel still use NaOMe as homogeneous basic catalyst.111 Base catalyzed reactions seem also to be more efficient for depolymerization of lignin than acid catalyzed reactions, which usually perform cross-linking reactions increasing the undesirable formation of coke and tar.112 Therefore, it is quite probable that research in the development of adapted or new solid basic catalysts will find renewed importance in biomass-based industries.
Metal and supported metal catalysts are also important materials employed in a number of reactions such as hydrogenation,113oxidation114 and C–H bond activation.115 The challenge in this field is to find catalysts resistant against the aggressive reaction media and harsh conditions usually found in biomass processing.68Leaching is the major issue,68,116 not only because it can decrease the catalyst lifetime, but also because the leached metal ions have to be remove from the products. Moreover, the leached metal ions, even at ppm levels, can also have a detrimental effect on the oxidative stability of biofuels.117
Bifunctionality, multi-step- and cascade reactions
In the preceding section it has already briefly been discussed, that multifunctional catalysts can be used for biomass processing, such as in the hydrogenolysis. Solid catalysts provide the possibility of combining in a unique catalyst several types of active sites, which would in principle be incompatible in homogeneous catalysis. For instance, solids can carry acidic and basic sites on their surface, because of the rigidity of the surface, which make these sites distant enough to allow their co-existence on the surface.118The potential bifunctionality of solid catalysts opens a window of possibilities to carry out complex transformations in a one-pot process. In principle, this means reduction of unit operations and intensification of processes.119 Furthermore, one-pot processes make possible the efficient utilization of cascade reactions and multistep conversions. As shown in Fig. 19, cascade conversions or one-pot reactions can circumvent the thermodynamic hurdles to reach the final product.119 For instance, the step-by-step production of D would face the unfavorable equilibrium constants of the transformations A → B and B → C. In one-pot conditions, these intermediates are consumed at the very time of their production, and the total yield of D is not restricted by the reaction thermodynamics of the steps A → B and B → C. Although the concept of cascade-reactions is very elegant, multi-step synthesis in one-pot can also result in the formation of small amounts of several byproducts. One has to therefore balance the costs of the process intensification against possible higher costs in the purification of the products.
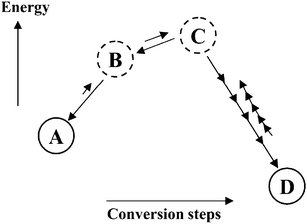 | ||
| Fig. 19 Potential power of cascade conversions to overcome thermodynamic hurdles in multistep syntheses. Reproduced with permission of ACS.119 | ||
Recently, Dumesic et al.120 showed an interesting example for such a kind of transformation, i.e. the reaction of sugars and polyols to hydrophobic alcohols, ketones, carboxylic acids, and heterocyclic compounds over Pt–Re catalysts. In this process, sugars and polyols are partially reformed to supply the hydrogen required for the partial deoxygenation of the remainder of the feed to monofunctional hydrocarbons. This elegant combination of endothermic reforming and exothermic deoxygenating reactions—in the same reactor—leads to the retention of more than 90% of the energy content of the polyol or sugar feed in the reaction products.120
Hydrophilicity/hydrophobicity and water resistance
Surface hydrophobicity and hydrophilicity of a solid determines the range of its potential catalytic applications since the key catalytic steps of adsorption and desorption depend strongly on these properties.121 Several surface groups present in solid surfaces, such as hydroxyl (–OH), carboxyl (–COOH), carbonyl (>CO), ether (R–O–R), alkyl, and ionic species (H+, Na+,Cl−, SO42−) strongly affect the surface polarity. In the majority of solid surfaces, not only are hydrophilic and hydrophobic sites are present, but both sites can coexist in close local proximity.121 Biomass transformation involves hydrophilic and hydrophobic molecules, or even both properties in different parts of the same molecule. Therefore, tuning of the surface properties is also necessary to optimize the adsorption and desorption processes and thus the overall catalytic transformation.
