First principles computational materials design for energy storage materials in lithium ion batteries
Ying Shirley
Meng
a and
M. Elena
Arroyo-de Dompablo
b
aDepartment of Materials Science & Engineering, University of Florida, Gainesville, 32611, USA
bDepartamento de Química Inorgánica, Universidad Complutense de Madrid, Madrid, 28040, Spain
First published on 8th April 2009
Abstract
First principles computation methods play an important role in developing and optimizing new energy storage and conversion materials. In this review, we present an overview of the computation approach aimed at designing better electrode materials for lithium ion batteries. Specifically, we show how each relevant property can be related to the structural component in the material and can be computed from first principles. By direct comparison with experimental observations, we hope to illustrate that first principles computation can help to accelerate the design and development of new energy storage materials.
 Ying Shirley Meng Ying Shirley Meng | Dr Shirley Meng received a PhD in Advance Materials for Micro & Nano Systems from the Singapore-MIT Alliance (National University of Singapore) in 2005. She then worked as a postdoc research fellow and research scientist at the Massachusetts Institute of Technology before joining the University of Florida as faculty. She has a bachelor degree in Materials Science and Engineering from the Nanyang Technological University of Singapore with First Class Honors. Dr. Meng's research focuses on the direct integration of experimental techniques with first principles computation modeling to develop new materials for electric energy storage. Her research investigates oxides and their electrochemical and thermoelectric applications to, processing – structure – property relations in functional nanomaterials and thermodynamic and transport properties of materials at nanoscale. |
 M. Elena Arroyo-de Dompablo M. Elena Arroyo-de Dompablo | M. E. Arroyo-de Dompablo received a PhD in Chemistry from the Universidad Complutense de Madrid in 1998. She then joined the Department of Inorganic Chemistry at the same university as Assistant Professor and was later appointed to Associate Professor. As a postodoctoral associate in the Department of Materials Science and Engineering at the Massachusetts Institute of Technology from 2000 to 2002, she undertook computational investigations in materials for energy storage. She subsequently held research scientist positions at CIDETEC-Centre for Electrochemical Technologies in San Sebastian (Spain) and Universidad San Pablo-CEU (Spain). Her research interests focus on the combination of experimental and computational techniques to investigate various areas of Solid State Chemistry, including materials for lithium ion batteries and transformations of solids under non-equilibrium (high pressure and/or high temperature) conditions. |
Broader contextNew and improved materials for energy storage are urgently required to make more efficient use of our finite supply of fossil fuels, and to enable the effective use of renewable energy sources. Lithium ion batteries are a key resource for mobile energy, and one of the most promising solutions for environment-friendly transportation such as plug-in hybrid electric vehicles (PHEV). This review introduces structure–property relations in electrode materials and presents an overview of the computational approach to design better electrode materials for lithium ion batteries. |
1. Introduction
The performance of current energy conversion and storage technologies falls short of requirements for the efficient use of electrical energy in transportation, commercial and residential applications.1 Materials have always played a critical role in energy production, conversion and storage, and today there are even greater challenges to overcome if materials are to meet these higher performance demands. Lithium ion batteries (LIB) have been used as a key component in portable electronic devices, and more importantly, they may offer a possible near-term solution for environment-friendly transportation and energy storage for renewable energies sources, such as solar and wind. Although LIB offers higher energy density and a longer cycle life than other battery technologies, such as lead-acid and nickel metal hydride (Ni–MH) batteries, to meet increasing energy and power demand advances in new materials for LIB are needed urgently.Electric energy storage (EES) materials used in rechargeable batteries are inherently complex; they are active materials that couple electrical and chemical processes, and at the same time, they have to accommodate mechanical strain fields imposed by the motions of the ions. To demonstrate interrelated chemical and physical processes happening in electrode materials under operating conditions, a schematic of a lithium ion cell is shown in Fig. 1. Mobile species Li+ is transported back and forth between the two electrodes. Electrical energy is generated by the conversion of chemical energy viaredox reactions at the anode and cathode. Multiple processes occur over different time and length scales; i.e. charge transfer phenomena, charge carrier and mass transport within the bulk of materials and cross interfaces, as well as structural changes and phase transformation induced by concentration change of Li.
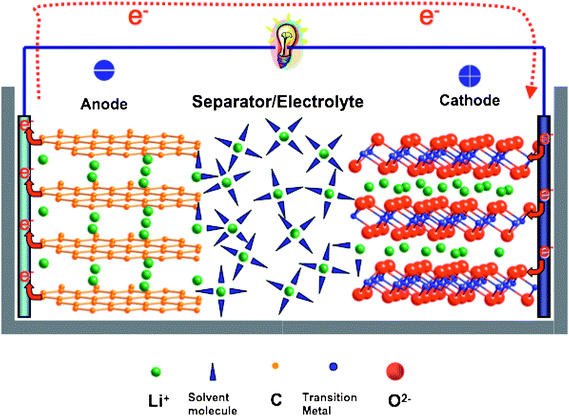 | ||
| Fig. 1 Illustration of the components in a lithium ion cell. | ||
To design and develop new materials for lithium ion batteries, experimentalists have focused on mapping the synthesis–structure–property relations in different materials' families. This approach is time/labor consuming and not very efficient due to the numerous possible chemistries. A longtime goal of scientists' is to be able to make materials with ideal properties, something which could be possible if the optimum atomic environments and corresponding processing conditions are known prior to synthesis. The primary challenge is that an understanding of the atomic environments cannot be easily obtained or measured except in the simplest systems. Various experimental techniques, such as X-ray/neutron/electron diffraction (XRD/ND/ED), nuclear magnetic resonance (NMR) and X-ray absorption fine structure spectroscopy (XAFS) etc., are capable of probing long-range or short-range atomic arrangement in complex structures, nevertheless, the interpretation on an atomic scale is often based on hypotheses and/or speculation. With modern computational approaches, one can gain useful insight into the optimal material (phase) for a specific use of the system under consideration and provide guidance for the design of experiments. First principles (ab initio) modeling refers to the use of quantum mechanics to determine the structure or property of materials. These methods rely only on the basic laws of physics such as quantum mechanics and statistical mechanics, hence they do not require any experimental input beyond the nature of the constituent elements (and in some cases the structure). Ab initio computation methods are best known for precise control of structures at the atomic level. It is perhaps the most powerful tool to predict structures and with computational quantum mechanics; many ground state properties can be accurately predicted prior to synthesis. More importantly the reliability and accuracy of the computational approaches can be significantly improved if experimental information is well integrated to provide realistic models for computation. Experiments and computation are complementary in nature. We believe that a combination of virtual materials design/characterization and knowledge-guided experimentation will have a significant impact on and change the traditional trial-and-true way of materials design, and so accelerate the pace and efficiency of development of new high energy high power density electrode materials for LIB.
In this review paper, we present an introduction to first principles methods based on density functional theory (DFT) and statistic mechanics (section 2), followed by an overview of the computation work aimed at designing better electrode materials (section 3). Specifically, we show how each relevant property is related to the structural component in the material that is computable, and we benchmark the computation results with experimental observations. By such direct comparison, we hope to demonstrate the complementary nature of computation and experiment. Finally, we present some of the key challenges faced by researchers in the field (section 4).
2. Brief overview of the theory behind ab initio modeling
2.1 Density functional theory
All first principles, quantum mechanical or ab initio, methods require a solution to the many-particle Schödinger equation. The exact solution of the full many-bodied Schrödinger equation describing a material is not solvable today, but by using a series of approximations, the electronic structure and so the total energy of most materials can be calculated quite accurately. The total energy of a compound is defined as ‘the energy required to bring all constituent electrons and nuclei together from infinite distance’ where they do not interact to form an aggregate.Density Functional Theory (DFT) is an approach to the quantum mechanical many-body problem, where the system of interacting electrons is mapped onto an effective non-interacting system with the same total density.2,3 Hohenberg and Kohn2 showed that the ground-state energy of an M-electron system is a function only of the electron density 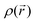 . In DFT the electrons are represented by one-body wavefunctions, which satisfy Schrödinger-like equations
. In DFT the electrons are represented by one-body wavefunctions, which satisfy Schrödinger-like equations
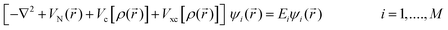 | (1) |
The first term represents the kinetic energy of a system of non-interacting electrons; the second is the potential due to all nuclei; the third is the classical Coulomb energy, often referred as the Hartree term; and the fourth, the so-called exchange and correlation potential accounts for the Pauli exclusion principle and spin effects. Vxc includes the difference between the kinetic energy of a system of independent electrons and the kinetic energy of the actual interacting system with the same density.
The exact form of the exchange–correlation potential, Vxc, is unknown. The simplest approximation to Vxc is the local density approximation (LDA), in which the exchange–correlation potential of a homogeneous gas of density 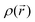 is used at each point. Therefore, the local density approximation is a good approximation for system with a slowly varying electron density. The first step beyond the LDA is a functional that accounts for gradients in the electron density
is used at each point. Therefore, the local density approximation is a good approximation for system with a slowly varying electron density. The first step beyond the LDA is a functional that accounts for gradients in the electron density 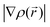 . The term generalized gradient approximation (GGA) denotes the variety of ways proposed for functions that attempt to capture some of the deviation of the exchange–correlation energy from the uniform electron gas result.4,5 It is well-accepted that GGA is more suitable in systems where the electronic states are localized in space. However GGA does not suffice for materials in which the electrons tend to be localized and interacting, such as transition metal oxides and rare earth elements and compounds. The DFT + U method, developed in the 1990s,6,7 extends the functional approach to deal with self-interacting electron correlations. DFT + U refers to the method itself without explicit reference to LDA or GGA (LDA + U or GGA + U). The method combines the high efficiency of LDA/GGA, and explicit treatment of correlation with a Hubbard-like model for subset of states in the system. Non-integer or double occupation of these states is penalized by the introduction of two additional interaction terms, namely, the on-site Coulomb interaction term U and the exchange interaction term J, by means of an effective parameter Ueff = U − J. The U value is different for each material, which brings the necessity of determining the appropriate U for each compound. The values of U can be determined through a recently developed linear response method that is fully consistent with the definition of the LDA + U Hamiltonian, making this approach for potential calculations fully ab initio.8 An alternative route consists of selecting these values so as to account for the experimental results of physical properties: magnetic moments, band gaps,9lithium insertion voltages,10 or reaction enthalpies.11 In section 3 the requirement of the U parameter to treat electrochemical properties of localized electron systems is highlighted. Along with GGA + U calculations that are widely used in combination with the plane wave basis sets, the non-local (the so-called DFT–HF hybrid) exchange–correlation functionals become useful for atomic basis set calculations.12,13 These hybrid functionals permit very accurate reproduction of atomic, electronic structure of insulators/semiconductors, including the gap which is strongly underestimated in DFT calculations.
. The term generalized gradient approximation (GGA) denotes the variety of ways proposed for functions that attempt to capture some of the deviation of the exchange–correlation energy from the uniform electron gas result.4,5 It is well-accepted that GGA is more suitable in systems where the electronic states are localized in space. However GGA does not suffice for materials in which the electrons tend to be localized and interacting, such as transition metal oxides and rare earth elements and compounds. The DFT + U method, developed in the 1990s,6,7 extends the functional approach to deal with self-interacting electron correlations. DFT + U refers to the method itself without explicit reference to LDA or GGA (LDA + U or GGA + U). The method combines the high efficiency of LDA/GGA, and explicit treatment of correlation with a Hubbard-like model for subset of states in the system. Non-integer or double occupation of these states is penalized by the introduction of two additional interaction terms, namely, the on-site Coulomb interaction term U and the exchange interaction term J, by means of an effective parameter Ueff = U − J. The U value is different for each material, which brings the necessity of determining the appropriate U for each compound. The values of U can be determined through a recently developed linear response method that is fully consistent with the definition of the LDA + U Hamiltonian, making this approach for potential calculations fully ab initio.8 An alternative route consists of selecting these values so as to account for the experimental results of physical properties: magnetic moments, band gaps,9lithium insertion voltages,10 or reaction enthalpies.11 In section 3 the requirement of the U parameter to treat electrochemical properties of localized electron systems is highlighted. Along with GGA + U calculations that are widely used in combination with the plane wave basis sets, the non-local (the so-called DFT–HF hybrid) exchange–correlation functionals become useful for atomic basis set calculations.12,13 These hybrid functionals permit very accurate reproduction of atomic, electronic structure of insulators/semiconductors, including the gap which is strongly underestimated in DFT calculations.
Most ab initio methods make use of functions called ‘pseudopotentials’ to replace nuclear potential and chemical inert core electrons with an effective potential, so that only valence electrons are explicitly included in the calculation.14,15 The pseudopotential approximation is valid as long as the core electrons do not participate in the bonding of the solid. Pseudopotentials are derived from atomic calculations that use atomic numbers as the only input. Because pseudo wave functions are smooth and modeless plane waves can be used as the basis set. A particular advantage of plane–wave calculations is that calculation of forces acting on atoms and stresses on unit cell is straightforward using the Hellmann–Feymann theorem. This opens the route to quantum ab initio molecular dynamic simulations to study the time development of a system.
