Nanoscale design to enable the revolution in renewable energy
Jason
Baxter
a,
Zhixi
Bian
b,
Gang
Chen
c,
David
Danielson
d,
Mildred S.
Dresselhaus
*e,
Andrei G.
Fedorov
*f,
Timothy S.
Fisher
*g,
Christopher W.
Jones
h,
Edward
Maginn
i,
Uwe
Kortshagen
j,
Arumugam
Manthiram
k,
Arthur
Nozik
l,
Debra R.
Rolison
m,
Timothy
Sands
n,
Li
Shi
*k,
David
Sholl
h and
Yiying
Wu
o
aDrexel University, Department of Chemical & Biological Engineering, Philadelphia, PA 19104
bUniversity of California at Santa Cruz, Department of Electrical Engineering, Santa Cruz, CA 95064
cMassachusetts Institute of Technology, Department of Mechanical Engineering, Cambridge, MA 02139
dGeneral Catalyst Partners, Cambridge, MA 02139
eMassachusetts Institute of Technology, Department of Physics and Department of Electrical Engineering and Computer Sciences, Cambridge, MA 02139. E-mail: millie@mgm.mit.edu
fGeorgia Institute of Technology, Woodruff School of Mechanical Engineering and Petit Institute of Bioengineering and Bioscience, Atlanta, GA 30332. E-mail: andrei.fedorov@me.gatech.edu
gPurdue University, Birck Nanotechnology Center and School of Mechanical Engineering, West Lafayette, IN 47907. E-mail: tsfisher@purdue.edu
hGeorgia Institute of Technology, School of Chemical & Biomolecular Engineering, Atlanta, GA 30332
iUniversity of Notre Dame, Department of Chemical & Biomolecular Engineering, Notre Dame, IN 45556
jUniversity of Minnesota, Department of Mechanical Engineering, Minneapolis, MN 55455
kThe University of Texas at Austin, Texas Materials Institute and Departments Mechanical Engineering, Austin, TX 78712. E-mail: lishi@mail.utexas.edu
lNational Renewable Energy Laboratory, Center for Revolutionary Solar Photoconversion, Golden, CO 80401
mNaval Research Laboratory, Surface Chemistry Branch, Washington, DC 20375
nPurdue University, Birck Nanotechnology Center, School of Materials Engineering and School of Electrical and Computer Engineering, West Lafayette, IN 47907
oOhio State University, Department of Chemistry, Columbus, OH 43210
First published on 10th March 2009
Abstract
The creation of a sustainable energy generation, storage, and distribution infrastructure represents a global grand challenge that requires massive transnational investments in the research and development of energy technologies that will provide the amount of energy needed on a sufficient scale and timeframe with minimal impact on the environment and have limited economic and societal disruption during implementation. In this opinion paper, we focus on an important set of solar, thermal, and electrochemical energy conversion, storage, and conservation technologies specifically related to recent and prospective advances in nanoscale science and technology that offer high potential in addressing the energy challenge. We approach this task from a two-fold perspective: analyzing the fundamental physicochemical principles and engineering aspects of these energy technologies and identifying unique opportunities enabled by nanoscale design of materials, processes, and systems in order to improve performance and reduce costs. Our principal goal is to establish a roadmap for research and development activities in nanoscale science and technology that would significantly advance and accelerate the implementation of renewable energy technologies. In all cases we make specific recommendations for research needs in the near-term (2–5 years), mid-term (5–10 years) and long-term (>10 years), as well as projecting a timeline for maturation of each technological solution. We also identify a number of priority themes in basic energy science that cut across the entire spectrum of energy conversion, storage, and conservation technologies. We anticipate that the conclusions and recommendations herein will be of use not only to the technical community, but also to policy makers and the broader public, occasionally with an admitted emphasis on the US perspective.
Broader contextA major scientific and societal challenge of the 21st century is the conversion from a fossil-fuel-based energy economy to one that is sustainable. The energy challenge before us differs in three ways from past large scale challenges: the first is the large magnitude and relatively short time scale of the transition (a predicted doubling of energy demand by mid-century and a tripling by the end of the century); the second is the need to develop CO2-neutral, renewable energy sources; and the third is the cost-competitive aspect of the transition (insofar as the cost of energy to the consumer must be competitive with the fossil fuel energy supply being replaced). What is clear is that the science and engineering research communities working with industry, and policy makers (government, economists, social scientists) will have to educate the citizenry and get them to function collaboratively and globally to enhance the quality of life and to preserve the environment of our planet for future generations. Our team has prepared a technical article on the role of nanotechnology in our energy future aimed at guiding both our own community of scientists and engineers and our policy makers who interface with the public. |
Introduction
This article is an outcome of the recent Workshop on Nanotechnologies for Solar and Thermal Energy Conversion and Storage held in conjunction with the 3rd Energy Nanotechnology International Conference (August 10–14, 2008) and sponsored by the US National Science Foundation. The participants of the workshop, who are authors of this paper, are a small but highly interdisciplinary group of scientists and engineers with diverse backgrounds and technical perspectives (materials science, chemical, electrical and mechanical engineering, physics and chemistry), representing academia, national and federal laboratories, and industry. This group consists of researchers who are all actively engaged in nanoscale research and technology development activities focused on energy. To this end, the reader should view the contents as an opinion paper that highlights basic research ideas in specific energy technologies and defines a small set of cross-cutting themes for focusing research in nanoscience and nanotechnology that will lay the groundwork for a renewable energy revolution. The resulting publication aims at stimulating research and development activities to promote revolutionary advances in solar, thermal, and electrochemical energy conversion, storage, and conservation technologies required to achieve the projected annual demand for sustainable and environmentally responsible energy. A series of reports from the Department of Energy Basic Energy Science (DOE BES) topical workshops (available online at http://www.sc.doe.gov/bes/reports/list.html) held in the last several years provides a comprehensive roadmap of research needs, both tactical and strategic, over the broad spectrum of energy technologies and spans an entire range of different power generation scales. In contrast to these comprehensive works, our goal in writing this article is to bridge a disciplinary divide between the various energy conversion, storage, and conservation technologies and to formulate a common vision, together with identifying cross-cutting research themes, for nanoscale-enabled technologies that offer the potential to lead the revolution in energy conversion, storage, and conservation.Scale of the energy problem, economics and environmental constraints
To establish the context for our discussion, we first briefly review the current energy situation and the projections for future needs, as well as the time constraints that define the dynamics of the energy problem; we then set a pace for developing the required technological advances. Current average global primary power consumption sits at approximately 14 terawatt (TW, 14 × 1012 Joules s−1), with more than 80% of this energy coming from the carbon-dioxide-emitting fossil fuel trio of oil, coal, and natural gas, and less than 1% coming from carbon-free renewable power such as geothermal, wind, solar power, and biofuels. Due in large part to the rapid industrialization of emerging economies, such as China and India, global power demand is expected to double to about 25–30 TW by 2050 assuming the “business as usual” development scenario.1 If the proportion of this power provided by fossil fuels is to remain at the current level of ∼80%, the concomitant carbon dioxide loading of the atmosphere will pose a grave environmental threat to human civilization.The Intergovernmental Panel on Climate Change (IPCC) analyzed the potential impact of various scenarios of CO2 emission trajectories associated with the use of carbon-based energy sources on climate change. The conclusions are summarized in a series of assessment reports with the latest one in 2007 (available online at http://www.ipcc.ch/ipccreports/assessments-reports.htm). While no specific tipping point in carbon dioxide level is explicitly identified, these reports provide strong evidence that if the global average temperature increases beyond 2.5–3.0 °C, then serious and likely irreversible negative consequences to the environment will result, with direct impact on agriculture, water resources, and human health.2 Recent publications indicate that this temperature increase will likely occur at average atmospheric concentrations of CO2 as low as 350 ppm,3 with a more conservative estimate from the European Union study suggesting a critical level to be approximately 550 ppm.4 Regardless of which specific prediction is most accurate, the bounds provided by these projections are already very tight and a stabilization or reversal cannot be achieved unless a rapid transition to renewable energy sources occurs on a global scale with approximately 20 TW coming from low-cost carbon-free power by 2050.1 Realization of such a change will require a massive effort and the discovery and development of disruptive new technology solutions. The carbon-free options available include hydropower, nuclear power, geothermal power, wind power, solar power, biofuels, fossil-fuel power generation with carbon dioxide capture and sequestration, and conservation measures, given that the technology enabling each of these options is already or will shortly become available.
A comprehensive, up-to-date summary of the world energy portfolio and costs can be found in the UN-commissioned World Energy Assessment Overview (the latest 2004 update is available online at http://www.undp.org/energy/weaover2004.htm). Hydropower currently provides only 2% of global primary energy, equivalent to 0.3 TW. For the United States, however, hydropower does not project as a scalable carbon-free power solution because the majority of economically viable sites have already been exploited. Nuclear power currently provides 7% of global primary energy, or the equivalent of 0.9 TW. Nuclear power, although currently a relatively expensive power option at 6–7 ¢/kW-hr installed (and with unresolved significant problems associated with nuclear weapons proliferation and long-term radioactive waste disposal), will very likely play an incremental role through the 2050 timeframe, and its deployment will vary greatly from one country to another. Global geothermal power capacity currently accounts for approximately 10 gigawatt (GW, 0.01 TW). The global generating capacity of wind power at the end of 2007 was approximately 100 GW with annual additional installed capacity growing at the likely unsustainable rate of 30% a year. Wind power is economical in regions with high average wind speeds (4–8 ¢/kW-hr), but because of its intermittent nature, wind can only provide approximately up to 20% of electric power demand while maintaining grid reliability. Solar power currently provides only a very small fraction of global electric power generation, with just under 10 GW of installed capacity globally as of 2007, but newly installed capacity is growing at approximately 30% per year and accelerating. Solar power remains significantly more expensive than other electric power sources, with an installed cost of approximately $8/W and an electricity cost of 25–40 ¢/kW-hr; furthermore, solar power suffers, as does wind power, from intermittency.
Fossil-fuel power generation with carbon dioxide capture and sequestration (CCS) represents a potentially promising strategy to eliminate carbon dioxide emissions with only minimal changes to the global energy infrastructure. However, the practical and economic problems related to CCS are formidable. Long-term geological sequestration has yet to be proven effective, and using the current generation of carbon dioxide capture technologies, electric power prices would at least double from their current levels. Further, up to 30% more fossil fuel would be consumed to meet the parasitic energy load of CO2 capture technologies.5 Eliminating carbon dioxide emissions from the transport sector will require a combination of widespread deployment of carbon-dioxide-neutral cellulosic biofuels, development of synthetic hydrocarbon fuels based on atmospheric or captured CO2 and water (e.g., using solar photocatalysis), hydrogen-driven vehicles and infrastructure, and/or a shift to electric-powered vehicles, with carbon-free power provided by the aforementioned technologies. Carbon dioxide capture and sequestration continue to present a daunting challenge from a scientific and technological perspective as well as from related political and economic issues encountered by energy industries working with government and global partnerships.
Need for a step change development mode in energy and the role of nanoscience and nanotechnology
To summarize, our civilization faces perhaps the most pressing challenge of the last century or more to develop an economic and viable route to globally sustainable energy. Ultimately, we need to capture vast amounts of dilute and intermittent, but essentially unlimited, solar energy to sustain our forecasted needs, and to scale its conversion to high power densities and readily storable forms. Most importantly, this transformation must be accomplished in an economically and environmentally feasible manner in order to achieve a positive global impact. The massive cost and latency associated with putting in place new energy conversion, distribution, and storage infrastructure on such a large scale and in such a short time implies that major scientific discoveries and engineering developments should occur on a time scale of 10–20 years, which is shorter than customary for discovery-to-technology transitions.This paper emphasizes the urgency of new investment in research and development to support both sustainable growth of technologically driven societies and improved prosperity throughout the world in the 21st century and beyond. Some countries, with Germany, Australia, and Brazil being notable examples among the world's major economies and the European Union on a larger scale, have already made strong commitments to rapid and large-scale conversion to renewable energy and lead in the global investment for a sustainable energy future. However, the environmental aspect of the energy problem, which cuts across the countries and continents, demands action on a global scale. Those countries that establish regulatory policies and market incentives to promote early adoption of this vision will enjoy technological leadership as well as significant financial rewards and benefits in the long-term, but ultimately the winner will be the entire world.
This paper is not intended to cover the entire spectrum of energy technologies, but rather, it focuses on an important subset that arguably has the greatest potential for a major impact in solving various aspects of the energy problem by exploiting advances in nanoscale science and technology. For example, revolutionary nanotechnology-enabled photovoltaic materials and devices are needed to satisfy the world demand in sustainable electricity at a price level less than 10 ¢/kW-hr. Nanotechnology-based disruptive devices for electric energy storage, such as batteries and supercapacitors, are required to enable massive deployment of solar power by mid-century. In addition to solar energy, nanotechnology-enabled energy storage solutions would bolster the wind power industry and allow it to expand rapidly beyond its current level of deployment. Importantly, not only must the storage capacity and energy density be significantly improved, but also the cost of battery technologies must plummet from the current $1,000/kW-hr to a level approaching $10/kW-hr to become cost competitive. The cost of cellulosic biofuels needs to decrease to 75 ¢/liter gasoline-equivalent, which requires the development of disruptive new catalysts designed at the nanoscale. In addition to these innovations on the energy supply side, achieving a sustainable energy future will also require much more efficient energy use and conservation. The development of disruptive nanotechnology-enabled low-cost/high-performance solid-state lighting, thermal insulation, and waste heat recapture technologies, amongst many others, is of paramount importance as well.