In lignocellulosic materials, cellulose and hemicelluloses are very hydrophilic components while lignin exhibits both hydrophilic and hydrophobic groups. If these materials are converted to intermediates or directly to liquid fuels, the oxygen content will decrease in the successive steps of the catalytic process (Fig. 17). Thus, the hydrophilic substrates will become more hydrophobic after a number of transformations. Generally, these transformations should take advantage of hydrophilic surfaces, as they can facilitate the approach of the substrates and the desorption of the less hydrophilic products. The tuning of the hydrophilicity/hydrophobicity is an important tool that can help to optimize two objectives simultaneously, catalytic activity and selectivity.
The reduction of the oxygen content in biomass by dehydration and hydrogenolysis is associated with the elimination of water. Water is also a convenient dispersant for lignocellulosic materials, since the accessibility of their functional groups is enhanced under sub- and supercritical conditions by partial dissolution of the cellulose fraction.122 However, severe poisoning of the acid sites by water is likely to occur.123 Therefore, development of new water-tolerant solid acids with tailored porosity and adjusted surface polarity could have a major impact on industrial applications involving biomass transformation.
Several examples of water-resistant catalysts are reported in the literature.123 Ratnasamy et al.124 showed that Fe–Zn double-metal cyanides are hydrophobic and thus water-tolerant Lewis acid catalysts for the simultaneous transesterification of triglycerides and esterification of the free fatty acids (FFA) present in unrefined and waste cooking oils and non-edible oils. Recently, Domen et al.125 reported that the layered oxide HNbMoO6 exhibits a remarkable catalytic performance on the hydrolysis of saccharides such as sucrose and cellobiose. The authors attribute the high catalytic activity to the water tolerance and the facile accessibility of saccharides into the strong acidic interlayer gallery of the solid. Using this catalyst, cellulose is converted to glucose and cellobiose at low yields (<10%).
Concluding remarks
Industrial chemistry of the 20th and the early 21st century was designed around crude oil, due to the high availability and low prices. However, presently this strongly focused raw materials base appears to become more fragile in the future, due to international financial and political factors. On June 11, 2008, the oil barrel price reached its historical peak at 139.85 US$. By the end of 2008, however, the price has dropped down again to around 40.00 US$ a barrel, leaving biomass economically less interesting on a short time-scale. However, a scenario based on crude oil is unsustainable at long term. Most researchers agree that peak oil, i.e. the point at which the production of oil worldwide can no longer be increased—will come, and many scenarios expect this peak around the year 2020. We have to prepare now for this period. The availability of crude oil will not dramatically drop from peak oil onwards (it is probably more a period of several years than a point in time), but prices will continue to increase. R&D is necessary now in this field, since—as discussed above for the example of cellulose utilization—we are just at the beginning, starting to formulate the relevant questions and working on providing answers. Many of the problems for academia and industry will be solved by catalysis, in the development or tuning of catalytic materials as well as in the development of novel catalytic processes involving unconventional solvents. In this article, we have discussed some of the points that need to be considered. Finding the answers will require both fundamental investigations and application oriented development.Acknowledgements
Financial support from the DFG Cluster of Excellence “Tailor made fuels from biomass”, in addition to the basic funding of the Max-Planck-Institut für Kohlenforschung, is gratefully acknowledged.References
- I. S. Goldstein, in Organic Chemicals from Biomass, ed. I. S. Goldstein, CRC Press, Florida, 1981, p. 144 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef.
- C. E. Wyman, Bioresour. Technol., 1994, 50, 3 CrossRef CAS.
- E. V. Sagadeev and V. P. Barabanov, Russ. J. Phys. Chem., 2006, 80, S152 CrossRef CAS.
- NIST Webbook, http://webbook.nist.gov/chemistry/.
- J. Marshall, New Sci., 2007, 2611, 28.
- S. Claude, Fett/Lipid, 1999, 101, 101 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13 RSC.