2.2 Cluster expansion
First principles calculation is a powerful tool for obtaining accurate ground state energies. Nevertheless, computing power limits the size of the unit cell to roughly 102 atoms. The inability of DFT-based ab initio computation to predict accurate energy at finite temperature also limits its application. Cluster expansion is one method of gaining knowledge about partially disordered states, and if combined with Monte Carlo techniques, enables information about the system at finite temperatures to be assessed. Such an approach has been successfully demonstrated in alloy systems16–20 as well as intercalation compounds.21–29 The principle of the cluster expansion method is that any property of a crystal that depends on the arrangement of atoms on particular sites (configuration) can be expanded in terms of polynomials of basis functions for each site. The key assumption here is that the contribution to free energy from the other degrees of freedom (e.g. vibrational, magnetic) is insignificant and can be coarse grained out. For a simple binary system (Li and vacancy) a general cluster expansion can be written as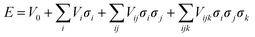 | (2) |
2.3 Monte Carlo simulation
Cluster expansion enables rapid calculation of the energy of systems that depend on arbitrary configurations within a given host. This feature makes it convenient for use in Monte Carlo simulation which is an efficient method to evaluate finite temperature behavior. First order transitions can be detected when the energy E or the slope of thermodynamic potential (Ω = E–µN) is discontinuous. Second-order transitions show continuous E at the transition point and are characterized by the peak in the fluctuation (such as heat capacity) that changes with system size.32 Free energy integration is necessary if phase transitions cannot be simply obtained from the energy or heat capacity calculated by Monte Carlo simulation, details of the thermodynamics and statistical mechanics are described in ref. 28. The most common algorithm is the Metropolis algorithm where if there are perturbations to the system, then(1) If Hold ≥ Hnew, then accept the perturbation
(2) If Hold < Hnew and exp[−(Hnew − Hold)/kT]<rand(0,1), then accept the perturbation
(3) Else, deny the perturbation
H old and Hnew are the Hamiltonian values of the original and perturbed systems, k is the Boltzmann constant, T is the temperature and rand(0,1) is a random number between 0 and 1 that is generated every time a perturbation is considered. Either fixed temperature or fixed chemical potential Monte Carlo simulations are conducted to scan T–µ phase space.
Fig. 2 shows a conceptual flow chart of the computational approach. While these first principles methods can calculate relevant properties of materials that could pertain to lithium ion batteries, inaccuracies may arise from both fundamental and computational limitations. For instance, the state-of-the-art DFT methods can predict many properties of non-strongly correlated material systems, but limitations including how to deal with strongly correlated materials are still not resolved. In addition, the cluster expansion method ultimately is a parameterization of quantum mechanical calculations and its predictive accuracy is therefore limited by the approximations made in solving the Schrödinger equations described above. Finally, the free energy obtained from Monte Carlo simulation usually only includes configurational entropy, other entropy mechanisms (including vibrational, electronic and magnetic) can be included systematically with significant computational expense.
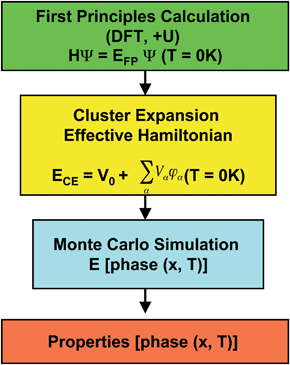 | ||
| Fig. 2 Conceptual flow chart of the computational approach based on DFT methods. | ||
3. Property prediction
This section focuses on lithium insertion electrode materials i.e. where the reaction that occurs at the positive electrode material is the insertion of lithium ions into the host during the discharge of the cell (spontaneous process), and the deinsertion of lithium ions from the host compound during the charge of the cell (non spontaneous process). Electrode reactions other than lithium insertion which might be of practical use for energy storage, will be briefly discussed in section 4.Fig. 3 shows the crystal structure and voltage-composition profiles of the most relevant positive electrode materials for Li-ion batteries. The structure of O3–LiCoO2 (α-NaFeO2 structural type, S.G. R-3m) can be viewed as an ‘ordered rocksalt’ in which alternate layers of Li+ and Co3+ ions occur in octahedral sites within the cubic close packed oxygen array. Lithium ions can be reversibly removed from and reinserted into this structure, creating or annihilating vacancies within the triangular lattice formed by Li ions in a plane. LiMn2O4 adopts the spinel structure, Mg[Al2]O4 (S.G. Fd-3m), with Li ions in tetrahedral 8a sites, Mn atoms in the octahedral 16d sites and the oxygen ions occupying the 32e sites arranged in an almost cubic close-packed manner. The resulting Mn2O4 framework of edge-sharing octahedra (16d and 32e sites) provides a three dimensional network of tunnels, where the Li ions are located, and throughout which the mobile Li ions can diffuse. The structure of olivine–LiFePO4 (S.G. Pnma) is usually described in terms of a hexagonal close-packing of oxygen, with Li and Fe ions located in half of the octahedral sites and P in one eighth of the tetrahedral positions. The FeO6 octahedra share four corners in the cb-plane being cross-linked along the a-axis by the PO4groups, whereas Li ions are located in rows running along the b-axis of edge-shared LiO6 octahedra that appear between two consecutive [FeO6] layers lying on the cb-plane described above. In LiCoO2 and LiFePO4 structures reversible specific capacity is limited to the maximum exchange of 1 Li ion per formula unit (Li1−xCoO2 and Li1−xFePO4 with 0 < x < 1), which correspond respectively to the redox active couples Co3+/Co4+ and Fe2+/Fe3+ In LiMn3+Mn4+O4 besides lithium removal (oxidation of Mn3+ to Mn4+), lithium ions can be inserted in the octahedral sites not occupied by Mn leading to Li2Mn2O4 (reduction of Mn4+ to Mn3+). The theoretical specific capacities of LiCoO2, LiMn2O4 and LiFePO4 are 273 mAh g−1, 297 mAh g−1 and 170 mAh g−1 respectively.
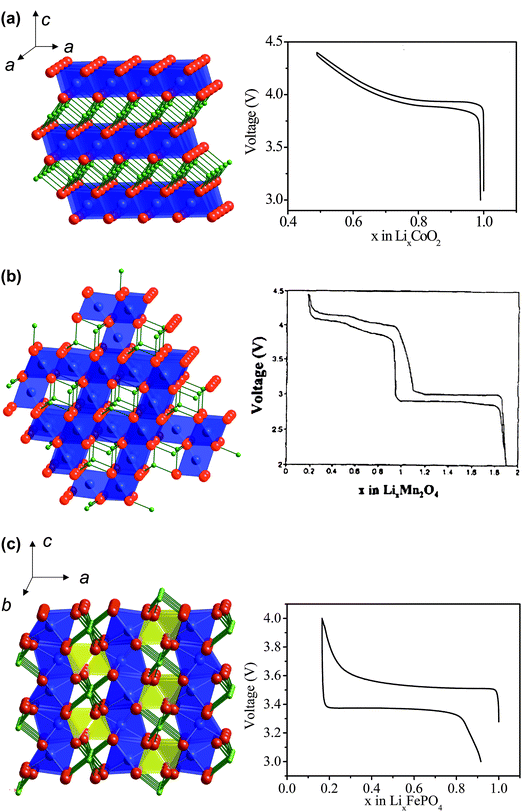 | ||
| Fig. 3 Crystalline structures and voltage–composition curves of (a) layered-LiCoO2 (R3-m S.G.)—oxygen (red) layers are stacked in ABC sequence, with lithium (green) and cobalt (blue) residing in the octahedral sites of the alternating layers; (b) spinel–LiMn2O4 (Fd-3m S.G.)—lithium (green) resides in the tetrahedral sites formed by oxygen stacking; and (c) olivine–LiFePO4 (Pnma S.G.)—phosphor (yellow) and oxygen form tetrahedral units linking planes of corner-sharing FeO6 octahedra. | ||
Along these lines we will show that extensive DFT investigations have been performed on the above described battery materials. DFT has also been successfully applied to investigate the electrochemical properties of many other host compounds Li2MSiO4,33–39NASICON–Li3M2(PO4)3,40,41 V2O5,42–44rutile–TiO2,45 anatase–TiO2,45,46 spinel–LiTi2O4,47,48 MoS2,49,50graphite,51,52 LixMPn4 (MPn = TiP, VP, VAs),53,54 spinel–Li4Ti5O12,55,56 VOPO4,57 and so forth.
For sake of clarity this section is organized to follow the lithium battery community's general way of thinking. Thus the layout is according to relevant properties that should be considered in the search for promising electrode materials. The starting point in section 3.1 is the modeling of the crystalline structure of the host materials. A good electrode for lithium ion batteries should display a nicely reversible lithium insertion process to favor long term cyclability, and this is intimately linked to the host structure and its possible phase transformations (section 3.1). One common objective for battery researchers is to have the appropriate tools to tentatively design a new lithium insertion compound, ideally displaying a high/low voltage for positive/negative electrode applications. As shown in section 3.2 DFT methods are a powerful tool to predict the lithium insertion voltage of electrode materials. It is very important to anticipate the polarization of the positive electrode since it directly governs the power rate capability that depends on the electrical conductivity of the active material. Information on intrinsic electronic conductivity can be directly inferred from the calculated electronic structure of a given electrode material. In sections 3.3 and 3.4 we show how more complex DFT–based investigations enable a further inspection of the electrical conductivity of the material. In many electrode materials the operating mechanism for electronic conductivity is not thermal excitation of electrons across the band gap but an electron hopping mechanism (section 3.3). Of crucial importance for the rate capability is ionic conductivity; lithium diffusion barriers are treated in section 3.4. Finally, the thermal stability of electrode materials and its relation to safety considerations is examined in section 3.5.
3.1. Crystal structure and phase transformations: capacity and cycling stability
On the other hand, one can fix the composition and evaluate the effect of crystal structures on the electrochemical properties. Furthermore, the relative thermodynamic stability of polymorphs can be explored by first-principles methods; in particular, pressure is an easily controllable parameter for DFT calculations in contrast to experiments. Fig. 4 shows the calculated energy vs. volume for various possible polymorphs of Li2MnSiO4.38 The Li2MSiO4 (M = Fe, Mn, Co, Ni) family is attractive as a positive electrode for lithium batteries due to the, at least, theoretical possibility to reversibly deintercalate two lithium ions from the structure.68,69 Li2MSiO4 compounds exhibit a rich polymorphism,70 adopting a variety of crystal structures built up from [SiO4], [LiO4] and [MO4] tetrahedral units.68,71 Synthesis conditions to isolate each Li2MnSiO4 polymorph can be inferred from Fig. 4; in particular, the denser Pmn21 polymorph can be obtained by treating, under pressure, the other polymorphs or their mixtures, as is confirmed experimentally.36,38 Polymorphism in many host compounds have been investigated by first-principles methods; LiCoO2,24,72,73 LiCoXO4 (X = P, As),65,66 V2O5,74,75 LiFeSiO4,39 TiO2,46,76 LiTiMO4 (M = Ti, V, Cr, Mn, Fe),77 MnO2,78,79 FePO4,60,80,81etc.
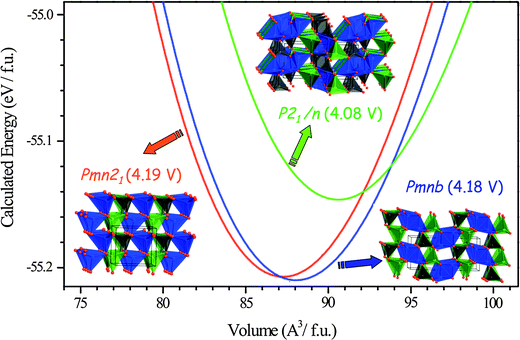 | ||
| Fig. 4 Calculated total energy vs. volume curves of Li2MnSiO4 polymorphs; Pmn21 (red), Pmnb (blue) and P21/n (green). DFT (GGA + U, Ueffect = 4 eV) data were fitted to the Murnagham equation of state. Calculated average voltage for the 2 electron process, host–Li2MnSiO4 ↔ host–MnSiO4 + 2Li, is given in parentheses. Adapted from ref. 38. | ||
DFT methods can handle periodic solids well, while in reality in many solids crystallographic sites show partial occupancies, or there is disorder between ions on a given crystallographic site. Nevertheless, an ordering scheme has to be imposed to simulate this type of materials. A good example is the spinel structure (Fig. 3b), AB2O4, where frequently several TM cations randomly occupy the octahedral 16d sites (B in A[B2]O4). This was experimentally found in Li[Mn1.5M0.5]O4 with M = Cu, Ni, Co,82 spinels that can be investigated by DFT methods imposing proper ordering models (M = Cu, Ni,83,84Co85). One can expect that at finite temperatures the real (disorder) solid gets stabilized with respect to the ordered one due to the contribution of mixing entropy to free energy.
There is, of course, the possibility of studying the relative stability of different ordering models at a fixed composition. As is discussed later, DFT methods are crucial to determine the ordering of TM and Li ions in the structure of LiMn0.5Ni0.5O2 and its implications to the electrochemical behavior.27
It should be stressed that DFT methods often treat ‘perfect solids’ while in reality defects are always present in ‘real’ solids. If desired, imperfections can be introduced in the computed structure. For example, it is possible to represent an impurity by studying a super-cell in which one atom is replaced by an impurity atom. Such a super-cell is repeated periodically and the concentration of the impurity depends on the size of the chosen super-cell. This procedure is however computationally very expensive, and empirical atomistic simulation methods, with short-range interatomic forces represented by effective pair potentials,86 are a superior way to anticipate the effect of doping and defects in electrode materials, as shown for olivine–LiFePO4,87 anatase–TiO288 and Li–Mn–Fe–O spinels.89 Contrary to quantum mechanical methods, such empirical methods do not provide any information of the electronic structure and redox potentials.