Lastly, the economic reality is such that a transition from the existing, largely carbon-based energy economy to fully renewable energy will not happen instantaneously, even if efficient and affordable technological solutions to the problem were available today. The energy challenge, though widely discussed, simply has not yet attracted sufficiently broad public attention to demand the timely release of resources—both human and capital—by countries and industries around the world such that the daunting scope of the energy problem is matched by investment in renewable energy technologies and supporting infrastructure. For example, low-cost, energy-efficient nanotechnology-based carbon dioxide capture technologies (e.g., nano-engineered sorbent and membrane materials and separation devices) will be much needed in the near- and mid-term to pave the way for widespread deployment of low-cost, fossil-fuel energy production with CCS. The carbon dioxide capture problem is very important not only from an environmental perspective, but also because of its effect of increasing total energy consumption if implemented on a large scale. Therefore, while the discussions at the NSF workshop tended to focus on renewable energy, related issues such as carbon dioxide capture are an essential elements of any sound strategy toward a sustainable environment and energy future.
Nanoscale design for the energy revolution
An understanding of the general characteristics of fundamental energy carriers is important in appreciating the connections between nanotechnology and energy. Table 1 summarizes the characteristic length and time scales for energy carriers in liquids, gases and solids. These scales define the space–time envelope within which, if accessible, the manipulation of matter should critically affect the energy carrier transport and conversion processes, thereby enabling drastic improvement in the performance of energy systems. Because the length scales in Table 1 are generally of the order 1 to 100 nm, this size regime naturally falls into the domain of nanoscale science and engineering by virtue of the match between the length scales of the energy carriers and the materials that control their transport. We assert that revolutionary improvements (i.e., an order of magnitude or greater enhancement when scaled to practical sizes) in the delivery of usable energy can be facilitated by advancing nanoscale design of both materials and associated energy conversion processes and systems.| Wavelength/nm | Mean free path/nm | Relaxation time/ns | |
|---|---|---|---|
| a refers to exciton electronic-vibration modes. b refers to skin penetration depth. c refers to de Broglie wavelength). | |||
| PHOTONS (solar/thermal radiation) | |||
| • in liquids/gases | ∼100–30,000 a | >1000 b | >10−6 |
| • in semiconductors | ∼25–30,000 a | >10 b | ∼10−7–10−6 |
| • in conductors/metals | — | ∼0.1–10 b | ∼10−10–10−9 |
| ELECTRONS | |||
| • in semiconductors/dielectrics | ∼1–50 | ∼1–500 | ∼(1–10) × 10−3 |
| • in conductors/metals | ∼0.1–1 | ∼1–10 | ∼(10–100) × 10−6 |
| PHONONS | |||
| • in semiconductors/dielectrics | ∼0.5–10 | ∼1–500 | ∼10−3–1 |
| MOLECULES/IONS | |||
| • in gas/plasma | 10−2–1 c | ∼103–107 | ∼1–100 |
| • in liquid/electrolyte | — | ∼0.1–1 | ∼10−3 |
| • in solid/electrolyte | — | ∼0.1–1 | ∼10−3 |
Obviously, not all renewable technologies can directly benefit from nanoscale design—wind, hydrothermal and hydroelectric technologies are notable examples. In the following sections, we focus on an important and large subset of renewable technologies, broadly placed in three overlapping categories (energy conversion, energy storage, and energy conservation; see Fig. 1), whose performance can be revolutionized by ‘nanoscale’ design. We concisely review salient physical principles underlying each technology and identify unique opportunities that are offered by understanding how energy is transferred and converted from one form to another, emphasizing the unique physicochemical features that only become accessible on the nanoscale, to achieve improved functionality and efficiency. Wherever possible, we define the thermodynamic limits and specify a transformational change(s) needed for each technology in terms of specific performance targets and economics.
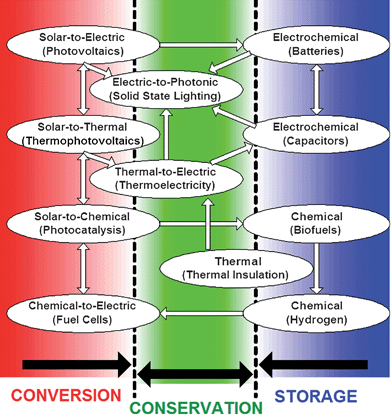 | ||
| Fig. 1 Portfolio of solar/thermal/electrochemical energy conversion, storage, and conservation technologies, and their interactions, that are the focus of the discussion. The electric grid is also shown in this figure as a network of connecting multiple elements (technology boxes) and allowing them to act a coherent whole. | ||
The technologies considered in this paper differ in the magnitude to which they will contribute to the supply of energy and their respective horizon for implementation, thereby impacting a carbon-neutral energy future on different time scales. In the near-term, the energy conservation technologies will likely have a major impact, especially for economically developed countries which are major energy consumers today, because every watt conserved diminishes demand by an equal amount. These conservation measures include the development and commercialization of advanced thermal insulation materials, waste heat utilization using thermoelectrics, and adaptation of transformational technologies, such as solid-state lighting. Also, biofuels will likely provide a sizable fraction of the renewable energy mix in the near-term, especially in some countries with an abundance of appropriate feedstocks with a rapid replication cycle (e.g., the very successful sugar-cane-based ethanol production in Brazil). In the mid-term, hydrogen fuel and electrochemical energy conversion devices (e.g., fuel cells) are expected to reach the point of being competitive in the energy market, especially for the transportation sector. For electric power generation, the second/third generation of solar cells (photovoltaics and thermophotovoltaics) coupled to high density and capacity electrochemical energy storage are expected to become price competitive, environmentally friendly and sustainable alternatives to today's fossil-fuel-based power generation. Finally, if we are successful, the very long-term energy future will rely on solar fuels as truly sustainable energy carriers, which would utilize only renewable feedstocks, such as water and carbon dioxide as reagents to produce high density, synthetic liquid fuels.
Transforming this vision of a long-term sustainable energy future into reality will require major breakthroughs in both understanding and implementation of solar-driven catalytic processes, such as photolysis/photocatalysis and artificial photosynthesis. This time-to-success categorization of the technology portfolio should guide the research community and decision makers in setting the priorities of R&D efforts (e.g., balancing fundamental research with development/demonstration activities), but it does not imply that we should prematurely narrow our options and invest only in a few of these technologies with an expected shorter-term payoff. The long-term consequences of such short-sighted decisions can negatively affect our ability to build a sustainable energy future. A coherent short-term plan and a long-term strategy are necessary because of the magnitude of the energy challenge and its urgent time scale.
In the following critical analysis for each energy conversion/storage/conservation technology, we first (1) describe the basic physical principles and essential energy carriers/interactions involved, then (2) identify the fundamental physical/chemical mechanism(s) defining the conversion and storage efficiencies and set the thermodynamic performance limits if known, and informed by the foregoing, (3) define nanotechnology-enabled critical research paths to revolutionary improvements in energy conversion, storage, and conservation. As an integral part of the discussion, we also identify the challenges and possible solutions to low-cost and scalable manufacturing of such nanotechnology-enabled technologies. Finally, we conclude each technology section by setting the critical research goals for each technology in the near-term (2–5 years), mid-term (5–10 years) and long-term (>10 years). Importantly, some of these goals set very specific performance targets, while others are broadly defined, allowing us to identify cross-cutting, fundamental research themes. Such exercises are necessary and should be periodically revisited, as progress is made and knowledge and experience are accumulated.
Energy conversion
From the above discussion, energy conversion on the most fundamental level can benefit enormously by tapping into a unique opportunity of matching/tuning the length and time scales of energy carriers involved in conversion processes when performed on the nanoscale (Table 1). At the same time, as the sizes of the energy conversion devices and active features shrink, the interface density (both internal/grains and external/heterojunctions) increases in importance and becomes the dominant factor defining the conversion efficiency. Understanding the mechanisms of nanoscale energy exchange between carriers and their transport across interfaces should lead to new creative approaches to the development of energy conversion devices of unprecedented efficiency, approaching thermodynamic limits. Important for successful commercialization are not only scientific insights into conversion mechanisms (new routes/approaches to managing energy exchange between the energy carriers), but also the engineering aspects of energy systems, including low cost and efficient synthesis/manufacturing techniques and their scalable deployment to yield products with a long life cycle.Solar photovoltaics: solar cells
Solar energy is abundant. In only 1.5 hours, the earth's surface receives as much energy as the world's population consumes in an entire year. Covering less than 1% of the earth's landmass with 10% efficient solar cells would provide 15 TW of electrical power,6 the projected need of carbon-free renewable energy by 2050, or half of the total energy consumption.1 However, electricity produced by direct solar-to-electricity conversion, also termed photovoltaics, is presently expensive, and the economic disincentives of utilizing solar electricity on a large scale remain overwhelming. For more than three decades, the cost of solar electricity has followed the “80%-learning curve”, as shown in the log-log plot of Fig. 2, according to which the photovoltaic module price drops by 20% for every doubling of the module production level. In order to reduce the cost per watt of solar electricity by a factor of ten and to achieve cost parity with coal-based electricity within the next decade (not accounting for the cost of carbon dioxide capture and sequestration), a revolution in photovoltaic technology as well as a breakthrough in the underlying science is needed to enable a radical disruption of the “80% learning curve.”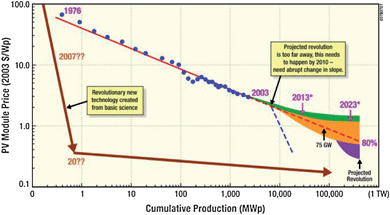 | ||
Fig. 2 Figure and caption reproduced from ref. 6: Learning curve for solar cells (solid red line). Since 1976, the module price has been dropping by 20% for every doubling of module production (80% learning curve). A few historical and predicted time points are labeled, indicating the pertinent year. Extrapolation of this historical trend into the future (dashed red line), plus a projected technological revolution at an annual production level of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 megawatt power (MWp) (purple curve) results in a prediction that $0.40/Wp would not be reached for another 20–25 years. Reaching $0.40/Wp sooner to accelerate large-scale implementation of PV systems will require an intense effort in basic science and associated technology development to produce a technological revolution that leads to a new, as-yet-unknown technology. This technological revolution requires a major reduction in the ratio of the PV module cost per unit area to the solar cell efficiency. The brown arrows show schematically the type of abrupt change in slope that is necessary. If this change were to begin now, it would be represented by the dashed blue line. 000 megawatt power (MWp) (purple curve) results in a prediction that $0.40/Wp would not be reached for another 20–25 years. Reaching $0.40/Wp sooner to accelerate large-scale implementation of PV systems will require an intense effort in basic science and associated technology development to produce a technological revolution that leads to a new, as-yet-unknown technology. This technological revolution requires a major reduction in the ratio of the PV module cost per unit area to the solar cell efficiency. The brown arrows show schematically the type of abrupt change in slope that is necessary. If this change were to begin now, it would be represented by the dashed blue line. | ||
Crystalline silicon photovoltaic modules have dominated the terrestrial market. However, diverse applications support a variety of current solar cell technologies, ranging from inexpensive organic devices with efficiencies of a few percent, to vacuum-deposited thin film technologies with efficiencies of ∼10–15%, to crystalline wafer-based cells with ∼20% efficiencies, to monolithic multi-junction solar cells with efficiencies up to ∼40%.7 Scientific breakthroughs in extracting more energy per photon and in coaxing cheap materials to perform as well as expensive ones will enable radical improvements over the past learning curve.
The basic principle of solar cell operation is the absorption of photons by a semiconductor to generate electron–hole pairs which are then transported to opposite polarity contacts to generate electrical current. Mismatch between the semiconductor bandgap energy and the solar spectrum results in losses, where carriers produced by photons with energy above the bandgap lose energy by thermalizing to the band edge, and photons with energy less than the bandgap are not absorbed at all. Considering these losses and the solar insolation spectrum, Shockley and Queisser calculated a theoretical maximum efficiency of 32% for a solar cell made from a single absorber.8 The absorber thickness is determined by the balance between light absorption (which requires a larger thickness) and charge collection (which requires a smaller thickness). High purity absorbers allow charge collection over lengths of 10 nm–50 µm, which is sufficiently large for efficient light harvesting; however, processing of such high purity materials is expensive. Dramatically reducing the cost per watt of photovoltaic devices will likely require a combination of improving solar cell conversion efficiencies and reducing the cost of their manufacture. Nanotechnology can make significant contributions on both fronts.