- G. Ertl, H. Knözinger, F. Schüth, J. Weitkamp, Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth, J. Weitkamp,Wiley-VCH, Weinheim, 2008, vol. 1, p. V Search PubMed.
- M. Schlaf, Dalton Trans., 2006, 4645 RSC.
- M. Beller, Eur. J. Lipid Sci. Technol., 2008, 110, 789 CrossRef CAS.
- A. A. Koutinas, R.-H. Wang and C. Webb, Biofuels, Bioprod. Bioref., 2007, 1, 24 Search PubMed.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777 CrossRef CAS.
- P. Gallezot, Green Chem., 2007, 9, 295 RSC.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS.
- P. Mäki-Arvela, B. Holmbom, T. Salmi and D. Y. Murzin, Catal. Rev., 2007, 49, 197 CrossRef.
- P. Gallezot, A. Kiennemann, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth, J. Weitkamp, Wiley-VCH, Weinheim, 2008, vol. 5, p. 2447 Search PubMed.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68 RSC.
- J. H. Clark, F. E. I. Deswarte and T. J. Farmer, Biofuels, Bioprod. Bioref., 2009, 3, 72 Search PubMed.
- J.-P. Lange, Biofuels, Bioprod. Bioref., 2007, 1, 39 Search PubMed.
- C. Judson King, in Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, 2007, electronic version DOI:10.1002/14356007.b03_01.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS.
- A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391 CrossRef CAS.
- W. Tischer and F. Wedekind, Top. Curr. Chem., 1999, 200, 95 CAS.
- R. A. Sheldon, Chem. Ind. (London), 1992, 903 CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 3352 RSC.
- J. F. Harris, A. J. Baker, A. H. Conner, T. W. Jeffries, J. L. Minor, R. C. Pettersen, R. W. Scott, E. L. Springer, T. H. Wegner, J. I. Zerbe, Two-stage, Dilute Sulfuric Acid Hydrolysis of Wood: An Investigation of Fundamentals, Gen. Tech. Rep. FPL-45, Madison, WI: U.S. Department of Agriculture, Forest Service, Forest Products Laboratory; 1985, 73 p Search PubMed.
- G. C. A. Luijkx, F. Rantwijk and H. Bekkum, Carbohydr. Res., 1993, 242, 131 CrossRef CAS.
- J. Goldemberg, Energy Environ. Sci., 2008, 1, 523 RSC.
- J. Goldemberg, Science, 2007, 315, 808 CrossRef CAS.
- B. Kamm, P. R. Gruber, M. Kamm, in Biorefineries - Industrial Process and Products, Wiley-VCH, Weinheim, vol. 1, 2006, p. 15 Search PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519 CrossRef CAS.
- D. Fengel, G. Wegener, Wood: Chemistry, Ultrastructure, Reactions, Berlin, 1984, 613 p Search PubMed.
- K. V. H. Prashanth and R. N. Tharanathan, Trends Food Sci. Technol., 2007, 18, 117 CrossRef CAS.
- M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1 CrossRef.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59 CrossRef CAS.
- P. M. Collins, R. J. Ferrier, Monosaccharides: Their Chemistry and Their Roles in Natural Products, John Wiley and Sons, Chichester, 1995, pp. 40–41 Search PubMed.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358 CrossRef CAS.
- P. Zugenmaier, Prog. Polym. Sci., 2001, 26, 1341 CrossRef CAS.
- M. Jarvis, Nature, 2003, 426, 611 CrossRef.
- R. Khan, Pure Appl. Chem., 1984, 56, 833 CAS.
- E. Adler, Wood Sci. Technol., 1977, 11, 169 Search PubMed.
- B. Saake, R. Lehnen, in Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, 2007, electronic version DOI:10.1002/14356007.a15 305.pub3.
- E. Dorrestijn, L. J. J. Laarhoven, I. W. C. E. Arends and P. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54, 153 CrossRef CAS.