Note that in some materials the computed variation of volume might be small, but severe distortions can occur at the local level. The predicted volume variation between Pmn21–Li2MSiO4 (Fig. 4) and the fully delithiated derivatives is about 2% for Mn and Co.37 However, the anisotropic variation of lattice parameters, together with the important structural rearrangements in the [SiMO4] corrugated layers, suggest that as Li is removed from Li2MSiO4 the structure of the host could become thermodynamically metastable with respect to other structures.38,39 Given that the structure is built up from [MO4] tetrahedral units, the crystal field stabilization effect constitutes a driving force for most M3+ and M4+ ions to change coordination upon lithium extraction and the structure of MSiO4 to transform into a more stable structure or to collapse. Indeed, joint computational and experimental work demonstrated that the Li2MnSiO4 collapses under lithium deinsertion.34 The authors found from first principles a new collapsed structure for MnSiO4 (S.G. C2/m) built by edge-sharing Mn4+ octahedra.
As in the latter example, many phase transformations of the host compound upon lithium insertion/deinsertion are associated with the electronic configuration of the TM ions and their crystal field stabilization energies. Since the TM oxidation state varies along the charge/discharge of the battery, the host compound can become metastable with respect to other crystal structures at intermediate lithium contents. A good example of such structural phase transformation following lithium removal is provided by the layered-to-spinel transformation that occurs in LiMnO2.93,94Ab initio calculations help to explain this transformation.95–97 The structures of spinel and O3–LiMO2 (Fig. 3a and b) both have the same close packed oxygen framework, although with distinct cation distribution in the interstitial sites. The transformation from the layered O3–Li0.5MO2 to the spinel phase can be done by cation migration from the TM layer to the Li plane. Fig. 5 shows the calculated formation energies for Li0.5MO2 within the spinel and layered structural types. For any TM investigated by DFT the spinel structure is more stable, indicating that there is a thermodynamic driving force for the layered → spinel transformation. First principles investigations demonstrated that due to the high activation barriers for cation migration the transformation at room temperature is kinetically impeded for TM other than Mn. The complex mechanism of the transformation96,97 involves the transport of the TM atom to the Li layer through tetrahedral sites, and in short, the particular tendency of Mn3+ to charge disproportionate (2Mn3+ → Mn4+ + Mn2+) creates Mn2+ ions (d5 configuration, no crystal field stabilization energy) with tetrahedral-site stability prompt to migrate.
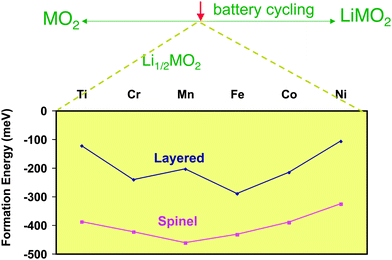 | ||
| Fig. 5 Formation energies of Li0.5MO2 of the layered (O3) and spinel structures for various transition metal cations. The formation energies are taken with respect to the layered forms of MO2 and LiMO2 (ΔfE = ELi0.5MO2 − 0.5ELiMO2 − 0.5EMO2). Adapted from ref. 95. | ||
The different electrochemical behavior of LixNiO2versus LixCoO2 also lies in the electronic nature of the transition metal cations.98–100Fig. 6 shows the calculated formation energies for LixCoO2, according to the reaction
| ΔfE = E − xELiCoO2 − (1−x)ECoO2 | (3) |
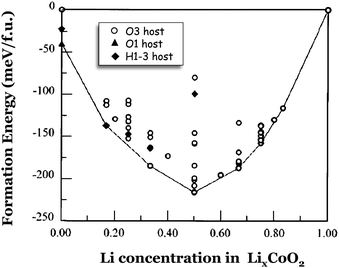 | ||
| Fig. 6 Calculated formation energies of LixCoO2 considering (i) 44 different Li-vacancy arrangements within the O3 host (○), (ii) five different Li-vacancy arrangements within the H1-3 (◆), and (iii) CoO2 in the O1 host (▲). Adapted from ref. 21. | ||
Note that in Fig. 6 the formation energies of LixCoO2 are calculated within 3 crystal structures, which are related by gliding of the oxygen planes. The O3 host is observed to be stable experimentally for Li concentrations between x = 0.3 and 1.0.104–106 The second host, referred as O1, was experimentally found when LixCoO2 was completely deintercalated (x = 0).106 Accordingly, DFT predicted O1 to be more stable than O3 at x = 0.22,23,107 The third host (H1-3), which was not identified experimentally at that time, was constructed by A. Van de Ven et al. and considered features of both O3 and O1.108 In Fig. 6 it can be seen that at x = 0.1666, the Li-vacancy arrangement in the H1-3 host is more stable than the two other Li-vacancy arrangements also considered on the O3 host at that concentration. Furthermore, the fact that it lies on the convex hull means that it is more stable than the two-phase mixture with overall Li concentration x = 0.1666 of any two other ordered Li-vacancy arrangements. This result indicates that the H1-3 host will appear as a stable phase in the phase diagram, resulting in a single phase region in the voltage–composition curve. The calculated phase stability of H1-3 and its crystalline structure21,108 are fully consistent with experimental results.105,106 The identification of this new H1-3 LixCoO2 phase underlines the capability of DFT to determine the relative stability of candidates (known or hypothetical) structures at a given composition, and to find new ground state structures, driven by a good knowledge of crystal chemistry.
LiNi1/2Mn1/2O2 represents a typical multi-electron redox system. Many of the desirable properties are derived from the synergetic combination of Mn4+ and Ni2+ in this material. Mn4+ is one of the most stable octahedral ion and it remains unchanged, stabilizing the structure when Li is extracted. As predicted by DFT,109Ni2+ can be fully oxidized to Ni4+, thereby compensating for the fact that Mn4+ cannot be oxidized.110 This material delivers 200 mAh g−1 reversible capacity between 3 to 4.5 V,111 contains no expensive elements and exhibits better thermal stability than that of LiCoO2.112 Although the average cation ion positions of LiNi1/2Mn1/2O2 form a layered O3 structure similar to that of LiCoO2, the more detailed cation distribution is shown to be complicated. There is always about 8–10% Li/Ni interlayer mixing observed in materials heat treated to around 900–1000 °C. We have devoted significant efforts to identifying the three-dimensional cation ordering in this system and how this ordering changes with the state of charge/discharge by a combined computational and experimental approach.27,113–118
Different structural models of the pristine LiNi1/2Mn1/2O2 have been proposed by various theoretical and experimental investigations, two lowest energy states are shown in Fig. 7. The intercalation potential and Li-site occupancies are calculated using both GGA and GGA + U (Fig. 8) within the flower-like structure as a function of Li.27,119 The simulation shows that early in the charge cycle, the Li ions that are part of the flower-like ordering in the transition metal layer are removed, freeing up tetrahedral sites which then become occupied by lithium. Tetrahedral Li requires a high potential to be removed and effectively lowers the attainable capacity of the material at practical voltage intervals. Using GGA + U approximations, the authors investigated phase transformations of layered LiNi1/2Mn1/2O2 at finite temperature.27 The simulation results suggest two phase-transition temperatures at approximately 550 °C and 620 °C. A partially disordered flower-like structure with about 8–11% Li/Ni interlayer mixing is found. The results from this work help explain many of the intricate experimental observations in LiNi1/2Mn1/2O2 with and without Li/Ni disorder.113
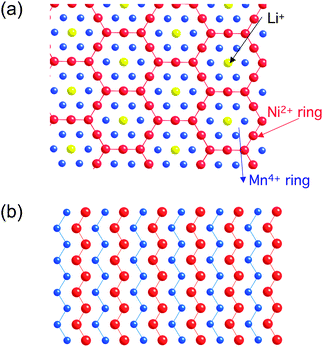 | ||
| Fig. 7 Structural details of LiNi0.5Mn0.5O2. (a) Flower-like pattern as proposed by ordering in the transition metal layer between Li, Mn and Ni. (b) Zigzag pattern proposed shows no Li in the transition metal layer. | ||
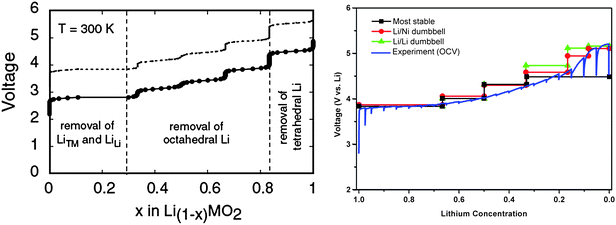 | ||
| Fig. 8 (a) GGA calculated voltage profile of LiNi1/2Mn1/2O2, note the dotted line is obtained by shifting the calculated profile by a constant amount (∼1 V). (From ref. 119.) (b) Comparison between the calculated voltage curves for different delithiation scenarios and the voltage profile during the first charge of a Li/Lix(Ni1/2Mn1/2)O2cell; charged to 5.3 V at 14 mA g−1 with intermittent OCV stands of 6 h. The calculated curves are obtained with GGA + U, there is no artificial shift of the curves. (From ref. 115.) | ||
At room temperature extraction of Li ions from LiFePO4 (Fig. 3c) proceeds via a biphasic process in which the final FePO4 structure (isostructural with heterosite) is obtained through minimum displacement of the ordered phosphor–olivine framework. Calculated volume variation within the DFT + U is 5.2% (see ref. 10 for GGA + U data), which is in reasonable accord with experimental data (6.9% from ref. 120). In order for phase separation to occur at room temperature, all intermediate LixFePO4 structures should have positive formation energy, large enough to overcome the potential entropy gain in mixing. It was found that both LDA and GGA qualitatively fail to reproduce the experimentally observed phase separation in the LixFePO4 system.60,121 Calculated formation energies of Li0.5FePO4 within the LDA + U become positive for U ≥ 3.5 eV. As explained by Zhou et al.121 the physics of the LixFePO4 is not well captured by LDA/GGA, as the self-interaction causes a delocalization of the d electrons, resulting in electronically identical Fe ions, that is to say, Fe2+ and Fe3+ coexist in the calculated intermediate LixFePO4 structures. The effect of the U term is to drive the Fe-3d orbital occupation numbers to integer 0 or 1, favoring charge localization and consequently reproducing the phase separation into Fe2+ and Fe3+ compounds. Therefore, DFT + U predicts that phase separation occurs at 0 K, but obviously at sufficient temperature the system should disorder forming a solid solution. In the LiFePO4 phase diagram an unusual eutectoid transition to the solid solution phase at about 400 °C was found experimentally (top panel (a) in Fig. 9). To compute phase stability above 0 K, one has to account for entropy, the most important of which is the configurational entropy due to Li-vacancy substitutional exchanges. The bottom panel (c) in Fig. 9 shows the calculated phase diagram of LixFePO4 constructed accounting only for this configurational ionic entropy, and which fails to reproduce the experimental results. The experimental phase diagram122,123 can only be reproduced when the configurational electronic entropy, it refers to the ordering of electrons and holes, is included in the simulation (middle panel (b) in Fig. 9). These computational results show that, surprisingly, the phase stability in the LixFePO4 system is dominated by configurational electronic entropy, rather than configurational ionic entropy as is usually the case.
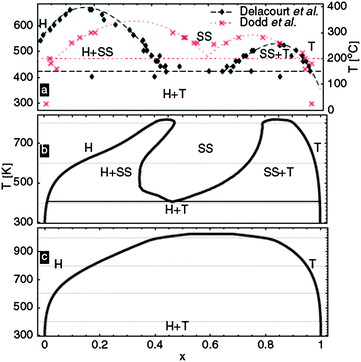 | ||
| Fig. 9 Phase diagrams for LixFePO4. Experimental (a) calculated considering both Li/vacancies and electron/holes orderings as source of configurational entropy, and (b) calculated accounting only for the ionic configurational entropy. (Taken from ref. 223.) | ||
We should stress that the computational investigation of the electrochemical properties of LiFePO4 is successfully accomplished within the DFT + U framework. Introducing the Hubbard like term, U, is also necessary to simulate the magnetic properties of delithiated olivine-like LixCoXO4 compounds; in CoXO4 (X = P, As) DFT + U methods anticipate that Co+3 ions (d6 configuration) are in high spin state (t2g4 eg2), while the DFT method predicts a non polarized state (t2g6 eg0).10,66,124 Experimental magnetic measurements confirmed the DFT + U predictions.125,126 Noteworthy, GGA is more appropriate than GGA + U to investigate systems without strong electron localization. This has been recently shown for the NaxCoO2 system (0.5 < x < 1),28 where within the GGA, holes are delocalized over the Co layer, while in GGA + U the charges on the Co layer completely localize, forming distinct Co3+ and Co4+ cations. Comparison with experimental results of ground states, c-lattice parameter, and distribution of Na within the distinct sites in the structure, consistently suggests that GGA is a better approximation for 0.5 < x < 0.8 than the GGA + U in NaxCoO2.