Among the physical barriers that limit solar cell efficiencies, the fast energy loss by hot carriers may be the most serious one.9,10 Hot carriers are produced when solar photons with energies significantly exceeding the bandgap energy of the active semiconductor material in a solar cell are absorbed to create an electron–hole pair. When the electron and/or hole are generated with energies significantly above their respective band edges, energy relaxation is very fast, typically on the order of picoseconds, and the excess energy is converted into lattice vibrations (phonons), leading to undesired heating of the solar cell whose operation is less efficient at a higher temperature. One approach to addressing this problem is to employ Si nanocrystals with different bandgap values to capture the full solar spectrum.11 Another mechanism that is believed to preempt the relaxation of hot carriers in semiconductor nanocrystals is the generation of two or more (up to n) electron–hole pairs in response to absorption of only a single photon with an energy of n times the bandgap energy of the nanocrystals. This mechanism, also known as multi-exciton generation (MEG), is particularly pronounced in quantum confined nanocrystals10 and has been demonstrated in a range of materials.12–15 Theoretical predictions indicate that MEG has the potential to enhance the conversion efficiency of solar cells up to 44%, which is well beyond the Shockley–Quiesser limit.16 At present, however, MEG has not yet been demonstrated in actual devices, and strategies for exploiting MEG need to be developed. The recent demonstration of “quantum cutting” in silicon nanocrystal arrays, an effect in which the excitation energy of an energetic photon is split to produce excitons in two separate nanocrystals, may present an interesting avenue.17
A different concept of exploiting hot carriers is based on the premise that hot carriers can be extracted from a solar cell before cooling to their respective band edges. Theoretical studies indicated that hot carrier utilization through energy selective contacts can significantly improve solar cell efficiencies by increasing the open circuit voltage of the solar cell.18,19 However, to date, experimental demonstrations of this effect in actual nanocrystal-based devices are absent.
An alternative, economically-plausible approach is to design nanostructured architectures that can reduce solar cell cost by using inexpensive materials and processing, while improving extraction of charge carriers. Charge-carrier recombination within the cell is a ubiquitous problem that degrades the photovoltaic efficiency. Recently, research has focused on using one-dimensional nanostructures, such as nanotubes and nanowires for increasing the carrier collection efficiency. Nanowire arrays have been used in dye-sensitized solar cells,20,21 extremely thin absorber solar cells,22 and radial p–n junction solar cells23 to decouple the direction of light absorption and charge separation. This design is more tolerant of defects and impurities in the solar cell materials than conventional planar p–n junction cells, thereby reducing processing costs. An example of a designed nanostructured architecture is the highly successful dye-sensitized solar cell (DSC), which uses a network of sintered titania nanoparticles as a transport medium for photo-generated electrons.24,25 Further improvement can be achieved by replacing the network of nanocrystals with an array of nanowires, which drastically reduces the number of grain boundaries impeding fast electron collection, so that the electron extraction rate can be increased by orders of magnitude and the probability of charge carrier loss through recombination can be reduced correspondingly. While the viability of this approach has been demonstrated, a challenge is to design nanowire structures with comparable surface areas as in nanocrystalline arrays or to design molecular or inorganic absorbers with higher extinction coefficients in order to achieve the same absorption as with chromophores attached to the nanoparticle film. The iodide/triioide redox couple has proven to be highly successful in dye-sensitized solar cells (DSC), but the long-term stability of the electrolyte will be a challenge because of both iodine's photoreactivity and the sensitivity of the dye molecules to water. Effective sealing to prevent water diffusion into the cell, as well as evaporation of the liquid solvent, is critical to long-term cell performance. Problems with sealing liquid DSCs can be mediated by replacing the liquid electrolyte with solid (or quasi-solid) hole-conducting media, such as polymeric, gel-type or solid electrolytes; but intimate interpenetration both of the sensitized oxide and the solid electrolyte is a major challenge and so far this strategy has resulted in lower efficiencies.26
Nanotechnology may provide attractive routes to reducing the cost of solar cell manufacture through novel methods of manufacturing thin film solar cells, which may reduce the manufacturing costs by a factor of 5–10.27 Nanocrystalline films can be produced with inexpensive solution processes and such films can achieve electronic properties that rival those of vacuum-deposited thin film semiconductors.28 With such techniques, “artificial semiconductor” films, i.e. films of quantum-confined yet interacting semiconductor nanocrystals, may be formed at lower cost and may provide the advantage of optical properties that are tunable through the size of the constituent nanocrystals. In a more pragmatic approach that emphasizes advanced manufacturability, “inks” of colloidal semiconductors may be used to exploit the low cost of commercial printing and coating techniques to produce thin film semiconductors that are subsequently transformed into conductive polycrystalline films by sintering.29–31
In the meantime, industry is moving fast forward. With recent discoveries of new, exciting physics in nanomaterials and the push being made by industry to capitalize on these discoveries, enhanced research support and intelligent incentive schemes, following those examples from Germany and Japan which have been shown to be successful in broadly deploying solar cell technologies, may transform photovoltaics into a major, environmentally friendly, safe, and affordable form of harvesting nuclear energy from the sun. In addition, in order for PV energy conversion to play an important role in meeting the global energy challenge, methods and infrastructure for storing the solar-derived electricity will need to be developed. Potential solutions are storage in the electric grid and in chemically based fuels, both of which will need to be investigated.
In conclusion, nanotechnology provides multiple exciting avenues to increase the efficiency of solar cells and significantly reduce their manufacturing cost. An important near-term research goal (2–5 years) is to assess which new physical phenomena such as MEG, “quantum cutting,” and enhanced carrier transport in nanostrutures may contribute to low-cost, high-efficiency solar cells. A mid-term research goal (5–10 years) is to explore which of these new physical effects can be realized in environmentally benign, widely abundant materials. An important long-term research goal (>10 years) is to translate scientific breakthroughs into novel manufacturing processes for low-cost, high efficiency “Third Generation” solar cells. Achieving this goal will require close collaboration between the research community and solar cell manufacturers for the smooth transition of technological advances.
Solar photocatalysis: solar fuels
Solar radiation can be used to drive heterogeneous electrochemical reactions at the surface of an optically active photocatalyst, resulting in the production of solar fuels, including hydrogenvia photolytic water splitting or hydrocarbons (methane, methanol, etc.) via CO2 photoreduction with water at low (ambient) temperature and pressure.32 This approach provides a more direct and therefore an economically attractive (more efficient and cheaper) way to produce sustainable fuels (e.g., hydrogen) as compared to electrolysis driven by photovoltaic cells. Further, when a hydrocarbon fuel is the ultimate product of a photochemical reaction driven with only solar energy input, carbon dioxide is consumed as a reagent, thus resulting in simultaneous energy conversion and storage (solar-to-chemical) with a negative CO2 footprint. This energy- and carbon- efficiency is not only a major attraction from an environmental prospective, but also appealing as a potentially viable long-term route to sustainable transportation, based on high-density synthetic liquid fuels while using a transportation and fuel-distribution infrastructure that are already in place.33Fig. 3 shows a schematic of the key processes involved in heterogeneous photocatalysis and their interactions. A photocatalytic reaction generally employs a semiconductor-based catalyst, which can be excited (i.e., promoted from the ground state to an excited state) by the absorption of a photon. Depending on the choice of the photocatalyst, visible (VIS) or near-ultraviolet (UV) light has sufficient photon energy to match the energy gap needed for excitation of the catalyst from its ground state to a much more interactive excited electronic state. Generally, catalytic oxidation–reduction reactions can proceed along one of three different and competing pathways: (1) radiative deactivation with emission of a quantum of light; (2) radiationless deactivation produced by vibrational relaxation of the lower state; and (3) chemical reactions involving reactant molecules and photogenerated electrons/holes on the catalyst surface.
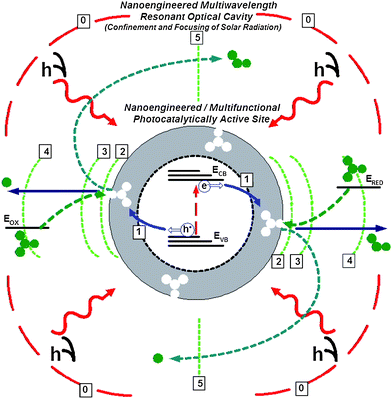 | ||
| Fig. 3 Schematic of an “idealized” photocatalytic active site with interacting energy conversion processes and key performance-controlling interfaces: (0) Semitransparent interface forming the light confinement cavity around the catalytically active site; (1) Charge (electron–hole) injection interface separating the light absorbing semiconductor (source of space-separated electron/holes) and the catalytically active surface layer (an interaction “hot” zone for electron/holes and surface-adsorbed/catalyst-coupled reduced/oxidized species); (2) Interface between the adsorbed (surface immobilized) reactant and the Debye double layer; (3) Interface between the Debye double layer and the reactant diffusion layer; (4) Interface between the diffusion layer and the external environment; and (5) Separation interface (e.g., chemically-selective membrane) to separate reaction products, thereby avoiding recombination. | ||
Until now, demonstrated photocatalytic efficiencies for hydrogen production are only at the few-percent level, and even lower for the synthesis of more complex hydrocarbon fuels. The ideal maximum thermodynamic efficiencies of direct solar photocatalysis and the solar photovoltaic (PV)-driven electrochemical route to fuel production should be the same (32% at one sun), and assuming 100% efficiency of electrolysis. However, practically speaking, the PV + electrolysis approach is a two-step process so the overall efficiency is the product of two lower numbers (∼20% for PV and ∼85% for electrolysis), resulting in ∼17% overall efficiency. Photocatalytic fuel generation (photoelectrolysis) is a single-step process and thus could, in principle, be higher in the actual efficiency if a photoelectrolysis system could be found that is photochemically stable and has an optimum bandgap and an excellent redox catalyst. Also, the photoelectrolysis system could, in principle, be less expensive and have a simpler engineering design with lower balance-of-plant (BOP) costs. While thermodynamically possible, such an efficient photoelectrolysis system does not yet exist, but these advantages should motivate significant basic research to make such a photoelectrolysis system economically viable for solar energy conversion and storage.
A particular challenge relevant to fuel production is to couple the oxidation and reduction reactions, which frequently require multiple electrons and holes, in order to drive an endothermic reaction that requires an energy input (such as solar-based H2O splitting). There have been significant efforts to develop new compounds/materials for the photoelectrochemical splitting of H2O to produce H2 and O2. One of the most important of these materials is TiO2, which has been widely studied since the discovery of the phenomena of photo-assisted H2O splitting in 1972.34 A continued focus on titania-based photocatalysts is due to the low cost of TiO2, its chemical inertness, and photostability, which are all essential prerequisites for the commercialization of this technology. When a TiO2 catalyst is illuminated by light of wavelength <370 nm (>3.2 eV in energy), the valence-band electrons of the TiO2 can be excited to the conduction band, creating highly reactive electron (e−) and hole (h+) pairs. These electrons and holes migrate to the TiO2 surface and are trapped at different sites facilitating surface (electro) chemical redox reactions involving adsorbed water molecules and resulting in the production of molecular hydrogen and oxygen.
However, whereas the anatase and rutile forms of TiO2 have many desirable features, they do not absorb solar photons well. Thus a number of researchers have focused on shifting the bandgap energy so as to create a better photocatalytic absorber of solar photons that can be used to efficiently split H2O.35 A very promising and relatively benign approach has been to dope TiO2 with nitrogen to form the oxynitride, TiO2−xNx, which can be made to absorb at longer wavelengths extending well past 500 nm (<2.4 eV) and be photocatalytic at these wavelengths.36 It has been shown that the nanoscale route is particularly effective to synthesize the visible-light-active oxynitride nanostructures.37,38 To facilitate the charge separation and reaction kinetics, these TiO2−xNxnanostructures are further seeded with noble metal-based clusters and transition metal ions to harvest the “reactive” electrons and to promote reduction and oxidation reactions, respectively.39 Creating a multi-phase nanocomposite (e.g., a smaller bandgap rutile phase mixed with a more reactive anatase phase of TiO2) has been shown to enhance the photocatalytic activity.40 Another example of such a nanocomposite photocatalyst is the reduced nickel oxide co-catalyst integrated with La-doped NaTaO3 which was shown to have >50% quantum yield of H2 production with >400 hours of stable operation.41 Finally, a micro/nano-structure of the catalyst support (e.g., zeolites) has been shown to greatly increase selectivity and efficiency of the reaction for methane/methanol synthesis from CO2 and H2O.42 In addition to enhancing the reaction kinetics, the ability to stimulate a preferential reaction pathway is of much interest. For example, it has been reported that water photooxidation on TiO2−xNx is not induced by the oxidation of a surface OH group with photogenerated holes but rather by the nucleophilic attack of an H2O molecule on a surface hole accompanied by oxygen–hydrogen bond breaking.43,44 Although significant advances have been made in using selective doping of semiconductor photocatalysts to modify their optical properties for enhanced visible-light response, a substantial improvement in catalytic properties is yet to be achieved—an improvement which would result in increased photochemical yield of reactions. Further, long-term chemical stability of doped-TiO2 photocatalysts remains to be demonstrated.