- A. Behr, A. Westfechtel and J. P. Gomes, Chem. Eng. Technol., 2008, 31, 700 CrossRef CAS.
- P. Calvini, Cellulose, 2005, 12, 445 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63 RSC.
- O. A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn and T. Heinze, Biomacromolecules, 2007, 8, 2629 CrossRef CAS.
- C. Li and Z. K. Zhao, Adv. Synth. Catal., 2007, 349, 1847 CrossRef CAS.
- C. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177 RSC.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS.
- R. Rinaldi and F. Schüth, non-published results.
- Q. Liu, M. H. A. Janssen, F. Rantwijk and R. A. Sheldon, Green Chem., 2005, 7, 39 RSC.
- M. Gamba, A. A. M. Lapis and J. Dupont, Adv. Synth. Catal., 2008, 350, 160 CrossRef CAS.
- Q. Wu, H. Chen, M. Han, D. Wang and J. Wang, Ind. Eng. Chem. Res., 2007, 46, 7955 CrossRef CAS.
- A. P. Abbott, P. M. Cullis, M. J. Gibson, R. C. Harris and E. Raven, Green Chem., 2007, 9, 868 RSC.
- S. Zhu, J. Chem. Technol. Biotechnol., 2008, 83, 777 CrossRef CAS.
- B. Kosan, C. Michels and F. Meister, Cellulose, 2008, 15, 59 CrossRef CAS.
- V. Yentekakis, R. M. Lambert, M. Konsolakis and N. Kallithrakas-Kontos, Catal. Lett., 2002, 81, 181 CrossRef.
- V. R. Choudhary and C. Samanta, J. Catal., 2006, 238, 28 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17 CrossRef CAS.
- P. J. Scammells, J. L. Scott and R. D. Singer, Aust. J. Chem., 2005, 58, 155 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2003, 81, 547 CrossRef CAS.
- E. Orglmeister, T. Mallat and A. Baiker, J. Catal., 2005, 233, 333 CrossRef CAS.
- P. Kritzer, J. Supercrit. Fluids, 2004, 29, 1 CrossRef CAS.
- M. Osada, T. Sato, M. Watanabe, M. Shirai and K. Arai, Combust. Sci. Technol., 2006, 178, 537 CrossRef CAS.
- P. E. Savage, S. Gopalan, T. I. Mizan, C. J. Martino and E. E. Brock, AIChE J., 1995, 41, 1723 CrossRef CAS.
- M. Osada, N. Hiyoshi, O. Sato, K. Arai and M. Shirai, Energy Fuels, 2008, 22, 845 CrossRef CAS.
- J. Connolly, J. Chem. Eng. Data, 1966, 11, 13 CrossRef.
- S. E. Ziemniak, J. Solution Chem., 1992, 21, 745 CrossRef CAS.
- J. A. Darr and M. Poliakoff, Chem. Rev., 1999, 99, 495 CrossRef CAS.
- H.-P. Hentze, M. Antonietti, in Handbook of Porous Solids, eds. F. Schüth, K. S. W. Sing, J. Weitkamp, vol. 3, 2002, p. 1964 Search PubMed.
- H.-P. Hentze and M. Antonietti, Curr. Opin. Solid State Mater. Sci., 2001, 5, 343 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- S. M. Csicsery, Zeolites, 1984, 4, 202 CrossRef.
- L. P. Ramos, Quim. Nova, 2003, 26, 863 CAS.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636 CrossRef CAS.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1 CrossRef CAS.
- T. J. Barton, L. M. Bull, W. G. Klemperer, D. A. Loy, B. McEnaney, M. Misono, P. A. Monson, G. Pez, G. W. Scherer, J. C. Vartuli and O. M. Yaghi, Chem. Mater., 1999, 11, 2633 CrossRef CAS.
- F. Schüth, in Handbook of Porous Solids, ed. F. Schüth, K. S. W. Sing, J. Weitkamp, Weinheim, vol. 1, 2002, pp. 535 Search PubMed.