In short, DFT investigation within a given host provides useful information about volume variation, structural distortions, stable lithium-vacancy orderings, or phase separation. A simple ‘two points’ calculation taking the fully lithiated and delithiated host can be a good starting point to anticipate structural changes and to screen for interesting materials. At the next level of complexity, computing structures with intermediate degrees of lithiation are very useful to calculate formation energies, and sketch the 0 K voltage–composition profile. Finally, a combination of DFT and cluster expansion with MC simulations allows the construction of a complete phase diagram. From the computational results researchers can evaluate the cycling stability of the material, and so anticipate possible failures due to instability of the host (amorphization, decomposition, or phase transformation). Examples of materials that transform to more stable crystalline phases upon lithium insertion/deinsertion have been provided. It is important to mention that finding the most stable structure (ground state) is often done by comparing the calculated energies of candidate structures (for instance layered against spinel in Li0.5MO2). This predicts the need to identify good candidate structures. In this context, several high-throughput methods for ab initio prediction of ground state structures are currently being developed.127–129
3.2. Electronic structure and lithium intercalation voltage
In an insertion reaction lithium ions are incorporated into the crystalline structure of the host compound and electrons are added to its band structure. Fig. 10 schematizes the relation between the equilibrium lithium insertion voltage (or Open Circuit Voltage) and the density of states of the host compound. A simplified view of the intercalation phenomena assumes that lithium ions are fully oxidized donating electrons to the unoccupied levels of the band structure of the host compound. In the electrode materials shown in Fig. 3 these levels arise from the d-states of the transition metal cations. The simplest approximation to the band structure of an intercalation compound is that of the host compound with the Fermi level moved up to accommodate the extra electrons. Determination of quantitative lithium intercalation voltages from the band-structure of host compounds is only valid under this simplest rigid band model, where it is assumed that the crystalline and electronic structures of the host material are minimally affected upon lithium insertion. However, as previously highlighted130–134 this simplified approximation works only in a very few cases. Li+ has an electrostatic effect over the host structure, thereby affecting both the crystalline and electronic structures of the host material (the screened-impurity rigid band model developed by Friedel132). In reality, transfer of the electron to the host is not complete, and strong interaction between the intercalated Li+ ion and the extra electron exits. Any rigid band model definitively breaks down in the case of late transition 3d elements, where the d band drops down into the anionic band and the charge is also transferred to the anion. In short, quantitative lithium intercalation voltage cannot be determined solely from the electronic structure of the positive electrode material.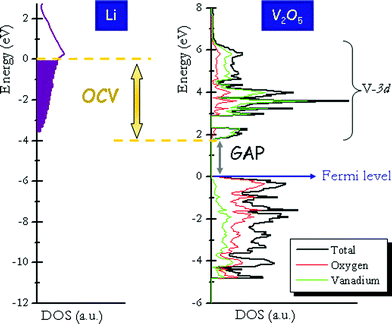 | ||
| Fig. 10 Density of states curves for the host compound (V2O5, positive electrode material) and Li metal showing the difference in chemical potentials and hence the origin of the cell voltage. (Calculated DOS of V2O5 is adapted from ref. 75.) | ||
The first investigation of lithium insertion voltages by means of first principles calculations dates from 1992, and deals with the LixAl system.135,136 It was only in 1997 that Ceder and co-workers demonstrated how the lithium insertion voltage of transition metal oxides can be inferred from the calculated total energies of the host compound and lithium metal.58,59,137 The intercalation reaction that occurs in the cathode material of a lithium cell can be expressed as
| (x2 − x1)Li(s) (anode) + Lix1host (cathode) → Lix2host (cathode) | (4) |
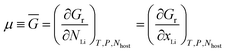 | (5) |
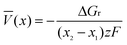 | (6) |
| ΔEr = Etotal(Lix2host) − [(x2 − x1) Etotal(Li) + Etotal(Lix1host)] | (7) |
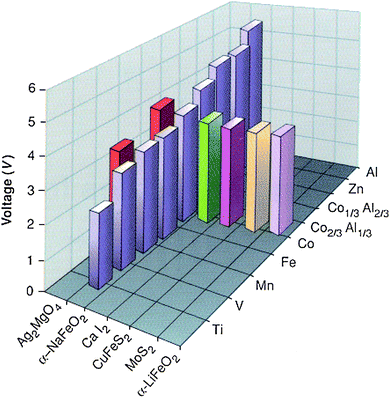 | ||
| Fig. 11 Independent effect of host crystal structure and composition on the predicted lithium intercalation voltage (vertical axis) in oxides (between MO2 and LiMO2) for use as positive electrode in lithium batteries. (Taken from ref. 73.) | ||
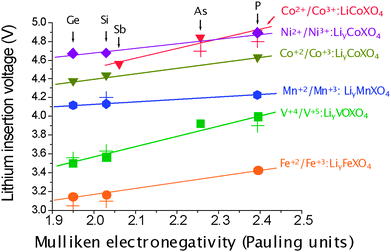 | ||
| Fig. 12 Calculated and experimental (crosses) average lithium insertion voltage for various polyoxianionic compounds vs. the Mulliken electronegativity of the central atom of the polyanion (X). The lines show the fit to respective linear functions. (Taken from ref. 37.) | ||
According to Eqn (4)–(7) the average lithium insertion voltage is calculated in between two lithium compositions, x1 and x2. In the first step x1 and x2 are taken as the fully lithiated and delithiated compounds; this is x = 0,1 in olivine LixMPO4 or layered LixMO2, x = 0,2 in LixMSiO4 and so forth. However, one can calculate the average voltage between any x1 and x2 value, provided that adequate crystallographic models of lithium/vacancy arrangements are constructed for those intermediate compositions. This is particularly interesting for materials containing several transition metal cations susceptible to varying oxidation states.142,143 As an example, Fig. 13 shows the experimental voltage–composition curve of LiNi1/3Fe1/6Co1/6Mn1/3O2 (a derivative of O3–LiCoO2) plotted together with the calculated potential curve.142 The average voltage profiles for LixNi1/3Fe1/6Co1/6Mn1/3O2 (0 < x < 1) were computed from the lowest energy lithium–vacancy arrangements in the six-formula supercell as function of lithium composition. The stepwise nature of the curves is artificial and due to the averaging of the potential over the specific composition interval, x2 − x1. The active redox couple at each oxidation process can be identified by analyzing the calculated DOS, or the net spin density distribution around the transition metal cations (see ref. 100,109,144 and 145). Fig. 13 illustrates examples of active redox couples inferred from the calculated DOS at x = 0, 1/3, 2/3 and 1.
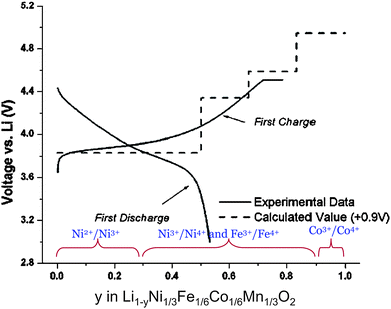 | ||
| Fig. 13 Comparison of experimental potential curve with potential curve predicted by DFT within GGA approximation. The calculated curve is shifted 0.9 V for a better comparison. Active redox couples at each compositional range are deduced from calculated DOS. (Adapted from ref. 142.) | ||
Beyond the step-like voltage curve, the complete voltage–composition profile of an electrode material can be modeled using a combination of first-principles energy methods and Monte Carlo simulations. We show above how such a combination allows the construction of a phase diagram of an electrode material as a function of the lithium content, with the energy dependence of the Li-vacancy configurational disorder parameterized with a cluster expansion. The voltage–composition curve contains the same information as the phase diagram, but it allows a better comparison with experiments. Monte Carlo simulations give the chemical potential as a function of concentration. The equilibrium potential of an electrochemical lithium cell depends on the chemical potential difference for lithium in the anode and cathode materials, expressed as
| V(x) = −[µLi cathode(x) − µLi anode(x)]/e | (8) |
Calculated voltages deviation with respect to experimental values as large as 1 V has been reported for NASICON–Li3M2(PO4)3,40,41 olivine–LiMPO4,10,66,124 or VOXO4 (X = P, As, S)57,64 compounds, within the LDA and GGA approximations. In 2004, the ability of the GGA + U method to precisely reproduce the electrochemical potential of a redox couple was proven for a variety of olivine–LiMPO4, layered-LiMO2 and spinel–LiM2O4 materials by Ceder and co-workers,10 who demonstrated that the lithium insertion voltages predicted with the GGA + U method differ from the experimental ones by only 0.1–0.3 V (see Table 1). This gives credibility to DFT methods to anticipate the voltage of hypothetical compounds even before they are synthesized. In this regard, the DFT + U predicted average voltages of lithium intercalation for LiNiPO4 (5.1 V63) and Li2CoSiO4 (4.4 V37) were subsequently confirmed by experiments; 5.1 V for LiNiPO4147 and 4.3 V for Li2CoSiO4.148
In summary, since 1997 DFT techniques have correctly modeled the energetics of lithium intercalation in many well-known compounds. These results have established the value and reliability of DFT to predict lithium insertion voltages, and nowadays ab initio methods are used as an almost routine method to screen for novel electrode materials with promising insertion voltages. This approach, while fascinating, should be handled with caution; predicting a promising average lithium insertion voltage does not necessarily mean than the material will be active once it is prepared. The calculated average voltage of the insertion reaction (Eqn (6)) is merely a measure of the relative thermodynamic stability of the inserted and deinserted materials. Obviously, more criteria besides of the average voltage are needed to determine ‘a priori’ whether a given compound will show an adequate response as an electrode material; electronic and structural factors should not be overlooked.
3.3. Electronic conductivity and hopping rate barrier: power and rate capability
The starting point to investigate the electronic properties of a potential electrode material is to determine its metallic or insulating character. Information about energy gaps between valence and conduction bands can be deduced from the calculated density of states (see Fig. 10). The LDA and GGA approximation underestimate the band gaps and, as occurs with other physical properties, the introduction of a U correction term in the GGA method could substantially improve the accuracy of calculated band gaps in systems with strong electron localization.7,10,11,38 Even if some reservations still exist about the quantitative prediction of band gaps, it is generally assumed that the general trends extracted from DFT/DFT + U calculations are reliable.Comparison between calculated band gaps and experiments is not always straight forward. Assuming a semiconductor intrinsic conductivity type, the extracted activation energy from conductivity measurements is half of the calculated band gap (ΔT = 2EA). Obviously, such a comparison does not work when other mechanisms for electrical conductivity predominate, other than the thermal excitation of electrons across the gap. The right experimental data to compare with the calculated band gap is the optical band gap. For instance, in olivine–LiFePO4 owing to a localized polaronic-type conductivity, the band gap extracted from the measured temperature dependence of the conductivity (ΔT = 2EA = 1eV149) is much smaller than the calculated gap within the GGA + U approximation (for U = 4.3 eV ΔT = 3.7 eV), while this calculated value is in good agreement with the measured optical band gap of 3.7–4.0 eV.150 Worth mentioning is the calculated gap within the GGA approximation, which is only 0.2 eV, showing that the Hubbard-like correction term (U) improves the accuracy of the calculated band gap. A similar situation is observed for V2O5, where the conductivity occurs by small polarons; the gap extracted from the measured activation energy (ΔT = 2EA = 0.46eV75) is substantially lower than the calculated band gap between the GGA (1.74 eV)75 or GGA + U (U = 3.1 eV, ΔT = 2.2 eV)11 approximations being the measured optical gap of 2.1 eV.151 Not surprisingly, in this case GGA and GGA + U values differ much less than those in the olivine–LiMPO4 compounds.
In addition to predicting band gaps from the calculated density of states, more complex DFT investigations offer the opportunity to explore the electronic conductivity by polaron hopping. When excess charge carriers, such as electrons or holes are present in a polar crystal, the atoms in their environment are polarized and displaced producing a local lattice distortion. The more the charge carriers are localized, the more pronounced the ion displacement becomes. The carrier lowers its energy by localizing into such a lattice deformation and becomes self-trapped. The quasiparticle formed by the electron and its self-induced distortion is called a small polaron if the range of the lattice distortion is of the order of the lattice parameter. In transition metal oxides it is generally accepted that charge carriers create small polarons.152 One of the fundamental concepts of polaron hopping is that the electronic carrier cannot transfer unless a certain amount of distortion is transferred. Maxisch et al. investigated the formation and transport of small polarons in olivine–LixFePO4 using first principles calculations within the GGA + U framework.153,154 The transfer of a single electron in FePO4 between a pair of two adjacent Fe atoms occurs by hopping between two equilibrium configurations FeA2+FeB3+ and FeA3+FeB2+, polaron migration is described by the distortion of the lattice deformation along a one-dimensional trajectory on the Born–Oppenheimer surface (Fig. 14). At the transition state, the total energy reaches a maximum value. The difference in energy between the transition state and equilibrium state defines the activation energy of polaron migration. It is also shown that in intrinsic (undoped) materials, excess charge carriers created by Li+ or vacancies, electrostatic binding or association energy between a positively charged Li ion and a negatively charged electron polaron is significantly large (500 meV for LiFePO4 and 370 meV for FePO4). Experimental values for the activation energy to electronic conductivity of pristine LiFePO4 are spread over a wide range (156 meV to 630 meV155) depending on the experimental setup. Removal of the self-interaction error with DFT + U or other self-interaction correction (SIC) methods create stable polarons in solids and open up an important field for ab initio studies on polaron hopping, providing a powerful pre-screening tool for evaluating new electrode materials.