The optimal match of time and length scales for energy carrier transport and conversion, which can be achieved via nanoscale design of the photocatalytic site, has the potential to eliminate the bottlenecks limiting the rate of photocatalytic reactions, thus vastly improving the prospect for high throughout solar fuel production once a suitable photocatalyst is perfected. At present, the chemical kinetics is the rate-limiting step for the photocatalysis process, both with respect to selectivity towards desired products as well as the reaction conversion/yield. In the long run, once a suitable catalyst (i.e., with long-term activity and fast kinetics/excellent selectivity) is developed, the reaction throughput may become photon-limited. In this case, the use of solar light concentrators would be beneficial. It is important to emphasize that controlling at the nanoscale both the intrinsic properties of materials (e.g., nitrogen doping of TiO2 to broaden its spectral response into the visible range; composite semiconductor/metal materials for efficient charge-separation and localization) as well as structural characteristics of the catalyst and its support are equally important in the effort to develop an improved photocatalytically active site (Fig. 3).
Finally, it is instructive to mention two other complementary-to-photocatalysis techniques for the production of solar fuels, which could also benefit significantly from advances in the nanoscale design of catalysts. One of these techniques is hydrogen production from seawater using electrolysis on Ni/Co/Zr-based catalysts followed by a methanation reaction consuming CO2 and yielding methane fuel.45 The other method is based on a two-step thermochemical cycle for hydrogen generation from water using ZnO/Zn or MnO/Mn redox chemistry in conjunction with large-scale solar energy concentration by an optical system to achieve a desired temperature of operation and to increase the ideal Carnot cycle efficiency.46 One practical disadvantage of both processes resulting in greater cost of equipment construction and operation is the above-ambient temperature of operation, which exceeds 523 K for electrolytic methane synthesis and as high as 2000 K for thermochemical cycles, further exacerbated by highly corrosive chemistry. A potentially significant advance has been recently reported for room-temperature water oxidation using a phosphate/Co-ion-based electrocatalyst driven by a photovoltaic cell operated in neutral (pH = 7) water.47 Yet another promising approach to artificial solar-driven hydrocarbon fuel photosynthesis from water and carbon dioxide is to decorate a large surface area of nanoporous silica scaffolds with well-defined inorganic polynuclear units (e.g., a bi-nuclear metal-to-metal charge-transfer chromophore), with a precisely controlled discrete molecular structure resulting in desired optical and charge transfer properties, which act as visible light electron pumps coupled to a multi-electron transfer catalyst.48,49
In summary, making fuels from water and the sun is a powerful approach that solves many energy and environmental problems all at once, if it can be made into a practical technology in terms of quantity and cost. This vision is compelling, but the implementation is difficult so it will take time. So far, we have made less progress in this area than in others (wind, hydroelectric, biofuels), yet photocatalysis may in the long-term have greater impact. We need new strategies that will contribute to advancement of the science and technology of photocatalytic fuel production on all time scales.
The near-term goal (2–5 years) is to greatly improve the electrocatalytic properties of semiconductor-based photocatalysts, aiming at a rational design of a reduction electrocatalyst selectively tailored to a specific fuel production reaction, especially those involving multiple electron transfer (i.e., going beyond hydrogen generation from water toward direct liquid hydrocarbon fuel production), and 2–3 orders of magnitude improvement in the catalyst activity and kinetics, which limit the reaction yield.
For the mid-term (5–10 years), the goal is to develop a theoretical strategy along with suitable materials including nanostructures for a much improved “active” site for broad-band-light capture and utilization, which includes the low-loss trapping/confinement strategy for solar radiation and an intimate integration with a wavelength-tunable photocatalyst to achieve light-to-charge conversion efficiencies approaching that of multi-junction solar cells.
The long-term goal (>10 years) is to economically implement the above-stated scientific advances, including development of simple and robust catalyst synthesis methods for solar photocatalysis via semiconductor nanoparticle modification with demonstrated long-term catalyst stability, desirably at ambient pressure and temperature, with manufacturability on a large scale at competitive costs.
Because this technology lies further into the future, we can expect that the detailed goals will change rapidly as this technology area develops. However, the near-to-long-term research goals have a common theme in the effort to design an optimal photocatalyst for renewable fuels: the exploration and exploitation of multiple co-existing functionalities of photocatalyst nanostructures by virtue of both tuning the material's chemical composition via doping and property modification by structural changes – to design an optimal photocatalyst for renewable fuels. Nature is largely based on photocatalysis, so part of the research process in this area should be devoted to studying how Nature does it and what we can learn from Nature that will help the human implementation process.
Solar thermal: Solar thermophotovoltaic & thermoelectric conversion
Solar radiation is a high temperature heat source, and hence one can think of converting solar radiation first into a terresterial heat source, and then into electricity. Current solar thermal-to-electricity conversion occurs via mechanical engines. However, it is also possible to use solid-state technology to convert the heat into electricity. One solid-state approach is solar thermophotovoltaic (TPV) cells (Fig. 4),50,51 which use solar photons to heat up an intermediate selective absorber (Fig. 4b), in order to re-emit photons near the bandgap of a PV cell, as shown in Fig. 4c and 4d. Such solar TPV cells have a theoretical maximum efficiency of 85.4%, nearly identical to that of infinite multijunction cells (86.8%), without the complication and associated high cost of growing multijunction PV cells. Instead, solar TPV cells shift the focus from the growth of multijunction PV cells to the design of selective absorber–emitters. Despite considerable promise, TPV research worldwide remains scarce.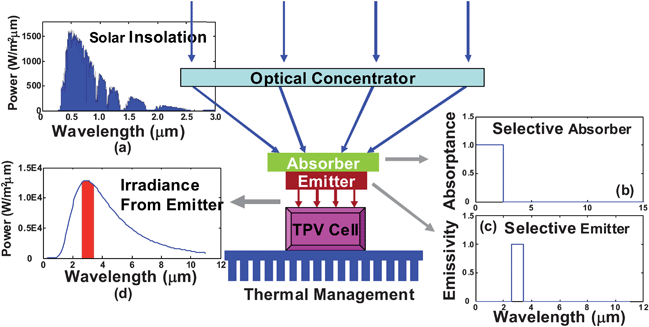 | ||
| Fig. 4 Illustration of a solar thermophotovoltaic (TPV) converter. Broadband solar insolation (inset a) heats up a selective absorber (inset b), which conducts heats to a selective emitter (inset c), which emits in a narrow band (inset d) into a PV cell to generate electricity. The theoretical limit of the efficiency of a single junction solar TPV converter is 85.4%. | ||
Another solid-state solar thermal-to-electricity conversion technology is solar thermoelectrics (TEs).52 In solar TEs, solar energy is absorbed by a spectrally selective surface that absorbs solar radiation but does not lose heat via thermal emission. The heat absorbed by the selective surface is coupled to the TE devices via direct contact to the devices. As discussed below, TE devices generate power via the Seebeck effect – a process whereby a temperature difference across a material generates a voltage,53 the most common examples being thermocouples used for temperature measurements. The efficiency of TE power generation is given as
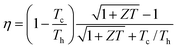 | (1) |
PV cell and TE materials, which are at the heart of solar TPV and solar TE, respectively, are discussed in detail in other sections of this paper. One common interest of solar TPV and solar TE is in the spectral control of photons. Both solar TE and TPV need surfaces that can absorb solar insolation, but do not lose heat via their own thermal emission. Although current selective solar absorbers used in hot-water systems can reach a solar absorptance of ∼0.95 and emittance of ∼0.05, the spectral control becomes more challenging in the medium-to-high temperature range. For solar TPV, spectral control is also needed to deliver photons emitted from the solar absorber to the photovoltaic cell. There has been more research in this area due to the broader interest in TPV other than solar TPV. Two strategies have been pursued in the past. One is direct control of emission by using selective emitters based on new materials,60,61 novel surface structures,62 interference filters63 and photonic crystals.64 All of these structures have characteristic length scales comparable or smaller than the wavelength of the maximum emitted power of solar radiation. The major difficulty with this approach lies in the high temperature operation of the emitters, under which materials can lose their stability. The other strategy is spectral control on the PV cell side, by reflecting back unwanted photons, mostly below bandgap photons. This approach has the advantage that spectral reflectors on the cell side are near room temperature operation. However, the spectral control structures, such as multilayer and plasmonic reflectors, can increase the optical losses on the PV cell side. Fundamental research in developing high-efficiency selective surfaces that can operate at high temperatures is needed in realizing the full potential of solar TE and TPV technologies. Photonic crystals and electromagnetic metamaterials are promising concepts to explore in developing selective surfaces, and these structures are usually in the tens to hundreds nanometer scale. Here, the key challenges are developing structures and materials that can be fabricated at a low cost, while at the same time, have highly desirable spectral characteristics and can operate reliably at high temperatures.
In the near-term (2–5 years) solar thermoelectric devices with an efficiency of ∼ 7% and solar TPV devices with an efficiency of ∼20% should be demonstrated to attract more government and private support for the development of the technologies. These tests should also show the low-cost and/or high efficiency potential of this technology. Low-cost solar selective surfaces with less than 3% emittance between room temperature and 250 °C should be developed.
For the mid-term (5–10 years), low-cost selective surfaces working in the mid temperature range (300–500 °C) with solar absorptance >0.9 and emittance less than 5% should be developed. Using such selective surfaces and advanced TE materials, solar TE with >15% efficiency should be demonstrated, and solar TPV should demonstrate an efficiency of up to 30%.
The long-term goal (>10 years) is to demonstrate solar TE systems with efficiency >20% and solar TPV with efficiency >50%. Long-term materials and structural reliability should be confirmed prior to scale-up.
Electrochemical: fuel cells
Fuel cells are direct electrochemical fuel-to-electrical energy conversion devices and they are attractive for a variety of applications ranging from portable to automotive to stationary power because they offer higher efficiency (50–60%) compared to conventional technologies such as internal combustion engines (∼35% efficiency).65,66 Among the various types of fuel cells, the proton-exchange membrane fuel cells (PEMFC), direct methanol fuel cells (DMFC), and solid oxide fuel cells (SOFC) are actively under research and development65,66 as they employ solid proton-conducting or oxide-ion conducting electrolytes that could make the operation and maintenance of fuel cells easier. While both PEMFCs and DMFCs employ a proton-conducting polymer such as Nafion™ as the electrolyte, SOFCs employ oxide-ion (O2−) conducting ceramics such as yttria-stabilized zirconia. Interestingly, the fuel cells can also be operated in reverse mode to produce fuels, e.g., DMFC could electrochemically react water (an abundant, renewable resource) with carbon dioxide (a greenhouse pollutant) to produce methanol (a synthetic liquid hydrocarbon fuel, albeit at rather low yield) with formic acid and formaldehyde as by-products; these by-products can be further catalytically converted (via a secondary step) into additional methanol to improve the reaction yield. To facilitate the electrocatalytic reaction of methanol synthesis, this process requires an external input of electric energy, which can in principle be supplied via a renewable route by the solar cells.Despite the appealing advantages of fuel cells, their commercialization is hampered by high cost, durability issues, and operability problems that are directly linked to severe materials challenges and systems issues. Nanomaterials and nanotechnology can play a critical role in advancing fuel-cell technologies and their commercialization prospects as materials are the bottlenecks in many instances. Development of more efficient, affordable materials and cost-effective processing will have a profound impact in the future of the fuel cell technologies. Some of the materials challenges are pointed out in Fig. 5. The need for the Nafion™membrane to be hydrated to allow proton conduction not only limits the operating temperatures of both PEMFCs and DMFCs to <100 °C, but also necessitates complex external humidification systems. Moreover, the Nafion™membrane is prone to degradation by peroxide intermediates formed during oxygen reduction at the cathode. In the case of DMFCs, high methanol permeability through Nafion™ not only wastes the fuel (low utilization) but also poisons the platinum catalyst at the cathode. Clearly, development of alternate membranes that can operate with low methanol permeability or at higher temperatures (∼150 °C) without requiring water will markedly enhance fuel-cell performance. Design of new polymeric membranes67,68 or nanocomposite membranes consisting of a polymer and proton-conducting inorganic nanoparticles69 offers great potential.
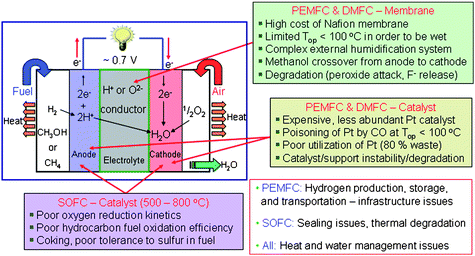 | ||
| Fig. 5 Challenges of fuel cell technologies. | ||
The low operating temperatures in PEMFCs and DMFCs necessitate the use of expensive electrocatalysts such as Pt and PtRu. Nearly half of the cost of the fuel cell is linked to the electrocatalyst cost, further compounded by the fact that there is not enough platinum available worldwide at current catalyst weight loadings to support widespread commercialization. As Pt is an industrial catalyst for many chemical reactions, its use in fuel cells could send prices through the roof and wipe out any cost savings one might anticipate through mass production of fuel cells.70 The standard impregnation approaches used to disperse nanoscopic Pt on the carbon support traps the metal within the micropores of the carbon where only the Pt atoms at the pore mouth are accessible to fuel or oxygen, thereby wasting nearly 80% of the expensive Pt-based catalysts.71 Redesign of the nanoparticle–carbon nanoscopic reaction zone to minimize micropore-filled “mines” of Pt would immediately lower the weight loadings of Pt necessary to reach comparable performance to current state-of-the art fuel cells.