- F. Schüth and W. Schmidt, Adv. Mater., 2002, 14, 629 CrossRef CAS.
- Y. Wan, H. F. Yang and D. Y. Zhao, Acc. Chem. Res., 2006, 39, 423 CrossRef CAS.
- Y. Wan, Y. F. Shi and D. Y. Zhao, Chem. Mater., 2008, 20, 932 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696 CrossRef CAS.
- M. Trueba and S. P. Trasatti, Eur. J. Inorg. Chem., 2005, 3393 CrossRef CAS.
- R. Rinaldi, F. Fujiwara and U. Schuchardt, Appl. Cat. A: Gen., 2006, 315, 44 Search PubMed.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, L. Cheng, D. Feng, Z. X. Wu, Z. X. Chen, Y. Wan, A. Stein and D. Y. Zhao, Chem. Mater., 2006, 18, 4447 CrossRef CAS.
- W.-C. Li, A.-H. Lu and F. Schüth, Chem. Mater., 2005, 17, 3620 CrossRef CAS.
- S. S. C. Ammal, S. Takami, M. Kubo and A. Miyamoto, Bull. Mater. Sci., 1999, 22, 851 Search PubMed.
- B. Smit and T. L. M. Maesen, Chem. Rev., 2008, 108, 4125 CrossRef CAS.
- K. Tanabe and W. F. Hölderich, Appl. Catal., A, 1999, 181, 399 CrossRef CAS.
- G. Busca, Chem. Rev., 2007, 107, 5366 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537 CrossRef CAS.
- U.S. DOE. 2006. Breaking the Biological Barriers to Cellulosic Ethanol: A Joint Research Agenda, DOE/SC-0095, U.S. Department of Energy Office of Science and Office of Energy Efficiency and Renewable Energy (available at http://www.doegenomestolife.org/biofuels/) Search PubMed.
- W. Holderich, M. Hesse and F. Naumann, Angew. Chem., Int. Ed., 1988, 27, 226 CrossRef.
- Y. Román-Leshkov., C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982 CrossRef CAS.
- B. Kasprzyk-Hordern, Adv. Colloid Interface Sci., 2004, 110, 19 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033 RSC.
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969 CrossRef CAS.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2001, 222, 247 CrossRef CAS.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 2008.
- U. Schuchardt, R. Sercheli and R. M. Vargas, J. Braz. Chem. Soc., 1998, 9, 199 CAS.
- T. J. McDonough, Tappi J., 1993, 76, 186 Search PubMed.
- A. Stanislaus and B. H. Cooper, Catal. Rev. Sci. Eng., 1994, 36, 75 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends and H. E. B. Lempers, Catal. Today, 1998, 41, 387 CrossRef CAS.
- R. Burch and M. J. Hayes, J. Mol. Catal. A: Chem., 1995, 100, 13 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- M. Mitterlbach and S. Schober, J. Am. Oil Chem. Soc., 2003, 80, 817 CrossRef.
- K. Tanabe, M. Misono, Y. Ono, H. Hattori, in New Solid Acids and Bases: Their Catalytic Properties, Elsevier, Amsterdam, 1989 Search PubMed.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622 Search PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417 CrossRef CAS.
- R. Gläser, J. Weitkamp, in Handbook of Porous Materials, ed. F. Schüth, K. S. W. Sing, J. Weitkamp, Weinheim, Vol. 1, 2002, p. 395 Search PubMed.
- Y. Ogihara, R. L. Smith Jr., H. Inomata and K. Ara, Cellulose, 2005, 12, 595 CrossRef CAS.
- T. Okuhara, Chem. Rev., 2002, 102, 3641 CrossRef CAS.
- P. S. Sreeprasanth, R. Srivastava, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2006, 314, 148 CrossRef CAS.
- A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363 RSC.
- L. Petrus and M. A. Noordermeer, Green Chem., 2006, 8, 861 RSC.
| This journal is © The Royal Society of Chemistry 2009 |