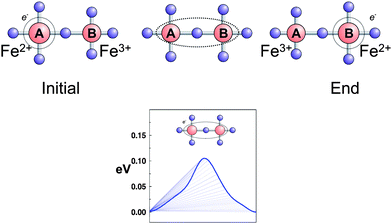 | ||
| Fig. 14 Simple illustration of the polaron conduction mechanism in LixFePO4. | ||
3.4 Lithium diffusion: power and rate capability
In rechargeable lithium ion batteries, high power requires that Li diffusion in and out of the electrode materials takes place fast enough to supply the electric current. There is no doubt that the engineering design of the porous electrode is an important factor for high power performance, nevertheless, lithium diffusion in the active material is an intrinsic property of the electrode material and is a necessary condition for high rate performance. Diffusion of ions in crystalline solids typically occurs by diffusion-mediating defects such as vacancies or interstitials. At the dilute limit (low defect concentration), the diffusivity can be written as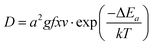 | (9) |
At the dilute limit, lithium diffusivity can be estimated using the equation above, by calculating the activation barrier, which is defined as the difference in energy at the activated state and the energy at the initial state of the ionic hop. In ab initio calculation, nudged elastic band (NEB)156 method can be used to determine the maximum energy along the lowest energy path between two neighboring lithium sites. In layered transition metal oxides LixMO2,157–159 olivine–LixMPO487,160 and spinel transition metal oxides LixM2O4,161 the atomistic lithium diffusion transition paths have been identified by DFT means. As shown in Fig. 15a, in the layered O3-type structure, lithium diffusion takes place in the lithium layer by hopping from one octahedral to another octahedral site through an intermediate tetrahedral site. In the spinel structure the lithium ion diffuses through the structure by moving from one 8a site to the neighboring empty 16c site and then to the next 8a site. Notice that such 8a-16c-8a diffusion paths are three-dimensionally interconnected.162 In the olivine structure, again, the transition state for lithium diffusion along the chain is the approximate tetrahedral site between the two octahedral sites (Fig. 15c). Li hopping between the chains is highly unfavorable at room temperature, with activation barriers more than 1 eV.160 The GGA calculated activation barriers for LixCoO2,159,163 LixMn2O4161 and LixFePO4160 when x is near to 1 show that the lithium diffusion barrier can be qualitatively estimated by first principles calculations. The intrinsic Li diffusion coefficient can be estimated from the atomistic scale behavior of the Li at the dilute limit.
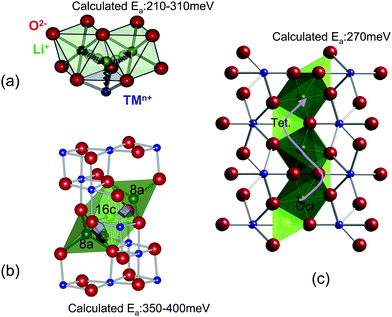 | ||
| Fig. 15 Diffusion paths and activation energies as determined by DFT methods in (a) layered structure, (b) spinel structure and (c) olivine structure. | ||
However, in lithium intercalation compounds nondilute diffusion is common, during the charging and discharging processes lithium ions are inserted and removed from the host undergoing a wide range of concentration changes. When the concentration of the carriers (vacancy or lithium) is sufficiently large, they interact, which complicates any analysis of diffusion. The migration ion will sample different local environments with different activation barriers. A more sophisticated formalism for nondilute systems have been well developed by Van der Ven et al.158 The approach makes use of a local cluster expansion to parameterize the environment dependence of the activation barrier. With minimal computation cost, one can then extrapolate energy values from a few configurations in a given crystal structure, to any ionic configuration within the same crystal structure. The results of such a local cluster expansion are then implemented in kinetic Monte Carlo simulation to investigate diffusion in a nondilute system. Model systems such as LixCoO2 and LixTiS2164 have been studied with first principles methods. There have been large quantitative discrepancies between the experimentally measured and ab initio calculated diffusion coefficients in these two systems, though the qualitative variations in diffusion coefficient vs.lithium concentration agree well. As shown in Fig. 16, a similar trend is observed in measured and calculated values, that is, low diffusion coefficients in the dilute limits (low vacancy concentration or low lithium concentration) and high diffusion coefficients at intermediate concentrations. A major source of this discrepancy can be attributed to the difference of the c-lattice parameter change between calculations and experiments. The calculated c-lattice parameter of the O3 host is systematically smaller than the experimentally observed value by approximately 4% and it drops more significantly in the composition region 0.15 < x < 0.5 than in the experimental results. In the calculated result, the c-lattice parameter changes from 13.8 to 12.9 Å21 when x decreases from 0.5 to 0.15. For the LixCoO2 thin-film experimental result, the c-lattice parameter changes from 14.42 to 14.31 Å165 when x decreases from 0.5 to 0.15. This large discrepancy is likely due to the inability of handling the Van der Waals forces in DFT.
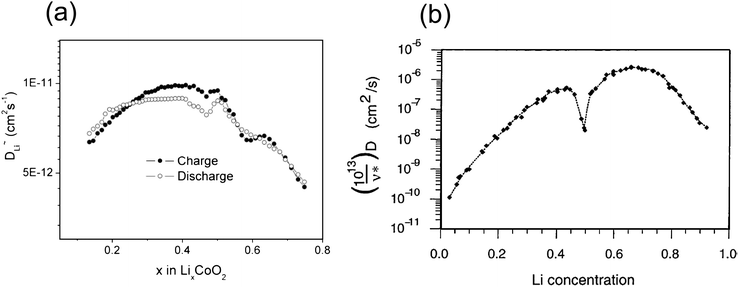 | ||
| Fig. 16 Lithium diffusion coefficient as a function of lithium concentration in LixCoO2. (a) Experimentally measured (from ref. 165) and (b) calculated (from ref. 159). | ||
Systematic study157 on factors that influence the activation barrier for Li diffusion in O3 layered oxides shows that the two dominant effects are the Li slab spacing (related to c-lattice parameter), and the electrostatic repulsion Li experience when it is in a transition state. Therefore, optimization of the layered oxides for high rate performance is conceptually straightforward; (i) create materials with large Li slab spacing over the relevant composition range and (ii) create the percolating network of transition state sites in contact with low valent transition metal cation. Such a strategy has led to the discovery of a high power cathode material LiNi1/2Mn1/2O2,113 as discussed in section 3.1.
Other simulation models have been applied to probe the lithium transport properties in pure ionic crystals. For example, using a potential model,87Li diffusion barriers are calculated to be 550 meV in LiFePO4 along the one dimensional diffusion channel, and more than 2 eV if inter-channel diffusion takes place, this reported trend is consistent with DFT studies.154,160 It should be stressed that most of the transition metal oxides have a significant degree of covalency, therefore it is arguable whether these methods that are designed for ionic compounds are well suited to quantitative property prediction in lithium intercalation compounds.
It is also important to point out that the direct comparison between experimentally measured diffusion data and calculated diffusion data should be exercised with caution. A common experimental method of measuring Li diffusion coefficients in electrode materials is with an electrochemical cell, where the electrode is made of active materials in powder form, polymer binder and conductive carbon additives. Such measurements, however, introduce large uncertainties since it is extremely difficult to quantify the geometrical dimensions of the active intercalation compound. For example, intercalation compounds like layered oxides and olivine materials are highly anisotropic materials, which means that the lithium diffusion coefficients in different crystallographic directions/planes are different. Diffusion coefficient measurement on the powder composite electrode is the average diffusion coefficient of the entire electrochemical cell, while in first principles calculation the intrinsic diffusion coefficient is investigated. In addition, the diffusion coefficient becomes irrelevant when the lithium intercalation process proceeds as a two-phase reaction, as is the case in olivine–LiFePO4. The kinetics of nucleation and growth of the second phase, as well as phase boundary movement have to be taken into consideration.
3.5. Thermal stability and safety considerations
As large scale applications of lithium ion batteries are on the horizon, safety issues have become an increased concern. Most cathode materials consist of oxygen and a transition metal, and they become highly oxidized and susceptible to degradation through exothermic and endothermic phase transitions. Few or no computational studies have been reported on understanding the stability of the electrode materials at a high state of charge. This is in part due to the difficulty of correctly predicting the energy of reduction reactions with standard DFT. Within the DFT + U scheme, a new method166 for predicting the thermodynamics of thermal degradation has been developed and demonstrated on three major cathode materials, LixNiO2, LixCoO2 and LixMn2O4. The general decomposition reaction of a lithium transition metal oxide can be expressed as| LixMyOz+z′ → LixMyOz + z′/2O2 | (10) |
It is shown that by constructing ternary Li–M–O2 phase diagrams, the reaction Gibbs free energy can be estimated by using entropy change ΔS from the oxygen gas released and by assuming that the temperature dependence of ΔH is much smaller compared to the −TΔS term. The entropy values for oxygen gas as a function of temperature are obtained from experimental database (JANAF)167 in this approach the thermodynamic transition temperature can be obtained by
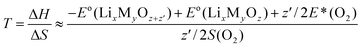 | (11) |
The overestimation of the binding energies of the O2 molecules is estimated to be −1.36 eV per molecule11 and is subtracted from the E*(O2) term. The correlation error in transition metals due to the localized d orbital is removed with the Hubbard U term, though a single U value for different valences of the transition metals is somewhat inadequate.
It is important to point out that in the case where the decomposition reaction is kinetically controlled, which means at the thermodynamic transition temperature the ions do not have high enough mobility, the kinetic transition temperature cannot be obtained through first principles computations. Modeling the kinetics of phase transformation from first principles is an unresolved problem in materials science.
4. Challenges
4.1 Other chemistries
Section 3 was devoted to classical electrode materials, where energy storage is possible thanks to a reversible insertion reaction in an inorganic host. In this section, paths for computational design of other classes of battery materials are introduced; first we extend the insertion reaction towards electrodes where organic components are present. Second, we refer to conversion reactions, in which the reversible reduction of transition metal ions permits chemical energy storage, without the need for an open framework with high electrical conductivity. Alloying reactions of metals with Li offer another interesting mechanism for energy storage.The rate limiting step of insertion electrodes based on inorganic frameworks is often found to be the diffusion of Li+ within the material. In addition, the electronic conductivity of an inorganic host is usually quite low, increasing the ohmic drop of the battery. In trying to overcome these limitations, organic materials such as conducting polymers (polypyrrol PPy, polyaniline PAni, polythiophene PTh) were studied and proposed as candidates for the development of rechargeable plastic lithium batteries,168–170 but they also present drawbacks, such as low specific energy. Recent trends propose sustainable organic-based batteries based on electrode materials made from biomass, active LixC6O6 organic molecules that can be prepared from natural sugars common in living systems, hypericine (an anthraquinone-derivative) present in St John's Wort, or the condensation polymers of malic acid have been suggested as potential high-capacity cathode materials.171 Recently, the bio-inspired Li-based organic salts Li2C8H4O4 (Li terephthalate) and Li2C6O4H4 (Li trans,trans-muconate), which have carboxylate groups conjugated with the molecule core as redox centres, have been shown to be attractive as negative electrode materials. Electrochemical investigation of these organic molecules has been successfully complemented with DFT calculations.172 Compared to a decade ago, it is now possible to study molecular species of polymers (hundred atoms in size) exclusively at DFT level. Despite successes, there seems to be important cases where current functionals reveal serious discrepancies.173 Simulation of polymers can be nicely accomplished combining DFT and Molecular Dynamics methods; typically DFT is used to investigate monomers and self-assembly of polymers with simple architectures, and MD simulation is used to explore microscopic properties of complex star-shaped and branched polymers.
Hybrid organic–inorganic materials, such as V2O5/PPy,174,175 V2O5/PTh,174 LiMn2O4/PPy176,177 or LiFePO4/PPy178,179 represent an opportunity to take advantage of the best properties of both organic and inorganic species. In these hybrid materials, the conductive polymer is either interleaved between the layers of the inorganic oxide lattice (as in V2O5 nanocomposites), or acts as a conductive matrix that connects the particles of the inorganic oxide (as in LiMn2O4 composites). These hybrid inorganic–organic composites do not fulfil initial expectations for applicability in commercial lithium batteries. Simulations of these materials have problems associated with the different approaches traditionally taken to model materials with different bonding characteristics. In addition, the large number of atoms in the unit cell, together with the complex nature of the physical–chemical interaction between the organic–inorganic components, leaves this class of electrodes hardly treatable by DFT calculations.
The recently reported electrochemical activity of [FeIII(OH)0.8F0.2(O2CC6H4CO2)].H2O180 opens new directions towards the possible utilization of metal–organic frameworks (MOFs) as an electrode for lithium batteries. MOFs can be defined as porous crystalline solids constructed from inorganic clusters connected by organic ligands. The simple geometric figures representing inorganic clusters, or coordination spheres, and the organic links constitute structural entities denoted as secondary building units (SBUs). It is the bridging organic ligands which allow for the large diversity in topologies and possible properties of these metal–organic coordination networks. (For reviews on this topic see ref. 181–184.) The application of MOFs as electrodes for lithium batteries is at a very early stage, and guidelines concerning ligands, metal centers, architectures, pore-size, etc., to produce electrochemically active MOFs have not yet been established. Therefore, this field is an unexplored challenge for computational research. DFT methods can be applied to investigate lithium insertion in a given MOF,180,185 though the large number of atoms in the unit cell makes this investigation computationally very expensive. An effective screening of MOFs for electrode applications might be achieved combining DFT methods with Monte Carlo simulations. Mellot–Drazniek and co-workers have shown how computational approaches may take advantage of the concept of SBUs, to produce both existing and as yet not-synthesized MOFs,186–188 Their method (Automated Assembly of Secondary Building Units, or AASBU) consists of three steps (i) calculation of the pre-defined building-block units that are usually met in existing compounds, (ii) parameterization of inter-SBUs interactions utilizing Lennard–Jones potentials and (iii) auto-assembly of the SBUs in 3D space through a sequence of simulated annealing and energy minimization steps (MC simulations). The AASBU method might be extended to the systematic investigation of possible MOFs with electrochemical activity through the DFT calculation of candidate lithiated SBUs.