This reliance on expensive platinum-group electrocatalysts for reasonable fuel-cell performance is a serious challenge, and needs to be addressed not just through better utilization of Pt within the electrode structure, but through development of less expensive, nanostructured electrocatalysts. For example, Pd-based nanoscale catalysts offer great promise for fuel-cell reactions72 and the cost of Pd is one-fifth of the cost of Pt. Core–shell structures consisting of an inexpensive metal or alloy at the core and Pt on the outer shell could also reduce catalyst cost. Moving from Pt-dominant electrocatalysts offers another advantage as the nanosize (∼3 nm) Pt particles desired for optimum PEMFC or DMFC cathode performance suffer from dissolution followed by precipitation and growth when the carbon support corrodes under the operating conditions of high temperature and potential.73 Decreasing the surface-to-volume ratio of the catalyst particles lowers fuel-cell performance. Nanotechnology can promote the development of more robust electrocatalysts and carbon architectures with optimized pore structures. In addition, development of carbon-free supports based on, for example, nanostructured oxide or carbide supports like TiOx and WC offer great promise. Optimized porous supports can not only enhance catalyst utilization but can increase catalytic activity through enhanced support–catalyst interactions.
In contrast to PEMFCs and DMFCs that rely on mobile protons, SOFCs operate above 500 °C in order to maintain adequate O2− conductivity. Higher operating temperatures provide two important advantages: the use of inexpensive, non-Pt-based oxide or metal-oxide composite cathodes and anodes; and direct oxidation of hydrocarbon fuels without requiring external reforming to produce hydrogen. The major challenge with SOFCs is slow kinetics of oxygen reduction at intermediate operating temperatures (500–800 °C) when using conventional cathodes. Poisoning of the anode by trace amounts of sulfur impurity present in the hydrocarbon fuel remains a problem in common with PEMFCs; in SOFCs, carbon deposits on the anode and degrades performance. Perovskite and perovskite-related intergrowth oxides containing Co offer high catalytic activity, but suffer from high thermal expansion mismatch with the electrolyte.74 Similarly, perovskite oxides based on Mo and Mn exhibit good catalytic activity for hydrocarbon fuel oxidation with a high tolerance to sulfur.75 Of particular promise is the design of new cathode and anode materials as well as nanocomposites in which one component offers high electronic conductivity and the other component offers high oxide-ion conductivity. It was recently demonstrated that ion wires can be designed into gadolinium-doped ceria aerogel nanoarchitectures to create grain-boundary free, macroscopically long paths for oxide-ion transport,76 which should improve the rate of fuel oxidation at the nanostructured, porous anode.
In addition to the challenges associated with the active components involved in fuel cells, economical and safe production, storage, and transportation of hydrogen are crucial for the success of PEMFCs. Innovative system design that offers efficient management of both heat and water is critical for the successful operation and durability of all fuel cells.
In the near-term (2–5 years), PEMFC and DMFC costs can be halved by judicious distribution of Pt electrocatalysts within the electrode structures with retention of comparable performance. For PEM fuel cells, another very important direction is the development of novel membranes having high proton conductivity at higher temperatures (>120 °C) without the need for humidification, which will effectively minimize or eliminate the major difficulties associated with the current state-of-the-art in PEMFC, such as CO-poisoning, water management, and thermal management.
In the mid-term (5–10 years), SOFCs will dominate the stationary and distributed power generation because of their high energy efficiency and excellent fuel flexibility, including a potential for direct utilization of hydrocarbons, coal gas, and renewable/synthetic fuels. As the SOFC operating temperature is continuously reduced by the development of new electrolyte and electrode materials/structures, SOFCs may find use in mobile applications such as electrical vehicles.
In the long-term (>10 years), carbon-free electrode structures and non-Pt-based electrocatalysts that provide sulfur-tolerance should yield order-of-magnitude improvements in performance-to-cost ratios for PEMFCs and DMFCs. Additionally, long-term goals need to focus on cost effective and efficient production of hydrogen fuel, for example, from an abundant and environmentally benign source like water, if the fuel cell technology is to be widely successful and competitive with other technologies.
Energy storage
The efficient storage of energy is an essential enabler of many energy conversion processes from sources that are intermittent (e.g., solar, wind) or whose costs change appreciably over short time scales. Energy storage technologies are generally categorized as chemical or electrical. Chemical storage approaches that offer the promise of nanotechnology-based breakthroughs include biofuels and hydrogen, but are not limited to these energy storage media. This category of storage technologies, in particular, lacks consensus and clarity regarding both fundamental limits of materials performance and practical issues ranging from cost to regional/national availability to impact on climate change. Similarly, electrical storage technologies, including batteries and capacitors, can be profoundly impacted by nanotechnology, yet they also face important and unresolved issues related to toxicity, durability, cost, and fundamental limits of materials performance. Further, the overarching electric power grid is a massive energy storage medium/network that ties together energy conversion and storage technologies. Nanotechnology has some potential influence on the grid itself (e.g., by nanoscale design of superconductors for power transmission). On the other hand, improvements in grid capacity could enable buffering and transmission of intermittent energy supplies from nanotechnology-enabled devices (e.g., solar cells).Often, the limiting factors in energy storage relate to practical considerations, such as environmental impact, safety, cost, and scalability. For example, many of these interrelated issues have received attention regarding the production of biofuels and associated environmental and economic impacts. Further, the prospects for the mass-production of affordable electric, advanced hybrid, and hydrogen-powered vehicles are closely tied to the cost of the energy storage system. The following subsections highlight various energy storage technologies that are currently under intense development and identify important challenges and opportunities for the research community.
Biochemical storage: biofuels
Biofuels will emerge as important energy storage media and will increasingly become a component of the transportation fuel pool in the coming decades.77 Currently, energy derived from the combustion of traditional and ‘modern’ biomass accounts for 10.7% of the global energy pool. As biomass is carbon-rich, it will necessarily become an important fuel feedstock as conventional fossil-fuel resources are depleted. An added benefit in the context of increasing fuel-derived carbon dioxide emissions is that CO2 produced from the combustion of biofuels is carbon dioxide neutral (although complete life cycle analysis shows that fossil-derived energy is invariably used in fuel production). Because CO2 is the only carbon building block for plants, CO2 released from biofuel combustion can be recaptured in plant growth to yield additional biofuel feedstock.Solar energy is captured by plants and stored in the form of biomass. Biomass can be harvested terrestrially (e.g., grasses, trees, sugar cane, corn, etc.) or from aquatic environments (e.g., algae) and used as a fuel. In ancient times or in less-developed areas of the globe, terrestrial biomass has traditionally been used as a fuel directly by combustion to generate heat. More recently, biomass has emerged as a feedstock for the creation of liquid or gaseous fuels that will fit within the modern energy infrastructure. In this context, plants utilize solar energy and atmospheric CO2 to generate biomass, which society can harvest and use as a feedstock that supplements or eventually replaces fossil compounds such as coal, natural gas, or petroleum, for fuel production.
Coal, petroleum, and natural gas are all hydrocarbons. These feedstocks can be used directly in combustion processes to generate energy, or they can be transformed into other hydrocarbon fuels (e.g., petroleum into gasoline and diesel) for combustion. Terrestrial biomass is uniquely different in that it is highly oxygenated (reducing its energy content in combustion relative to hydrocarbons), and it is generally a solid feedstock. Thus, the existing technology suite designed for the transformation of conventional hydrocarbon feedstocks that are fluids is not optimally designed for the conversion of biomass. The solid nature of biomass places a particularly inconvenient constraint on its utilization for transportation fuels. Whereas fluids like gas and oil can be easily transported to centralized processing facilities that benefit from economies of scale, such scenarios for biomass conversion are less likely, owing to the enormous transportation costs associated with moving solid biomass.
To this end, “the central and surmountable impediment to more widespread application of biocommodity engineering is the general absence of a low cost processing technology,” as stated by Lynd et al.78 Plant biomass for use in biofuel production is widely available, but it exists in dispersed locations and must be collected and converted to liquid or gaseous fuels. Economical processes to facilitate this conversion are generally lacking. The technological needs can be broken into two categories: (i) processes for the conversion of raw biomass into processable fluid media, and (ii) methods for upgrading the liquefied or solvated biomass into transportation fuels.77 Advances in nanotechnology can directly impact both groups.
The central nanotechnology involved in the conversion of fossil feedstocks into conventional fuels or biomass feedstocks into biofuels is the application of nanostructured solids to catalyze the desired chemical transformations. The application of solids as catalysts, commonly referred to as heterogeneous catalysis, is perhaps the oldest commercially practiced field of nanotechnology. For at least fifty years, and some would argue for up to 100 years, the nanoengineering of catalytic solids has been practiced as a means to control chemical reactions.
The two fundamental parameters that must be controlled in the design of an effective catalyst are: (i) creation of the active center and (ii) transport of reactants to and products away from the active center, respectively. Both of these aspects can be controlled by manipulation of the catalyst structure on the nanoscale. A good example is the engineering of crystalline, microporous zeolitic solids to fundamentally alter catalytic cracking technology.79,80 Since the initial discovery, the crystal size (101–104 nm) as well as pore size and shape (0.3–50 nm) have been engineered to alter the distribution of reaction products during the catalytic cracking of large petroleum-derived hydrocarbons.81 Advances in the development of heterogeneous catalysts with the aid of new tools of nanotechnology continue to be made today, and it is reasonable to expect that heterogeneous catalysis will solve many of the problems associated with the economical processing of biomass, as described above. The following discussion will focus solely on the largest readily available source of non-food biomass, lignocellulosic biomass.
In the initial processing of raw biomass, technologies have been developed to transform biomass into fluid media that can readily be converted in subsequent steps into fuels.77 Biomass can be gasified to yield synthesis gas (syngas mixture of H2/CO), which is an important and versatile feedstock for conversion into other fuels, as discussed below. A current primary challenge in biomass gasification is the removal or minimization of condensable tars (condensable organic species) from the gasified media.77,82 This might be addressed through design of new, efficient nanostructured catalysts. For instance, catalysts can be added directly to the gasification reactor to alter the reaction chemistry and limit the formation of condensable species.83,84 As an alternative, tars can be removed in a post-gasification conditioning step, and in this case different catalyst designs might be required as compared to the case of tar reduction during gasification.85 Typical tar destruction catalysts include metal oxide supported metal nanoparticles, and it is expected that the evolving ability to synthesize metal nanoparticles with diverse sizes and shapes may impact the development of new, more efficiently supported metal catalysts.86
Processing of raw biomass to create liquid media that can be further upgraded downstream can also benefit from advances in catalyst nanotechnology. For the fermentation of carbohydrate biomass into common transportation fuels such as alcohols, an efficient pre-treatment technology is needed to hydrolyze the polymeric sugars into fermentable, monomeric units.87Hydrolysis can be achieved, for example, via solution-phase acid catalysis using mineral acids, or by a rapid pretreatment of the biomass with acids followed by enzymatic hydrolysis of the solublized biomass using cellulase enzymes. However, subsequent fermentation or other downstream processing with heterogeneous catalysts (see below) can be adversely affected by the presence of fermentation inhibitors such as sugar degradation products or residual soluble acids. The nanoscale design of new catalysts for biomass hydrolysis may offer solutions to these problems. Common fermentation inhibitors are produced in a set of classic series reactions, whereby the intermediate products, monomeric sugars, are desired and the secondary product (e.g., furfurals), are to be avoided. As such, manipulation of the relative reaction rates using catalysts offers a classical approach to maximize the desired product. However, these series reactions are complicated by the fact that the feedstock is being transformed from a solid slurry into a liquid solution. As a result, the solid catalyst must be designed to have nanoscale features that are consistent with these constraints. For instance, one could envision engineering the appropriate combination of nanostructured catalysts to achieve biomass hydrolysis with near-complete catalyst recovery, leading to more efficient biomass processing with minimization of downstream process-inhibiting catalyst residues in the products.88 Or, one could employ a two-stage pretreatment of solid, crystalline cellulose with a non-porous, nanostructured catalyst that can potentially provide efficient solid–solid (catalyst reagent) contacting that is needed for initial break down of the solid feedstock. Such a catalyst could be a superparamagnetic spinel ferrite that is surface functionalized with acid groups.88–90 As a consequence of the nanoscale dimensions of the catalyst and its composition, it may provide effective catalyst–reagent interactions and it can be recovered from the liquid media after reaction via application of a magnetic field.91 In a subsequent step, a solid catalyst with designed macroporosity and/or mesoporsity can be used to further catalyze the breakdown of the oligomeric sugar fragments into fermentable monomeric sugars. In this case, the porosity would need to be engineered on the nanoscale to allow efficient conversion of the bulky oligomeric reactants.92,93
Pyrolysis or liquefaction has also been used to break down biomass into liquid media that are amenable to further processing.77 Of the pyrolytic methods, usually fast pyrolysis is used to produce liquefied biomass as a ‘bio-oil’ that is composed of a mixture of oxygenated hydrocarbons and water.94,95 Bio-oils can also be produced by liquefaction, a process involving both high temperatures and pressures, with liquefaction producing bio-oils having lower oxygen content.77,96 Both of these approaches can be carried out in the presence of solid catalysts, and manipulation of the catalyst structure at the nanoscale can lead to altered reaction selectivities. For example, adding zeolite catalysts directly to the pyrolysis process can lead to significantly altered reaction selectivities and thus different bio-oil products, compared to conventional pyrolysis.97–99 In this context, Huber has recently used acidic ZSM-5 zeolites to produce significant concentrations of aromatics during biomass pyrolysis, a trend that is typical of the use of zeolites as additives to pyrolysis reactions.100
The above sections describe the catalytic pretreatment of biomass to yield fluid media that can be subsequently processed into traditional fuels. Solid, nanostructured catalysts are also critical in the secondary conversion to fuels. Syngas has been converted on the commercial scale to either methanol or a mixture of alkanes (Fischer–Tropsch synthesis) over solid catalysts for a number of years.101 Recent studies have shown that the use of bifunctional zeolite catalysts102 can produce significant fractions of aromatic hydrocarbons from syngas. Use of syngas derived from biomass would lead to renewable methanol and renewable diesel fuel synthesis. An emerging target is the synthesis of ethanol and propanol from syngas, as these products could directly impact the transportation fuel pool (as gasoline blending components) as well as the massive plastics industry (viacatalytic dehydration of the alcohols to ethylene and propylene, followed by polymerization).101,103 Efficient routes to these higher alcohols might be developed by the nanoengineering of new solid catalysts. Advanced computational techniques, such as first principles electronic structure calculations and ab initio molecular dynamics simulations are playing an increasing role in these efforts.104Catalysts for the conversion of syngas to higher alcohols are complex, multiphase solids with numerous different types of surface sites including metallic, oxide, carbide, nitride, phosphide and sulfide surfaces.101 Manipulation of the catalyst composition and structure on the nanoscale will undoubtedly lead to more active and selective catalysts for higher alcohol synthesis.
Liquefied biomass obtained by pyrolysis, liquefaction or hydrolysis also must be catalytically upgraded into fuels with a lower oxygen content.77,105 Deoxygenation yields a larger energy density to the biofuels and renders them miscible with conventional fossil-based transportation fuels. Two primary approaches have been evaluated to achieve this transformation: catalytic treatment with hydrogen and hydrogen producing reagents (hydrodeoxygenation) or catalytic dehydration, primarily using zeolite catalysts.77Catalytic hydrodeoxygenation (HDO)106 is conceptually similar to hydrodesulfurization (HDS), a field that has witnessed tremendous growth in the last decades. Hydrotreating catalysts are now optimized for HDS reactions, and future years will see development of new nanostructured materials specifically for HDO.
A drawback of HDO is that it consumes a significant amount of hydrogen, as oxygen is liberated in the form of water. In contrast, dehydration using zeolite catalysts offers the potential for deoxygenation of many oxygenated, biomass-derived feedstocks without the use of hydrogen.107 As noted above, the reactivity of zeolite catalysts is dictated largely by the catalyst nanostructure, including pore size and shape, as well as particle size. Roughly twenty years ago, zeolites were evaluated extensively in the upgrading of bio-oils. However, at that time, only a handful of zeolites were available, and even the modern literature is dominated by old reports using well-established, commercial zeolites, such as H-ZSM-5, H–Y and related commercially available structures.108 However, today, there are over 170 different molecular sieve topologies, meaning there is the distinct possibility to use catalysts of different nanostructures to affect catalytic deoxygenation in profoundly new ways.109 New classes of nanostructured materials, such as metal–organic framework compounds (MOFs)110 and polyoxometallate compounds (POMs),111 have been synthesized recently and their catalytic utility in these reactions remains largely unexplored. As these and other new materials come under increased scrutiny, the potential for breakthroughs in chemical conversion of biomass looks bright.
The foregoing paragraphs describe many of the key technical problems that advances in catalyst nanotechnology might address. However, it should be noted that there are broader issues beyond simply biomass transformation into energy-dense fuels that must be solved in parallel. In particular, the constraints imposed by solid biomass as a feedstock will necessitate growth of many small fuel processing facilities dispersed about the globe, rather than solely increased utilization of large centralized refineries. However, as centralized refineries represent highly efficient processing facilities that have evolved over 100+ years of technical innovation, significant efforts in the development of biofuels that are compatible with existing refinery infrastructure are needed to leverage this resource. In the short term, the biofuels that will be developed and used are based on a few select molecules that are fundamentally different from traditional fossil-derived transportation fuels. Ethanol is the leading example of such a biofuel for gasoline blends, while fatty acid methyl esters (FAMEs or biodiesel) are the key example for diesel replacement. These fuels are fundamentally different from traditional fuels and their structures generally require blending with conventional fuels at the fuel terminal, rather than the refinery. In the longer term, it is therefore necessary to develop new, cost-effective technologies that allow for the transformation of biomass into what is sometimes referred to as ‘green gasoline’ or ‘green diesel,’ which are molecules that are identical to those found in traditional petroleum fuels.
In the context of catalyst nanotechnology for biofuel production, this leads directly to a series of research directions:
More efficient production of known biofuels. Fermentation based approaches to ethanol production are likely to be the dominant technologies commercialized for gasoline blending. As such, catalyst nanotechnologies are especially needed in the first stage of biofuel production–conversion of solid feedstocks into processable, fermentable liquid media.88
Commercial development of new single molecule biofuels and start of the transition to green gasoline and green diesel. New catalytic routes to single molecule, replacement biofuels will be developed, such as the recent suggestions for dimethylfuran112 and γ-valerolactone.113,114 To transition to the production of green gasoline, catalyst nanotechnologies are needed that allow for efficient production of liquefied or gasified biomass on the smaller scales that will be associated with distributed processing. These include catalysts for gasification, liquefaction and pyrolysis, as well as hydrodeoxygenation for bio-oil stabilization. For diesel fuels, catalysts for the decarboxylation of fatty acids for the production of green diesel that is easily blended with traditional diesel are needed, as green diesel usage bypasses many of the problems associated with biodiesel (blending ratios or engine modifications).
New catalysts for refinery conversion of mixed petroleum and liquefied biomass streams. As a larger fraction of the total transportation fuel pool is made up of biofuels, contributions from all sectors will be required. Ethanol and perhaps other single molecule biofuels will contribute, but larger scale production utilizing petroleum refineries via co-feeding stabilized, liquefied biomass with petroleum is needed.
The continued development and refinement of heterogeneous catalysts through the manipulation of their nanostructure undoubtedly will play a large role in the pace of development of biofuels as energy carriers. As noted here, heterogeneous catalysts, especially those used for fuel production, represent one of the oldest commercialized forms of nanotechnology. Without question, they will continue to play a central role in maintaining stable energy supplies in the coming century.
Chemical storage: hydrogen
Hydrogen has been identified as an attractive energy carrier for future energy systems and has the potential to reduce greenhouse emission and pollution that have been plaguing mankind since the beginning of the industrial revolution. Molecular hydrogen is an ultra-high energy density ‘green’ fuel, and its availability in a high purity state is essential to enable a transition from conventional thermal power generation technologies, such as internal combustion and gas turbine engines, to high-efficiency electrochemical engines, such as fuel cells. This transition is especially important because CO2 capture from distributed power sources is particularly challenging. Unlike fossil fuels, hydrogen is not naturally available as a primary energy source. It is instead a carrier of energy produced from primary sources, including fossil fuels in the short term, and solar, geothermal, and nuclear in the long-term. As an energy carrier, hydrogen offers the advantage of clean and complete conversion to water, which condenses in the form of rain and hence is environmentally benign. However, the major disadvantage of hydrogen is the need for large storage volumes when carried in its molecular form. Therefore, the viability of the hydrogen economy depends on scientific and technological breakthroughs in storage (and by association, distribution) of hydrogen.The storage of hydrogen at sufficiently high mass and volumetric densities in a readily accessible state, at reasonably low cost, and with sufficient efficiency represents a major technological challenge and perhaps the primary barrier to the use of hydrogen as a clean energy carrier for transportation.115 On-board hydrogen storage systems must meet stringent requirements of gravimetric and volumetric energy densities. According to the US Department of Energy's targets,116 the gravimetric density should be at least 6 wt.% (i.e., 6 kg H2 in a 100 kg tank), the volumetric density should be at least 45 g H2/L by 2010, and even more aggressive targets have been set for future years. Hydrogen storage technologies that meet performance and cost requirements are crucial for the success of a hydrogen economy. The candidate hydrogen storage material/method should offer not only high volumetric storage capacity and gravimetric density, but also fast kinetics of hydrogen charge/discharge (a fast hydrogen charging process is needed to meet the expectation of consumers, and the targeted fueling rate is 1.5–2.0 kg of H2 per minute). These requirements call for materials with low release/dissociation/desorption temperatures, moderate release/dissociation/desorption pressures, and also low heats of formation in order to minimize the energy necessary for hydrogen release. Further, the use of abundant raw materials, chemical stability under cyclic loading, and low manufacturing cost are equally important. No material or process has yet been developed that is capable of meeting all of these functional requirements and the DOE performance targets for storage capacity and volume.
Compressed hydrogen gas is commercially available, but it requires extremely high pressures even to approach the DOE target for volumetric density. Cryogenic liquid hydrogen tanks (operating at a temperature of 20 K) are also available, but they suffer from the large energy consumption required for liquefaction and the phenomenon of hydrogen boil-off during storage. Here, we focus on alternatives to high-pressure gas and low-temperature liquid storage that may exploit nanoscale phenomena to achieve breakthroughs that would satisfy the various technological goals for hydrogen storage.
Complex hydrides, high-pressure metal hydrides, and cryosorbents represent promising classes of solid-state reversible hydrogen storage materials, but current materials exhibit problems such as low hydrogen gravimetric density, slow hydrogen release, and difficulty of regeneration.117 For hydride materials, advanced multi-component alloys will likely be essential to achieve the DOE-established targets for gravimetric and volumetric energy densities.116 Another possibility is physisorption on high surface area solids, which offer intrinsically fast charge/discharge kinetics not involving an electron transfer or chemical bond formation. Metal organic frameworks (MOFs) have given the most consistently promising results for this class, but they still fall short of the DOE capacity requirements.118 Lighter carbon-based materials, including carbon nanotubes (CNTs), have also been examined, but the results are inconclusive in part because of difficulties in repeatable synthesis and high-yield separation of appropriate material types.119,120
Considerable progress has been made in recent years in identifying light solids with improved bulk thermodynamic properties for reversible H2storage.121,122 A variety of nanotechnology approaches are being explored to improve these characteristics, from nanostructuring metal hydride powders123–125 to embedding catalytic clusters126 to synthesizing new cage-like MOFs for cryosorption.127 However, nanostructuring of the storage medium can also decrease effective thermal conductivity, to the detriment of the rate of hydrogenation of the metal in the fueling process.128,129 The motivating factor underlying the desire for good heat transfer properties in solid-state hydrogen storage materials is the need for rapid vehicle fueling. Finding a suitable balance between competing goals of rapid reaction rates and high thermal effective thermal conductivity represents a significant challenge.
For example, with a projected fueling time of 3 to 5 minutes, a total hydrogen mass of 6 kg, and a heat of reaction in the metal hydride hydrogenation process of 20–50 kJ mol−1 of H2,130 the corresponding heat load ranges from 200 to 800 kW during the fueling period. If the thermal properties and cooling of the metal hydride system are inadequate, temperatures will rapidly rise such that the reaction rate will decrease, and the fueling time will extend beyond the target. In many applications involving nanoscale geometric features, heat transfer is dominated by transport across interfaces. Thermal properties can be governed by either electrons or phonons, depending on the electrical conduction type, which varies from metallic to semiconducting for different compounds.131 For electrically conductive metal hydrides, electrons are expected to be the dominant thermal energy carriers, while for semiconductors phonons are dominant. At moderate temperatures, the mean free paths are typically on the order of 10–100 nm for both electrons and phonons in bulk materials, but powder forms of these materials are necessary to achieve high reaction rates. Particle beds with effective particle diameters less than 1000 nm and/or particle-to-particle contact spot sizes less than 100 nm can experience a significant decrease in effective thermal conductivity because the carrier mean free path is shortened by boundary and interface scattering.132 Further, when the contact spot size approaches that of the carrier wavelengths, wave effects can substantially impede the flow of heat.133
Along with equilibrium storage capacity and intrinsic kinetics of H2adsorption/desorption by the material, the fundamental latency of the system for hydrogen uptake and release also depends on a packaging solution for the hydrogen storage medium. The development of a suitable package for the storage materials will require: (1) packaging materials that are compatible with the storage medium, (2) a means for rapid hydrogen charge/discharge to/from its full capacity, (3) exclusion of contaminant species, and (4) low latency thermal response for rapid charge/discharge. In particular, DOE has set stringent targets for high fuel purity. Levels of NH3 and total hydrocarbons, for example, must be <0.1 ppm and <2 ppm, respectively.116 We also note that alternative approaches that allow these constraints to be relaxed could have a dramatic impact on the field. For example, on-board purification systems that accept higher impurity levels but release H2 at acceptable purities would have broad applicability if they also were lightweight, durable, and inexpensive.134 Further, we note that the packaging system must also meet stringent safety standards,135,136 and many issues related to long-term reliability and crashworthiness remain to be addressed on topics ranging from cryogenic hydrogen storage137 to the use of composite storage vessels.138
To summarize, the challenges in developing practical hydrogen storage technologies are significant and require major advances in both basic science and engineering research. Looking toward the future, the following set of goals and activities are recommended to stimulate research efforts:
In the near-term (2–5 years), research should focus on the discovery and development of a set of materials compositions and structures that simultaneously meet the key requirements for hydrogen storage capacity, density, and intrinsic sorption/desorption kinetics. High-throughput screening of materials with both experimental and theoretical tools should be a priority to establish materials libraries suitable for down-selecting promising materials for intensive development activities. Concurrently, efforts to characterize the fundamental phenomena controlling H2 uptake and release in prototypical materials should continue.