To date, the specific capacity delivered by lithium ion batteries using intercalation electrode materials is limited to the exchange of one electron per 3d metal. One way to achieve higher capacities is to use electrode materials operating in a conversion reaction, where the metal–redox oxidation state can reversibly change by more than one unit. The general expression for such a conversion reaction can be expressed as
| Mz+Xy + zLi+ + ze− → M0 + yLiz/yX | (12) |
Equally as with intercalation reactions, the thermodynamics of any conversion reaction can be investigated from first principles methods by computing the total energy of the involved compounds. Furthermore, intermediate species that may occur in the course of the complete reduction of Mz+Xy can also be investigated by DFT techniques. For instance, in the case of FeP first principles computations reveal that a thermodynamically stable LiFeP intermediate phase is achievable upon reduction of the FeP electrode.196 Experiments support that a two-step insertion/conversion reaction (FeP + Li → LiFeP and LiFeP + 2Li → Li3P + Fe0) occurs for the FeP electrode, after the one-step conversion reaction (FeP +3Li → Li3P + Fe0) in the first discharge.196 The complex electrochemistry of other electrodes involving conversion reactions have been extensively investigated combining experimental and computational methods, MnP4,197 NiP2,198Cu3P,199 FeF3,200Ti/Li2O,201Cu/LiF202 and so forth.
It has been well known for decades that the reduction of many Mz+Xy compounds by lithium is thermodynamically favorable (Ellingham diagrams203.) What is surprising is that the reaction is almost reversible, in spite of the poor reactivity and/or transport properties of massive Li2O. The main limitation of conversion electrodes is the large hysteresis found between the discharge and charge of the cell.190,195,204 The kinetics of conversion electrodes has been shown to be controlled by particle size; the highly divided and large surface area of the nanoparticles formed during the first discharge of the Li cell facilitates the reverse reaction. With reversibility of the conversion reaction governed by the nanostructure of the materials, the next obvious step for computational design of novel conversion electrodes is to account for nanoscale effects.
Unlike transition metal oxides, some metals (Al, Sn, Si, Sb, In, …) form alloys with Li, delivering high specific capacities.205 DFT methods have been successfully utilized to investigate some of these reactions135,206 though important cases, such as the Si anode, remain unexplored to date.
4.2. Nano size effects
A first principles study of nanosize effects in conversion electrodes is hindered by several major hurdles. One is obviously the limiting computation power, a simple 2 nm Pt nanoparticle consists over 250 atoms, which is already a highly intensive calculation. In experimental observations, an assembly of 1–5 nm metal nanoparticles is visible in converted materials after first discharge.207 Secondly, a creative methodology has to be established for modeling a complex oxides/fluorides/oxyfluroides nanocomposite with extreme chemical heterogeneity. The kinetics of conversion reactions involves the simultaneous diffusion of an inserted element and a displaced element coupled with phase transformations among multiple phases. Thirdly, the transport properties and phase transformation mechanisms during conversion reactions are currently not well understood either experimentally or computationally. It will take rigorous theoretical and experimental efforts that combine first principles calculations, statistical mechanical techniques, continuum modeling of diffusion and phase transformation, as well as various experimental techniques for nanomaterials to systematically elucidate the conversion/reconversion mechanisms and transport kinetics. A recent work200 on FeF3 successfully implemented the nano-size effect on the calculated voltage profile. It was found that when 1 nm Fe particles form, the potential for a conversion reaction (from FeF3 to LiF and Fe) is considerably reduced from the bulk value, in good agreement with the experimental observations.Nanostructured materials designed for improved electrochemical properties are critically needed to overcome the existing bottleneck for energy storage materials. Advances in nanosynthesis have opened the potential for providing synthetic control of materials architectures at nanoscale. However, little is known about how the electrochemistry of nanoparticles, or nanotubes/nanofibers vary with size and whether these effects are thermodynamic in nature or purely kinetic. For example, in nano-LiFePO4, several models have been proposed to elucidate the ultra-fast delithiation processes.208–212 One of the mechanisms proposed by first principles calculations within the GGA + U framework investigated by the authors involved several surface properties of olivine-structure LiFePO4. Calculated surface energies and surface redox potentials were found to be very anisotropic, shown in Fig. 17.208 The two low-energy surfaces (010) and (201) dominate in the Wulff (equilibrium) crystal shape and make up almost 85% of the surface area. Another study213 based on the atomistic potential method predicted a similar particle morphology. More interestingly in ref. 208, the Li redox potential for the (010) surface was calculated to be 2.95 V, which is significantly lower than the bulk value of 3.55 V. This study revealed the importance of controlling both the size and morphology of nano LiFePO4, and pointed towards the relevance of thermodynamic factors in the electrochemistry of nanomaterials. Based on such insights gained from computational modeling, ultra-fast rate (9 s discharge) in modified LiFePO4 was recently successfully demonstrated by Kang and Ceder.214
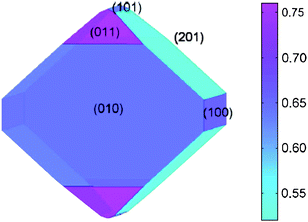 | ||
| Fig. 17 Wulff shape of LiFePO4 using the calculated surface energies in nine directions. The color scale bar on the right gives the energy scale of the surface in units of J m−2. (From ref. 208.) | ||
It is generally believed that using nanostructured electrodes, better rate capabilities are obtained because the distance over which Li ion must diffuse in the solid state is dramatically decreased. Such experimental efforts made over the last decade using one dimensional nanofiber/nanotube as electrode materials have achieved considerable success. Nevertheless, optimization of the size and chemistry of nanostructured electrodes are still mostly carried out in the traditional trial-and-true way. Ab initio studies on carbon nanotubes are prevalent, computational studies on inorganic nanotubes (such as MX2, M = Ti, Co, Mn, etc. X = S or O) are less common, largely owing to the size of the supercell (nearly 100 atoms in a supercell for a 1 nm nanotube). Several computational studies have been performed using density functional tight binding method (DFTB), which allows calculation of larger nanotubes but with less accuracy than DFT. By modeling the nanotube surface as a curved surface in DFT,215 it is found that for TiS2nanotube radii (5–25 nm), the Li diffusion activation barrier is 200 meV smaller than in the bulk material, which could result in improved mobility of Li by thousand-folds at room temperature. More interestingly, the activation barrier was found to increase for small nanotube (radii less than 5 nm) as a result of stronger electrostatic repulsion and less relaxation of S atoms. This prediction implies that experimental effort should not be made to further reduce nanotube size, though its validity remains untested. It is worth mentioning that experimental investigation of lithium diffusion property on nanotube or free surface is still in its infant stage, little is known about how the surfaces of the nanotube/nanofiber look on an atomistic scale. It is also very difficult to perform controlled experiments where lithium diffusion on a single isolated nanotube can be measured. However, with important advances being made in nanomaterials' manipulation and characterization, we believe it is possible that these efforts will be successful, providing better synergy between experiments and ab initio computation modeling.
4.3. Interphase effects
A relevant interphase effect occurs in M/Liz/yX nanocomposites obtained by conversion reactions (M = transition metal, X = O or F). At low voltages, Li+ ions are stored in the oxide side of the interface while electrons are localized on the metallic side resulting in charge separation (pseudo-capacitive behavior with high rate performance).216 This novel interfacial mechanism of additional Li storage, which relies on the presence of nanoparticles, was recently experimentally observed201,217 and theoretically proven.202,216,218,219 To understand the mechanistic details of this lithium storage anomaly, Zhukovskii et al. performed comparative ab initio calculations on the atomic and electronic structure of the nonpolar Cu∕LiF (001) and model Li∕LiF (001) interfaces.Electrochemical energy storage systems often operate far below extreme condition of the organic electrolyte, which being thermodynamically unstable cause the electrolyte to decompose. The phase that forms as a reaction layer between the electrode and electrolyte (the solid electrolyte interface, or SEI) is critical to performance, life and safety of lithium ion batteries. As nanomaterials and/or higher voltage materials are developed to enhance the rate capability and/or energy density of the electrodes, the interphase becomes increasingly important. It has been identified as one of the grand challenges for science—to predict and manipulate the structure of the electrochemically formed interphase between the electrode and electrolyte under large variations in potentials, as well as to quantify the electron and/or ion transport through the interphase layer. Little is actually understood about SEI composition except for the graphite/carbon anode and many fundamental questions remain unanswered. Some effort has been dedicated to understand the SEI formation mechanism on carbon using quantum chemistry (B3PW91, a hybrid DFT + HT functional) methods, reviewed in detail by Wang and Balbuena.220 For non-carbon based new anode materials, such as Si, Sn based materials, such effort is still lacking. To understand the transport property of the SEI interphase, the apparent physical and chemical complexity of the SEI has to be broken down into discrete molecular-level problems, where DFT-based methods have limitations. MD simulations that include electric field effects and chemical reactivity may be particularly well suited to address this challenge. MD simulations221,222 have been applied to understand some electrolyte properties, such as free energy for ion transport. To establish a computational model that can generate a priori predictions of the dynamic behavior of SEI requires integrated experimental/computational approaches, and new innovative in situ experimental techniques are needed to provide the important physical insights necessary to formulate realistic computation models. Advances in understanding of interfacial effects in nanocomposite electrodes are critical to the development of new energy storage materials, and ab initio computation will surely play a critical role in such pursuits.
Conclusions