In the mid-term (5–10 years), intensive development of promising materials should take place in the context of complete storage systems to seek engineering-based solutions for multifunctionality via device integration and intensification of charging/discharging processes. Aggressive screening of new materials should continue, with emphasis likely shifting to catalysts and other additives for optimizing storage capacity and cycling kinetics while using materials that are abundant, inexpensive, and environmentally benign and that meet the target performance metrics.
The long-term goal (>10 years) is to develop sustainable and scalable engineering, manufacturing, and recycling routes for new materials and packaging solutions that address complete hydrogen storage systems, including integration with all auxiliary components. Implementation of optimized storage systems will require full integration of the storage device(s) with the engine/fuel cell in which the hydrogen will be used and the large-scale infrastructure that will be used to deliver hydrogen to individual vehicles, necessitating a holistic view of thermal management, control of impurities, and safety issues.
Electrochemical storage: batteries
Batteries provide the electrons that perform electrical work because of the difference in chemical energy in the materials in the cathode and anode of the battery. To convert stored energy into power, a physicochemical balancing act is required among multiple reactions: mass transport; electron transport; ion transport; and the kinetics of the (electro)chemical transformations. Just as one is accustomed to gridlock at the three-phase boundary in heterogeneous catalytic processes, including those at the electrocatalysts that establish reasonable current densities in fuel cells, the reactions that are operative in a battery can suffer charge–discharge rate restrictions that limit performance.139It is in this multifunctional arena in which a design perspective guided by arranging matter and function on the nanoscale offers the chance to move to a new performance curve, particularly for lithium-ion batteries. These batteries are the rechargeable power source of choice because they offer higher energy density arising from the high electromotive force inherent to the electroreaction of Li0/+ and from the use of nonaqueous electrolytes, which offer a wider electrochemical stability window than aqueous electrolytes. Although lithium-ion technology, presently based on a layered LiCoO2 cathode and graphite anode, revolutionized the portable electronics market, its adoption for plug-in hybrid electric vehicles (PHEV) and ultimately electric vehicles (EV) is hampered by several issues as seen in Fig. 6. The cathode suffers from high cost and safety concerns arising from chemical instability of LiCoO2 at deep charge, which restricts the practical capacity of LiCoO2 to 50% of its theoretical capacity. The high capacity, inexpensive anode suffers from safety concerns when scaling to large cells because of the unavoidable formation of a solid–electrolyte interfacial (SEI) layer via reaction of the graphite surface with the electrolyte. These difficulties have prompted the search for alternative electrode materials, electrolytes, and design strategies.
 | ||
| Fig. 6 Challenges of lithium-ion battery technology. | ||
Improving energy density requires moving beyond an operational 0.5 electrons per transition metal center. Energy-storing reactions that are difficult to achieve with micron-sized versions of active battery materials, particularly multielectron reactions, often become feasible, reversible, and kinetically facile for the same material on the nanoscale. Oxides, phosphates, halides, silicates, and intermetallics that were once written off as candidate materials for the active phases in battery electrodes can now be reexamined as nanoscale materials, especially if a cost-effective manufacturing process can be devised that yields such materials on the nanoscale with consistent and desirable properties.
Nanoscale approaches have already yielded fruit for lithium-ion batteries. Applying nanoscale redesign to LiMn2O4 by cationic and anionic doping as well as surface modification with nanoscopic oxides (e.g., Al2O3 and TiO2) suppresses Mn dissolution140 and improves cyclability of spinel cathodes.141,142 Reducing the particle size of LiFePO4 to ∼30 nm converts the mechanism of lithium-ion intercalation from a two-phase reaction with a miscibility gap to a single-phase solid-solution reaction.143 Coating the nanoscopic LiFePO4 with conductive carbon overcomes its electronic and lithium-ion conductivity limitations144 yielding a cathode with high rate capability and excellent cyclability.145,146 Similarly, multielectron conversion reactions with metal oxyfluorides that are ill-behaved on the macroscale, become viable on the nanoscale.147
The intriguing developments with nanostructured LiFePO4 have prompted renewed interest in lithium-ion insertion hosts that are generally poor electrical conductors, such as materials with Nasicon structures, which exhibit fast alkali metal ion conduction. Architectural designs, including three-dimensional strategies,139,148 are necessary in order to integrate the poor mixed-conducting active material with its electron path (e.g., by using networked carbon nanotubes or massively parallel 3D current collectors using nanofoams) and to minimize solid-state transport lengths for ions and electrons. Other olivines such as LiMnPO4, LiCoPO4, and LiNiPO4 (with respective discharge voltages of 4.2, 4.8, and 5.2 V vs 3.4 V for LiFePO4) are appealing as next-generation cathodes, but several challenges remain to be addressed, not the least of which is developing electrolyte compositions that are stable to 5.2 V. Surface modification of the nanoscopic high-voltage cathode material with nanostructured oxides149 or even with ultrathin solid-state electrolytes150 offers a means to confer kinetic control while also eliminating or modifying undesired SEI layers to achieve a much-improved rate capability. While most of the attention has been on lithium-ion batteries, alternative battery chemistries are under consideration using sodium, magnesium, or aluminum as the insertion ion. The physical stress generated upon inserting these larger cations appears to be ameliorated in nanomaterials.151
When one succeeds in improving the energy-storing capacity of the cathode, the next target becomes increasing the capacity of the anode while maintaining operational safety. To move beyond carbon-based anodes, efforts to alloy Li with other elements such as Si, Ge, and Sn permit access to higher storage capacities. The major drawback in this strategy lies in the huge volume change occurring during the charge–discharge process; nano-enabled strategies in order to control the mechanical disruption are underway by suppressing the stress/strain using nanowires or embedding the electrochemically active nanoclusters in a matrix152–154 or limiting solvent access to the alloy through an ultrathin solid-state electrolyte.155 The recent demonstration of Li/LiOx reversibility in the lithium–air battery156 markedly increases the capacity of the anode while the proposed use of MnO2-painted 3D carbon nanofoams157 offers one way to accommodate the generation of a solid compound within the anode structure.158
The design of new nanostructured insertion hosts and 3D-architecturally configured electrode structures coupled with compatible electrolytes is key in realizing the full potential of such alternate battery chemistries. These strategies could help to significantly increase the energy density in the near term. However, nanostructured materials could potentially aggravate unwanted side reactions between the electrodes (cathodes and anodes) and electrolyte, and this problem needs to be judiciously addressed by developing compatible electrolyte–electrode combinations. Moreover, cathodes operating at higher voltages (>4.5 V vsLi/Li+) or anodes operating at voltages close to Li/Li+ tend to form undesired solid–electrolyte interfacial (SEI) layers, which not only reduce the charge–discharge rate but also introduce safety problems.
In the near-term (2–5 years), Li-ion battery technology is likely to be the technology of choice. On the basis of the breakthrough with LiFePO4-based batteries, a nanomaterials-enabled exploration of low-cost, high capacity, stable cathode materials and architectures should permit doubling the energy density of lithium-ion batteries. Research that emphasizes strategies to fabricate ultrathin (submicron) solid-state separator/electrolyte phases will be necessary in order to enable three-dimensional battery designs. The three-dimensional redesign of the battery on the nanoscale should improve the rate capability or power density; however, it is unlikely to enhance the energy density or the number of lithium ions per transition metal center. To increase the energy density, exploration of new chemistries and novel mico/nanostructures of electrode materials will be required. In concert with addressing these basic science questions, robust and low-cost, manufacturable approaches, such as use of (nano)composites/conductive coatings, should be further developed to mass-produce high electrical conductivity electrodes.
For the mid-term (5–10 years), the search for new electrolytes with high ionic conductivity, particularly at low temperatures, and increased voltage stability will be of paramount importance to pave the way to high operating voltage (>5 V) batteries. More robust electrolytes will be necessary, including an assessment of SEI issues at Li-alloyed anodes with traditional and new electrolyte compositions. Research will be needed into the nature of point defects that increase energy storage in cathode materials, including nanoengineering in appropriate additives to pin the defect states during the energetics and physicochemical changes accompanying charge–discharge of the battery. Cost-effective manufacturability of nanoscale-redesigned batteries, including three-dimensional architectures, will need to be established. Shelf life, a wide temperature window of efficient operation, and low losses/high system efficiency are additional performance targets that should be emphasized.
For the long-term (>10 years), as the science and art of making high energy storage capacity and high power density batteries is matured, environmental friendliness of the materials and processes enabling such high performance battery technology should gain an increasing importance, leading to engineering approaches towards a sustainable product life cycle management starting from battery manufacturing to battery disposal and ultimately recycling.
Electrochemical storage: capacitors
Capacitors offer another option to store electrical energy, but with a different balance of performance metrics: energy density is an order of magnitude lower than that of batteries, while power density is several orders of magnitude higher.159 Electrochemical capacitors (ECs, designated as ultracapacitors and supercapacitors) offer significantly higher energy density than conventional electrostatic capacitors, making them attractive for peak power demands, which require a combination of energy and power densities.160 Unlike electrostatic capacitors, electrochemical capacitors store energy in a layer of ions that assemble at the interface between the electrolyte and the electrode (with its excess or deficit of surface electronic charge), thereby creating an electrical double layer. In addition to the capacitance of the electrical double-layer, which tracks the surface area of the electrified interface, some materials exhibit pseudocapacitance arising from surface redox reactions. The magnitude of energy that can be stored per area or volume or mass of the electrode concomitantly increases by the addition of stored electronic charge to yield higher specific capacitance. Although faradaic processes occur, the current–voltage characteristics mimic the featureless curves seen for double-layer capacitance.Because the electrodes in ECs are necessarily high-surface-area objects, they are designed as porous structures. As ions need to approach and be expelled from the electrified interface during charge–discharge, the rate of electrolyte mass transport through the pore network of the electrode structure plays a critical role in determining the magnitude of the normalized capacitance, the frequency of the capacitive response, and thus the power density. Electrochemical capacitors in which the double-layer capacitance dominates the charge–storage mechanism typically are designed using high-surface-area carbons and these devices exhibit electrochemical reversibility for 105 cycles.160,161 A morphological variety of carbons including powders, nanofoams (such as aerogels), nanofibers, nanotubes, cloths, and nanosheets have been investigated in aqueous and nonaqueous electrolytes: capacitance values as high as 200 Fg−1 have been realized.161–163 The stability of porous, high surface-area carbons in the presence of corrosive electrolyte environments (e.g., H2SO4 and KOH) remains a critical consideration for long-term cyclability and durability of carbon-based ECs.