In this review, we have illustrated how first principles computation can accelerate the search for energy storage electrode materials for lithium ion batteries. New electrode materials exhibiting high energy, high power, better safety and longer cycle life must be developed to meet the increasing demand of energy storage, particularly in transportation applications such as plug-in HEV. We have demonstrated achievements in predicting relevant properties (including voltage, structure stability, electronic property etc.) of electrode materials using DFT-based first principles methods. In summary, these capabilities establish first principles computation as an invaluable tool in the design of new electrode materials for lithium ion batteries. However, despite these capabilities, it is important to recognize that many challenges have still to be resolved—predicting new mechanisms (other than intercalation), new properties of nanoscale materials, and atomistic understanding of surface and interphase remain challenges for first principles computation.Acknowledgements
M. E. Arroyo-de Dompablo acknowledges financial support from Spanish Ministry of Science (MAT2007-62929, CSD2007-00045) and Universidad Comlutense de Madrid (PR34/07-1854, PR01/07-14911). Y. Shirley Meng would like to express her gratitude to University of Florida for the new faculty startup funding.References
- “Basic research needs for electric energy storage” (United State Department of Energy, 2007) Search PubMed
.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B, 1996, 54, 16533 CrossRef CAS
- F. Herman, J. P. Van Dyke and I. B. Ortenburger, Phys. Rev. Lett., 1969, 22, 807 CrossRef
- V. I. Anisimov, I. V. Solovyev, M. A. Korotin, M. T. Czyzyk and G. A. Sawatzky, Phys. Rev. B, 1993, 48, 16929 CrossRef CAS
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B, 1991, 44, 943 CrossRef CAS
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B, 2005, 71, 16
- L. Li, W. Yu and C. Jin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 174115 CrossRef
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B, 2004, 70, 8
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B, 2006, 73, 6
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS
-
M. L. Cohen, V. Heine, in Solid State Physics, Ed. H. Ehrenreich, F. Spaepen, F. Seitz, D. Turnbull, Academic Press, New York, 1970, pp. 37 Search PubMed
- J. C. Phillips and L. Kleinman, Phys. Rev., 1959, 116, 287 CrossRef CAS
- J. M. Sanchez, F. Ducastelle and D. Gratias, Physica A, 1984, 128, 334 CrossRef
- C. Wolverton and D. Defontaine, Phys. Rev. B, 1994, 49, 8627 CrossRef
-
D. De Fontaine, in Solid State Physics, Ed. D. T. Frederick Seitz, Henry Ehrenreich, Academic Press, New York, 1994, vol. 34, pp. 74 Search PubMed
- G. Ceder, A. Van der Ven, C. Marianetti and D. Morgan, Model. Simul. Mater. Sci. Eng., 2000, 8, 311 CrossRef CAS
- M. Asta, C. Wolverton, D. Defontaine and H. Dreysse, Phys. Rev. B, 1991, 44, 4907 CrossRef CAS
- A. Van der Ven, M. K. Aydinol, G. Ceder, G. Kresse and J. Hafner, Phys. Rev. B, 1998, 58, 2975 CrossRef
- C. Wolverton and A. Zunger, Phys. Rev. B, 1998, 57, 2242 CrossRef CAS
- C. Wolverton and A. Zunger, Phys. Rev. Lett., 1998, 81, 606 CrossRef CAS
- C. Wolverton and A. Zunger, J. Electrochem. Soc., 1998, 145, 2424 CrossRef CAS
- M. E. Arroyo de Dompablo, A. Van der Ven and G. Ceder, Phys. Rev. B, 2002, 66, 064112 CrossRef
- Y. S. Meng, Y. Hinuma and G. Ceder, J. Chem. Phys., 2008, 128, 8
- Y. Hinuma, Y. S. Meng, K. S. Kang and G. Ceder, Chem. Mater., 2007, 19, 1790 CrossRef CAS
- Y. Hinuma, Y. S. Meng and G. Ceder, Phys. Rev. B, 2008, 77, 16
- D. Carlier, A. Van der Ven, C. Delmas and G. Ceder, Chem. Mater., 2003, 15, 2651 CrossRef CAS
- A. Van de Walle and G. Ceder, J. Phase Equilib., 2002, 23, 348 CrossRef CAS
- P. D. Tepesch, G. D. Garbulsky and G. Ceder, Phys. Rev. Lett., 1995, 74, 2272 CrossRef CAS
-
M. E. J. Newman, G. T. Barkema, Monte Carlo Methods in Statistical Physics, Oxford University Press, Oxford, 1999 Search PubMed
- P. Larsson, R. Ahuja, A. Nyt and J. O. Thomas, Electrochem. Commun., 2006, 8, 797 CrossRef CAS
- A. Kokalj, R. Dominko, G. Mali, A. Meden, M. Gaberscek and J. Jamnik, Chem. Mater., 2007, 19, 3633 CrossRef CAS
- S. Q. Wu, J. H. Zhang, Z. Z. Zhu and Y. Yang, Curr. Appl. Phys., 2007, 7, 611 CrossRef
- M. E. Arroyo y de Dompablo, U. Amador, J. M. Gallardo-Amores, E. Moran, H. Ehrenberg, L. Dupont and R. Dominko, J. Power Sources, 2009, 189, 638 CrossRef CAS
- M. E. Arroyo-de Dompablo, M. Armand, J. M. Tarascon and U. Amador, Electrochem. Commun., 2006, 8, 1292 CrossRef
- M. E. Arroyo-de Dompablo, R. Dominko, J. M. Gallardo-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik and E. Moran, Chem. Mater., 2008, 20, 5574 CrossRef CAS
- M. E. Arroyo y de Dompablo, J. M. Gallardo-Amores, J. Garcia-Martinez, E. Moran, J. M. Tarascon and M. Armand, Solid State Ionics, 2008, 179, 1758 CrossRef CAS
- D. Morgan, G. Ceder, M. Y. Saidi, J. Barker, J. Swoyer, H. Huang and G. Adamson, J. Power Sources, 2003, 119, 755 CrossRef
- D. Morgan, G. Ceder, M. Y. Saidi, J. Barker, J. Swoyer, H. Huang and G. Adamson, Chem. Mater., 2002, 14, 4684 CrossRef CAS
- J. S. Braithwaite, C. R. A. Catlow, J. D. Gale, J. H. Harding and P. E. Ngoepe, J. Mater. Chem., 2000, 10, 239 RSC
- X. Rocquefelte, F. Boucher, P. Gressier and G. Ouvrard, Chem. Mater., 2003, 15, 1812 CrossRef CAS
- Z. Y. Li and Q. H. Wu, ChemPhysChem, 2008, 9, 300 CrossRef CAS
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Solid State Ionics, 2002, 152–153, 189 CrossRef CAS
- M. V. Koudriachova and N. M. Harrison, J. Mater. Chem., 2006, 16, 1973 RSC
- M. Anicete-Santos, L. Gracia, A. Beltran, J. Andres, J. A. Varela and E. Longo, Phys. Rev. B, 2008, 77, 085112 CrossRef
- M. Wagemaker, A. Van Der Ven, D. Morgan, G. Ceder, F. M. Mulder and G. J. Kearley, Chem. Phys., 2005, 317, 130 CrossRef CAS
- X. Rocquefelte, I. Bouessay, F. Boucher, P. Gressier and G. Ouvrard, J. Solid State Chem., 2003, 175, 380 CrossRef CAS
- X. Rocquefelte, F. Boucher, P. Gressier, G. Ouvrard, P. Blaha and K. Schwarz, Phys. Rev. B, 2000, 62, 2397 CrossRef CAS
- K. R. Kganyago, P. E. Ngoepe and C. R. A. Catlow, Solid State Ionics, 2003, 159, 21 CrossRef CAS
- J. S. Filhol, C. Combelles, R. Yazami and M. L. Doublet, J. Phys. Chem. C, 2008, 112, 3982 CrossRef CAS
- V. Mauchamp, P. Moreau, L. Monconduit, M.-L. Doublet, F. Boucher and G. Ouvrard, J. Phys. Chem. C, 2007, 111, 3996 CrossRef CAS
- M. P. Bichat, F. Gillot, L. Monconduit, F. Favier, M. Morcrette, F. Lemoigno and M. L. Doublet, Chem. Mater., 2004, 16, 1002 CrossRef CAS
- P.-E. Lippens, M. Womes, P. Kubiak, J.-C. Jumas and J. Olivier-Fourcade, Solid State Sci., 2004, 6, 161 CrossRef CAS
- C. Y. Ouyang, Z. Y. Zhong and M. S. Lei, Electrochem. Commun., 2007, 9, 1107 CrossRef CAS
- M. Launay, F. Boucher, P. Gressier and G. Ouvrard, J. Solid State Chem., 2003, 176, 556 CrossRef CAS
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B, 1997, 56, 1354 CrossRef CAS
- M. K. Aydinol, A. F. Kohan and G. Ceder, J. Power Sources, 1997, 68, 664 CrossRef CAS
- P. Tang and N. A. W. Holzwarth, Phys. Rev. B, 2003, 68, 165107 CrossRef
- P. Deniard, A. M. Dulac, X. Rocquefelte, V. Grigorova, O. Lebacq, A. Pasturel and S. Jobic, J. Phys. Chem. Solids, 2004, 65, 229 CrossRef CAS
- J. M. Osorio-Guillen, B. Holm, R. Ahuja and B. Johansson, Solid State Ionics, 2004, 167, 221 CrossRef CAS
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144 CrossRef CAS
- M. E. Arroyo-de Dompablo, P. Rozier, M. Morcrette and J. M. Tarascon, Chem. Mater., 2007, 19, 2411 CrossRef CAS
- U. Amador, J. M. Gallardo-Amores, G. Heymann, H. Huppertz, E. Moran and M. E. Arroyo-de Dompablo, Solid State Sci., 2009, 11, 343 CrossRef CAS
- M. E. Arroyo-de Dompablo, U. Amador and F. Garcia-Alvarado, J. Electrochem. Soc., 2006, 153, A673 CrossRef CAS
- M. E. Arroyo y de Dompablo, U. Amador and J.-M. Tarascon, J. Power Sources, 2007, 174, 1251 CrossRef CAS
- R. Dominko, M. Bele, M. Gaberscek, A. Meden, M. Remskar and J. Jamnik, Electrochem. Commun., 2006, 8, 217 CrossRef CAS
- A. Nyten, A. Abouimrane, M. Armand, T. Gustafsson and J. O. Thomas, Electrochem. Commun., 2005, 7, 156 CrossRef CAS
- A. R. West and F. P. Glasser, J. Solid State Chem., 1972, 4, 20 CrossRef CAS
- V. V. Politaev, A. A. Petrenko, V. B. Nalbandyan, B. S. Medvedev and E. S. Shvetsova, J. Solid State Chem., 2007, 180, 1045 CrossRef CAS
- D. Carlier, L. Croguennec, G. Ceder, M. Menetrier, Y. Shao-Horn and C. Delmas, Inorg. Chem., 2004, 43, 914 CrossRef CAS
- G. Ceder, Science, May, 1998, 280, 1099 Search PubMed
- P. Balog, D. Orosel, Z. Cancarevic, C. Schön and M. Jansen, J. Alloys Compd., 2007, 429, 87 CrossRef CAS
- J. M. Gallardo-Amores, N. Biskup, U. Amador, K. Persson, G. Ceder, E. Moran and M. E. Arroyo-de Dompablo, Chem. Mater., 2007, 19, 5262 CrossRef CAS
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Phys. Rev. Lett., 2001, 86, 1275 CrossRef CAS
- A. Kuhn, Pilar Diaz-Carrasco, Maria Elena Arroyo y de Dompablo and Flaviano Garcia-Alvarado, Chem. Inform., 2007, 38 Search PubMed
- D. Balachandran, D. Morgan, G. Ceder and A. Van de Walle, J. Solid State Chem., 2003, 173, 462 CrossRef CAS
- D. Morgan, D. Balachandran and G. Ceder, Mater. Res. Soc. Symp. Proc., 2003, 755, 43
- M. E. Arroyo-de Dompablo, J. M. Gallardo-Amores, M. T. Azcondo, G.A.F. and U. Amador, J. Phys. Chem. Solids, 2006, 67, 1243 CrossRef CAS
- M. E. Arroyo-de Dompablo, J. M. Gallardo-Amores and U. Amador, Electrochem. Solid-State Lett., 2005, 8, A564 CrossRef CAS
- P. Strobel, A. Ibarra Palos, M. Anne and F. Le Cras, J. Mater. Chem., 2000, 10, 429 RSC
- M. E. Arroyo y de Dompablo and J. Morales, J. Electrochem. Soc., 2006, 153, A2098 CrossRef CAS
- N. Biskup, J. L. Martinez, M. E. Arroyo-de Dompablo, P. Diaz-Carrasco and J. Morales, J. Appl. Phys., 2006, 100, 093908 CrossRef
- J. S. Braithwaite, C. R. A. Catlow, J. H. Harding and J. D. Gale, Phys. Chem. Chem. Phys., 2000, 2, 3841 RSC
-
C. R. A. Catlow, Computer Modelling in Inorganic Crystallography, Academic Press, San Diego, 1997, vol. 61, pp. 1795–1803 Search PubMed
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., Oct, 2005, 17, 5085 Search PubMed
- C. L. Olson, J. Nelson and M. S. Islam, J. Phys. Chem. B, 2006, 110, 9995 CrossRef CAS
- S. M. Woodley, C. R. A. Catlow, P. Piszora, K. Stempin and E. Wolska, J. Solid State Chem., 2000, 153, 310 CrossRef CAS
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769 CAS
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461 CrossRef CAS
- A. Van der Ven, C. Marianetti, D. Morgan and G. Ceder, Solid State Ionics, 2000, 135, 21 CrossRef CAS
- S.-J. Hwang, H.-S. Park, J.-H. Choy and G. Campet, Chem. Mater., 2000, 12, 1818 CrossRef CAS
- Y. Shao-Horn, S. A. Hackney, A. R. Armstrong, P. G. Bruce, R. Gitzendanner, C. S. Johnson and M. M. Thackeray, J. Electrochem. Soc., 1999, 146, 2404 CrossRef CAS
- G. Ceder and A. Van der Ven, Electrochim. Acta, 1999, 45, 131 CrossRef CAS
- J. Reed and G. Ceder, Chem. Rev., 2004, 104, 4513 CrossRef CAS
- J. Reed, G. Ceder and A. Van Der Ven, Electrochem. Solid-State Lett., 2001, 4, A78 CrossRef CAS
-
A. Van Der Ven, G. Ceder, Lithium Batteries: Science
and Technology, Ed. G. Nazri, G. Pistoia,Kluwer Academic Publishers, 2004 Search PubMed
- M. E. Arroyo-de Dompablo and G. Ceder, J. Power Sources, 2003, 119, 654 CrossRef
- M. Arroyo y de Dompablo, C. Marianetti, A. Van der Ven and G. Ceder, Phys. Rev. B, 2001, 63, 9
- M. E. Arroyo y de Dompablo and G. Ceder, Chem. Mater., 2003, 15, 63 CrossRef
- C. Chazel, M. Menetrier, L. Croguennec and C. Delmas, Inorg. Chem., 2006, 45, 1184 CrossRef CAS
- C. Delmas, J. P. Peres, A. Rougier, A. Demourgues, F. Weill, A. Chadwick, M. Broussely, F. Perton, P. Biensan and P. Willmann, J. Power Sources, 1997, 68, 120 CrossRef CAS
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091
- T. Ohzuku and A. Ueda, J. Electrochem. Soc., 1994, 141, 2972 CAS
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114 CAS
- C. Wolverton and A. Zunger, J. Power Sources, 1999, 81, 680 CrossRef
- A. Van der Ven, M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1998, 145, 2149 CAS
- J. Reed and G. Ceder, Electrochem. Solid-State Lett., 2002, 5, A145 CrossRef CAS
- W. S. Yoon, S. Iannopollo, C. P. Grey, D. Carlier, J. Gorman, J. Reed and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A167 CrossRef CAS
- T. Ohzuku and Y. Makimura, Chem. Lett., 2001, 744 CrossRef CAS
- Z. H. Lu, L. Y. Beaulieu, R. A. Donaberger, C. L. Thomas and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A778 CrossRef CAS
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS
- Y. S. Meng, G. Ceder, C. P. Grey, W.-S. Yoon and Y. Shao-Horn, Electrochem. Solid-State Lett., 2004, 7, A155 CrossRef
- J. Breger, Y. S. Meng, Y. Hinuma, S. Kumar, K. Kang, Y. Shao-Horn, G. Ceder and C. P. Grey, Chem. Mater., 2006, 18, 4768 CrossRef CAS
- J. Breger, M. Jiang, N. Dupre, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575 CrossRef CAS
- Y. S. Meng, G. Ceder, C. P. Grey, W. S. Yoon, M. Jiang, J. Breger and Y. Shao-Horn, Chem. Mater., 2005, 17, 2386 CrossRef CAS
- H. H. Li, N. Yabuuchi, Y. S. Meng, S. Kumar, J. Breger, C. P. Grey and Y. Shao-Horn, Chem. Mater., 2007, 19, 2551 CrossRef CAS
- A. Van der Ven and G. Ceder, Electrochem. Commun., 2004, 6, 1045 CrossRef
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CAS
- F. Zhou, C. A. Marianetti, M. Cococcioni, D. Morgan and G. Ceder, Phys. Rev. B, 2004, 69, 4
- C. Delacourt, P. Poizot, J.-M. Tarascon and C. Masquelier, Nat. Mater., 2005, 4, 254 CrossRef CAS
- J. L. Dodd, R. Yazami and B. Fultz, Electrochem. Solid-State Lett., 2006, 9, A151 CrossRef CAS
- O. Le Bacq, A. Pasturel and O. Bengone, Phys. Rev. B, 2004, 69, 245107 CrossRef
- H. Ehrenberg, N. N. Bramnik, A. Senyshyn and H. Fuess, Solid State Sci., 2009, 11, 18 CrossRef CAS
- M. E. Arroyo-de Dompablo, U. Amador, M. Alvarez, J. M. Gallardo and F. Garcia-Alvarado, Solid State Ionics, 2006, 177, 2625 CrossRef CAS
- S. Curtarolo, D. Morgan, K. Persson, J. Rodgers and G. Ceder, Phys. Rev. Lett., 2003, 91, 4
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef
- C. C. Fischer, K. J. Tibbetts, D. Morgan and G. Ceder, Nat. Mater., 2006, 5, 641 CrossRef CAS
- J. Rouxel, Mater. Res. Soc. Symp. Proc., 1989, 135, 431 CAS
-
W. R. McKinnon, in Chemistry Physics of Intercalation, Ed. A. P. L. a. S. Flandrois, Plenum Press, New York 1987, vol. B172, pp. 181–194 Search PubMed
- J. Friedel, Adv. Phys., 1954, 3, 446
- C. Umrigar, D. E. Ellis, D.-S. K. Wang and H. M. Posternak, Phys Rev. B., 1982, 26, 4935 CrossRef CAS
- C. M. Julien and M. Balkanski, Mater. Res. Soc. Symp. Proc., 2003, 293, 27
- J. N. Reimers and J. R. Dahn, Phys. Rev. B, 1993, 47, 2995 CrossRef CAS
- J. N. Reimers, J. Power Sources, 1995, 54, 16 CrossRef CAS
- M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1997, 144, 3832 CAS
- G. Ceder, Y. M. Chiang, D. R. Sadoway, M. K. Aydinol, Y. I. Jang and B. Huang, Nature, 1998, 392, 694 CrossRef CAS
- A. K. Padhi, V. Manivannan and J. B. Goodenough, J. Electrochem. Soc., 1998, 145, 1518 CAS
- A. K. Padhi, K. S. Nanjundaswamy, C. Masquelier and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 2581 CAS
- A. K. Padhi, K. S. Nanjundaswamy, C. Masquelier, S. Okada and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1609 CAS
- Y. S. Meng, Y. W. Wu, B. J. Hwang, Y. Li and G. Ceder, J. Electrochem. Soc., 2004, 151, A1134 CrossRef CAS
- M. S. Islam, R. A. Davies and J. D. Gale, Chem. Mater., 2003, 15, 4280 CrossRef CAS
- K. Kang, D. Carlier, J. John, E. M. Arroyo, G. Ceder, L. Croguennec and C. Delmas, Chem. Mater., 2003, 15, 4503 CrossRef CAS
- B. J. Hwang, Y. W. Tsai, D. Carlier and G. Ceder, Chem. Mater., 2003, 15, 3676 CrossRef CAS
- A. Van der Ven and G. Ceder, Phys. Rev. B, 1999, 59, 742 CrossRef CAS
- J. Wolfenstine and J. Allen, J. Power Sources, 2005, 142, 389 CrossRef CAS
- C. Lyness, B. Delobel, A. R. Armstrong and P. Bruce, Chem. Commun., 2007, 46, 4890 Search PubMed
- S. Y. Chung, J. T. Bloking and Y.-M. Chiang, Nat. Mater., 2002, 1, 123 CrossRef CAS
- F. Zhou, K. S. Kang, T. Maxisch, G. Ceder and D. Morgan, Solid State Commun., 2004, 132, 181 CrossRef CAS
- N. Kenny, C. R. Kannewurf and D. H. Whitmore, J. Phys. Chem. Solids, 1966, 27, 1237 CrossRef CAS
-
P. A. Cox, The Electronic Structure and Chemisty of Solids,Oxford University Press, Oxford, 1987 Search PubMed
- T. Maxisch and G. Ceder, Phys. Rev. B, 2006, 73, 4
- T. Maxisch, F. Zhou and G. Ceder, Phys. Rev. B, 2006, 73, 6
- Y. N. Xu, S. Y. Chung, J. T. Bloking, Y. M. Chiang and W. Y. Ching, Electrochem. Solid-State Lett., 2004, 7, A131 CrossRef CAS
-
H. Jonsson, G. Mills, K. W. Jacobsen, Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions, Ed. B. J. Berne, G. Ciccotti, D. F. Coker, World Scientific Publishing Company, River Edge, NJ, 1998 Search PubMed
- K. Kang and G. Ceder, Phys. Rev. B, 2006, 74, 7
- A. Van der Ven, G. Ceder, M. Asta and P. D. Tepesch, Phys. Rev. B, 2001, 64, 17
- A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2000, 3, 301 CAS
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30 CrossRef CAS
- B. Xu and Y. S. Meng, 2009, in preparation.
- M. Wakihara, Electrochemistry, 2005, 73, 328 CAS
- K. Kang and G. Ceder, Phys. Rev. B, 2006, 74 Search PubMed
- A. Van der Ven, J. C. Thomas, Q. Xu, B. Swoboda and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 104306 CrossRef
- H. Xia, L. Lu and G. Ceder, J. Power Sources, 2006, 159, 1422 CrossRef CAS
- L. Wang, T. Maxisch and G. Ceder, Chem. Mater., 2007, 19, 543 CrossRef CAS
-
NIST, JANAF Thermochemical Tables ( 1998) Search PubMed
- M. D. Levi, Y. Gofer and D. Aurbach, Polym. Adv. Technol., 2002, 13, 697 CrossRef CAS
- E. Spila, S. Panero and B. Scrosati, Electrochim. Acta, 1998, 43, 1651 CrossRef CAS
- P. Novak, K. Muller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207 CrossRef CAS
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribiere, P. Poizot and J.-M. Tarascon, Nat. Mater., 2009, 8, 120 CrossRef CAS
- R. J. Meier, Comput. Mater. Sci., 2003, 27, 219 CrossRef CAS
- G. R. Goward, F. Leroux and L. F. Nazar, Electrochim. Acta, 1998, 43, 1307 CrossRef CAS
- F. Leroux, G. Goward, W. P. Power and L. F. Nazar, J. Electrochem. Soc., 1997, 144, 3886 CAS
- S. Kuwabata, S. Masui and H. Yoneyama, Electrochim. Acta, 1999, 44, 4593 CrossRef CAS
- A. Du Pasquier, F. Orsini, A. S. Gozdz and J. M. Tarascon, J. Power Sources, 1999, 81–82, 607 CrossRef CAS
- Y.-H. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237 CrossRef CAS
- Y.-H. Huang, K.-S. Park and J. B. Goodenough, J. Electrochem. Soc., 2006, 153, A2282 CrossRef CAS
- G. Férey, Franck Millange, Mathieu Morcrette, Christian Serre, Marie-Liesse Doublet, Jean-Marc Grenèche and Jean-Marie Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259 CrossRef CAS
- C. Janiak, Dalton Trans., 2003, 2781 Search PubMed
- L. J. Stuart, Chem. Soc. Rev., 2003, 32, 276 RSC
- J. L. C. Rowsell and O. M. Yaghi, Micropor. Mesopor. Mater., 2004, 73, 3 CrossRef CAS
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780 Search PubMed
- C. Combelles and M. L. Doublet, Ionics, 2008, 14, 279 CrossRef CAS
- C. Mellot Draznieks, J. M. Newsam, A. M. Gorman, C. M. Freeman and G. Férey, Angew. Chem., Int. Ed., 2000, 39, 2270 CrossRef CAS
- C. Mellot-Draznieks, J. Mater. Chem., 2007, 17, 4348 RSC
- C. Mellot-Draznieks, J. Dutour and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6290 CrossRef CAS
-
J. M. Tarascon, S. Grugeon, S. Laruelle, D. Larcher, P. Poizot, The key role of nano particles in reactivity of 3d metals oxides towards Li. Ed. G.-A. Nazri, G. Pistoia, in Lithium Batteries: Science and Technology, Springer, 2009 Search PubMed
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS
- Y. Oumellal, A. Rougier, G. A. Nazri, J. M. Tarascon and L. Aymard, Nat. Mater., 2008, 7, 916 CrossRef CAS
-
L. F. Nazar, O. Crosnier, in Lithium Batteries: Science and Technology, Ed. G. Nazri, G. Pistoia, Springer, 2009 Search PubMed
-
G. G. Amatucci, N. Pereira, in Lithium Batteries: Science and Technology, Ed. G. Nazri, G. Pistoia, Springer, 2004, pp. 247–269 Search PubMed
- H. Li, P. Balaya and J. Maier, J. Electrochem. Soc., 2004, 151, A1878 CrossRef CAS
- P. Poizot, S. Laruelle, S. Grugeon and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A1212 CrossRef CAS
- S. Boyanov, J. Bernardi, F. Gillot, L. Dupont, M. Womes, J.-M. Tarascon, L. Monconduit and M. L. Doublet, Chem. Mater., 2006, 18, 3531 CrossRef CAS
- F. Gillot, L. Monconduit and M. L. Doublet, Chem. Mater., 2005, 17, 5817 CrossRef CAS
- F. Gillot, S. Boyanov, L. Dupont, M. L. Doublet, M. Morcrette, L. Monconduit and J. M. Tarascon, Chem. Mater., 2005, 17, 6327 CrossRef CAS
- B. Mauvernay, M. L. Doublet and L. Monconduit, J. Phys. Chem. Solids, 2006, 67, 1252 CrossRef CAS
- R. E. Doe, K. A. Persson, Y. S. Meng and G. Ceder, Chem. Mater., 2008, 20, 5274 CrossRef CAS
- P. Balaya, A. J. Bhattacharyya, J. Jamnik, Y. F. Zhukovskii, E. A. Kotomin and J. Maier, J. Power Sources, 2006, 159, 171 CrossRef CAS
- Y. F. Zhukovskii, E. A. Kotomin, P. Balaya and J. Maier, Solid State Sci., 2008, 10, 491 CrossRef CAS
- H. Ellingham, J. Soc. Chem. Ind., 1944, 63, 125 Search PubMed
- P. L. Taberna, S. Mitra, P. Poizot, P. Simon and J. M. Tarascon, Nat. Mater., 2006, 5, 567 CrossRef CAS
-
R. A. Huggins, in Lithium Batteries: Science and Technology, Ed. G.-A. Nazri, G. Pistoa, Springer, 2009, pp. 270–296 and references therein Search PubMed
- S. Naille, R. Dedryvere, D. Zitoun and P. E. Lippens, J. Power Sources, 2009, 189, 806 CrossRef CAS
- I. Plitz, F. Badway, J. Al-Sharab, A. DuPasquier, F. Cosandey and G. G. Amatucci, J. Electrochem. Soc., 2005, 152, A307 CrossRef CAS
- L. Wang, F. Zhou, Y. S. Meng and G. Ceder, Phys. Rev. B, 2007, 76, 11
- P. Gibot, M. Casas-Cabanas, L. Laffont, S. Levasseur, P. Carlach, S. Hamelet, J. M. Tarascon and C. Masquelier, Nat. Mater., 2008, 7, 741 CrossRef CAS
- C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 2008, 7, 665 CrossRef CAS
- N. Meethong, H. Y. S. Huang, W. C. Carter and Y. M. Chiang, Electrochem. Solid-State Lett., 2007, 10, A13 CrossRef CAS
- N. Meethong, H. Y. S. Huang, S. A. Speakman, W. C. Carter and Y. M. Chiang, Adv. Funct. Mater., 2007, 17, 1115 CrossRef CAS
- C. A. J. Fisher and M. S. Islam, J. Mater. Chem., 2008, 18, 1209 RSC
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS
- K. Tibbetts, C. R. Miranda, Y. S. Meng and G. Ceder, Chem. Mater., 2007, 19, 5302 CrossRef CAS
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215 RSC
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621 CrossRef CAS
- Y. F. Zhukovskii, P. Balaya, M. Dolle, E. A. Kotomin and J. Maier, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 235414 CrossRef
- Y. F. Zhukovskii, P. Balaya, E. A. Kotomin and J. Maier, Phys. Rev. Lett., 2006, 96, 058302 CrossRef
-
Y. Wang, P. B. Balbuena, in Lithium Ion Batteries: Solid-Electrolyte Interphase, Ed. P. B. Balbuena, Y. Wang, Imperial College Press, London, 2004 Search PubMed
- O. Borodin, G. D. Smith and P. Fan, J. Phys. Chem. B, 2006, 110, 22773 CrossRef CAS
- O. Borodin, G. D. Smith, O. Geiculescu, S. E. Creager, B. Hallac and D. DesMarteau, J. Phys. Chem. B, 2006, 110, 24266 CrossRef CAS
- F. Zhou, T. Maxisch and G. Ceder, Phys. Rev. Lett., 2006, 97, 4
- C. Delmas, M. Menetrier, L. Croguennec, S. Levasseur, J. P. Peres, C. Pouillerie, G. Prado, L. Fournes and F. Weill, Int. J. Inorg. Mater., 1999, 1, 11 CrossRef CAS
- A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 2001, 148, A224 CrossRef CAS
- T. Ohzuku and A. Ueda, Solid State Ionics, 1994, 69, 201 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2009 |