Among the various pseudocapacitive materials investigated,164 X-ray amorphous, hydrous ruthenium oxide (RuO2·xH2O) offers the highest capacitance value at >700 Fg−1 (for x ∼ 0.5). The capacitance value of RuO2·xH2O depends sensitively on the water content and the degree of crystallinity.165 The excellent performance of hydrous ruthenium oxide as a pseudocapacitive material arises because it is an innate nanocomposite with a high mobility phase for electron transport, due to metallic conduction along a percolation network of nanocrystalline rutile RuO2 (present even for x > 2, which has a specific capacitance >500 Fg−1) coupled to high proton mobility along water structured at the rutile network.166 Operational stability of the device is further enhanced by the chemical stability of RuO2·xH2O in acidic and basic electrolytes. The major issue with RuO2·xH2O is the prohibitively high cost and limited availability of Ru (Fig. 7).
 | ||
| Fig. 7 Challenges of electrochemical capacitor technology. | ||
With an aim to lower cost while increasing the gravimetric storage capacity relative to carbon-based ECs, a number of transition metal oxides in which the metal ions can support multiple valences have been pursued to replace RuO2·xH2O. Some examples are amorphous or nanostructured MnO2 and V2O5 with capacitance values of up to 400 Fg−1.167,168 Substitution of less expensive elements such as V and Cr for Ru in RuO2·xH2O as well as composites consisting of RuO2·xH2O on amorphous WO3·xH2O and Na0.37WO2·xH2O have been found to exhibit 550–700 Fg−1 at a reduced cost.169,170 As with carbon-based ECs, the physicochemical nature of the nanoarchitectured materials and the quality of the pore structures are dictated by the synthetic and processing approaches that are used and they affect the magnitude of the capacitance and the charge–discharge rate. Nanohybrid systems consisting of conducting polymers and inorganic oxides or carbon materials with capacitance values of up to 450 Fg−1 have also been reported, but long-term chemical stability of conducting polymers during EC operation (resulting in an inferior cycle life) remains a concern.162
Applying the structural lesson imparted by hydrous ruthenium oxide, i.e., incorporate a massively parallel current collector in three dimensions (3D) and on the nanoscale in order to maximize electron and proton mobility166 affords an immediate improvement when using less expensive materials. This design approach has already led to marked improvement of the system capacitance of carbon–manganese dioxide hybrids in which ∼10 nm thick birnessite MnO2 is painted via self-limiting electroless deposition onto the interior carbon walls of ultraporous carbon nanofoams simply by immersing the carbon electrode in neutral aqueous solutions like sodium permanganate.157 The area-normalized capacitance of the hybrid structure is 1.5 Fcm−2, which is orders of magnitude higher than the areal capacitance typically reported for other C–MnO2 composites at 10–50 mFcm−2. Using nanoscale materials as the “walls” in nanoarchitectures processed with optimal pore structures171,172 can significantly impact the ability to realize high capacitance values at an affordable cost.
One important finding has appeared for pseudocapacitive oxides such as manganese and vanadium oxides when structured in well-designed ultraporous nanoarchitectures: they demonstrate an ability to express both pseudocapacitive behavior and the ion-insertion reactions characteristic of batteries.172,173 In that many applications require pulse power (from capacitors) and baseline power (from batteries), so electrical circuit design is necessary to condition the responses from these two time-dissimilar devices. Nano-enabled battery–capacitor hybrids should simplify the nature of power conditioning in future devices.
Overall, the nanoarchitectures, pore structures, surface area, chemical stability in the electrolyte environment, and accessibility of redox couples within the electrochemical stability window of the electrolyte control the properties and performance of electrochemical capacitors.
In the near-term (2–5 years), redesign of materials on the nanoscale and novel chemical synthesis and processing approaches to control the nanostructure will have a significant impact in advancing the commercial feasibility of electrochemical capacitor technology. One goal will be to reach capacitance values that approach those for hydrous ruthenium oxide in the next five years using optimized carbon–manganese oxide hybrids, which decrease the materials cost relative to ruthenium-based ultracapacitors by orders of magnitude. Finally, robustness of the system design and materials (stability to multiple charge/discharge cycles and long-term performance degradation) needs to be addressed, especially for the novel pseudocapacitor-based systems.
In the mid-term (5–10 years), development of new composite material/structure combinations should be pursued. Many transition metal oxides exhibit high pseudocapacitance, in combination with electrochemically active polymers, and can be combined with the high surface area micro/nano-structured carbon frameworks providing high electrical conductivity. Optimization of the electrode nanostructure and interfacial chemistry should be pursued to allow record higher capacitance and fast ion transport in the system for high energy density and fast charge/discharge times.
In the long-term (>10 years), controlled 3D integration of opposing electrified interfaces (i.e., cathode and anode), structured ionic layers, and mobile ions within redesigned nanoarchitectures will create battery–capacitor hybrids that should achieve power densities typical of carbon-based ECs and energy densities comparable to the best lithium-ion batteries. Environmental friendliness of the materials and processes and product life cycle management should become an integral part of the system design and manufacturing.
Energy conservation
Energy consumption in the United States per capita is currently about 7800 kg of oil equivalent per year. This number is approximately 10 times higher than that of developing countries, which have nearly eighty percent of the world population. It can be argued that it will be almost impossible to meet the global energy demand if the entire world population acquires the same quality of living as that in the US at the current rate of energy consumption per capita in the US. Hence, we stress that advances in energy conservation technologies for controlling the demand for energy are as critical as those in energy generation for increasing the energy supply. Currently, about 37% of global energy consumption is for industrial use, 20% is for commercial and personal transportation, 11% is for residential homes, 5% is for commercial buildings, and the other 27% is lost in energy transmission and generation. For residential homes, almost half of the average energy consumption is used for space heating. Another 17% is used for water heating, 6% is for cooling rooms, and 5% is for refrigeration. Lighting represents about 30% of the total electric energy used within residential and commercial buildings. Nanotechnologies can play an enabling role to greatly improve the energy efficiency in lighting, heating and cooling, transportation, etc. For example, solid state lighting can potentially cut lighting energy use by more than 30%, as discussed below. Advances in fuel cell and battery technologies can greatly reduce the transportation energy use, which has been discussed in the preceding sections and will not be repeated here.Thermoelectrics
The conversion of waste heat to electrical power arguably represents the greatest opportunity for energy conservation. Energy usage accounts for a significant fraction of the cost of aluminum, chemicals, glass, metal casting, petroleum refining, and steel. For example, energy costs account for ∼8–12% of the total for glass production, and about 15% for steel. The waste heat from these processes is of a relatively high grade, often with temperatures in the range of 500–1000 °C. In a more distributed form, about 40% of the energy supplied from gasoline to power automobiles is lost as exhaust heat, and another 30% is lost through engine cooling.One promising approach to improve the energy efficiency is to employ solid-state thermoelectric (TE) devices to recover part of this waste heat and to convert it directly into electricity by utilizing the Seebeck effect (Fig. 8a).174 Compared to conventional conversion processes between thermal energy and electricity employing a working fluid, TE conversion is attractive because it does not need moving parts and requires little maintenance. Besides waste heat recovery, TE conversion has the potential to make two other major impacts on both global energy demand and preservation of the environment. One of them is to replace current refrigeration and temperature control units based on chlorofluorocarbon and hydrochlorofluorocarbon refrigerants with cleaner solid state Peltier devices (Fig. 8b) that do not emit greenhouse gases. Moreover, as discussed previously in connection with solar–thermal energy conversion, solar TE systems are potentially simpler and more cost-effective than photovoltaic (PV) conversion, and can also be used to complement PV conversion by utilizing the long wavelength spectrum of solar radiation with energy less than the bandgap of a PV material.175
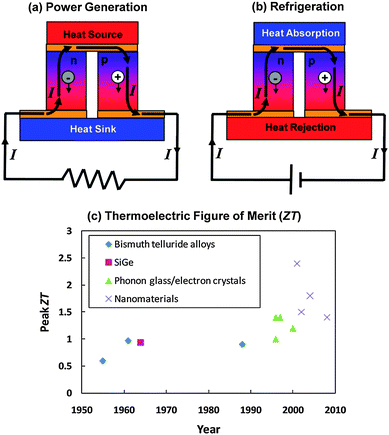 | ||
| Fig. 8 TE power generation and refrigeration. (a) When a temperature gradient is established between two junctions made of an n-type semiconductor and a p-type semiconductor, electrons in the n-type segment and holes in the p-type segment diffuse along the temperature gradient, producing an electrical current through the Seebeck effect. (b) When an electrical current is run across the two TE junctions, electrons in the n-type segment and holes in the p-type segment absorb heat at one junction and reject heat at the other based on the Peltier effect. (c) Progress in the reported peak ZT values of different materials for the past 55 years. | ||
Currently, the low efficiency of TE devices limits their applications to niche markets, such as cooling of car seats and providing electrical power for deep space probes and for data communication equipment in remote oil pipelines.176 The energy efficiency of TE devices is determined by the dimensionless TE figure of merit (ZT), as discussed previously. At the present time, commercially available TE materials have a rather small ZT value, close to unity, and only over a narrow temperature range. TE devices based on such materials have been demonstrated with thermal-to-electrical conversion efficiency greater than 12% with the hot side at 550 °C.177 In order to increase the efficiency further to 20%–30% so that TE conversion becomes superior to other existing energy conversion technologies, the ZT of the materials must be increased to above 3 over the entire temperature range between the cold and hot sides, and the effective ZT of the system, taking into account various loss mechanisms, must be increased to 2 or higher.
ZT depends on the Seebeck coefficient (S), electrical conductivity (σ), thermal conductivity (κ) according to ZT = S2σT/κ. The thermal conductivity consists of a lattice or phonon contribution (κl), an electronic contribution (κe) that is proportional to σ according to the Wiedemann–Franz law, and a bi-polar contribution (κe−p) due to the thermal diffusion of electron–hole pairs that does not contribute to net charge transport or σ. A good TE material should have a large Seebeck coefficient to increase the electromotive force, a high electrical conductivity to reduce the loss due to Joule heating, and a small thermal conductivity to suppress the parasitic thermal leakage from the hot to cold junctions. The electrical conductivity can be increased by increasing the doping concentration in TE materials. However, doing so will usually result in a decrease in the Seebeck coefficient because of the increased symmetry of electron conduction around the Fermi energy,178 as well as an increase of κe. It has been a significant challenge to increase the ZT of bulk materials in the past 55 years (Fig. 8c) because of the interdependence of the three properties that enter into the ZT expression.
In the early 1990s, it was suggested that the dilemma due to the coupling of S and σ might be overcome by employing the enhanced electron density of states (DOS) in quantum-confined nanostructures.179,180 These theoretical works have stimulated intense experimental studies of two-dimensional (2-D) quantum wells and 1-D quantum wires. While the quantum enhancement effect in realistic nanowire and thin film superlattice structures is often masked by other non-ideal effects, such as interface roughness and surface states, the same principle of power factor enhancement via an asymmetric DOS178 has been recently demonstrated by doping bulk PbTe with Tl that modifies the DOS near the Fermi energy.181 In parallel, it has been suggested that the power factor (S2σ) can be increased drastically by using solid-state thermionic emission and electron energy filtering in heterostructures.182,183
The pursuit of power factor enhancement in low dimensional systems has been interweaved with the successful reports of large suppression of the lattice thermal conductivity due to the scattering of phonons by abundant interfaces in thin film superlattices and nanocomposites.57,181,184–186 Heat flow and charge transport usually take the same path in a solid. Consequently, both of them encounter more resistance if the conducting medium consists of a larger interface density. However, it is possible to decouple the resistances to heat and charge transport because electron transport depends on the electric potential, while phonon transport is influenced by inter-atomic potentials. For example, alloys composed of atoms with similar electric potentials but quite different masses can scatter phonons effectively without affecting the electron mobility significantly. Another example of reducing the thermal conductivity with little effects on electron transport is the so-called phonon-glass electron-crystal approach,187 in which the cage-like unit cell is filled or partially filled with loosely bonded atoms whose “rattling” motion scatters phonons. As discussed later, in nanostructured materials the lattice thermal conductivity can be greatly suppressed because the abundant heterointerfaces in nanostructured materials scatter a wide spectrum of phonons more effectively than alloy scattering, which usually affects only the short wavelength phonons.188Electron scattering in these nanostructures can be minimized by reducing interface states and using a homogeneous electric potential in the material.189 The simultaneous decrease in thermal conductivity and increase in power factor were demonstrated experimentally in two examples of ball-milled nanocomposites.186,190
These new approaches to controlling electron and phonon transport have led to ZT enhancement into the 1.4–2.4 range,57,181,184–186 as shown in Fig. 8c, as well as Peltier devices with about a 20% improvement in cooling performance from commercial devices because of a 40% increase in ZT in one of the legs of the thermocouple.186 However, ZT > 2 has not been reproducibly demonstrated in the laboratory, and will require fundamental research in TE materials that is focused on the simultaneous manipulation of both the numerator and the denominator in the ZT expression. To this end, an enhancement of the power factor can potentially be realized together with the suppression of the thermal conductivity by the combination of interface scattering of phonons with an asymmetric electronic DOS in confined dimensions, via doping, or energy-selective filtering of charge carriers. Moreover, it remains to be explored whether the Lorenz number (L ≡ κe/σT) can be suppressed below that found in typical TE materials191 or whether electron transport and electronic thermal transport can be decoupled by violating the conditions governing the Wiedemann–Franz law.
In addition to improving the efficiency, TE conversion can be made to be cost effective by reducing the thickness of TE elements so as to minimize the use of materials, some of which, such as tellurium and germanium, are costly because of limited availability. In addition, further improvements in efficiency require breakthroughs in the reduction of parasitics (including electrical contact resistances <10−8 Ω cm2), as well as the heat dissipation capability of 1000 W cm−2 or higher at the cold side. Fundamental studies of the manipulation of the thermal conductivity at the nanoscale, and of the nature of the electrical interface resistance are essential, together with major progress in the design and engineering of heat sinks that can benefit from the use of nanomaterials breaking the existing limits of thermal transport properties.192
For TE technologies to contribute significantly to meeting the global energy challenge, major scientific discoveries and technological advances need to be made in order to drastically increase the energy efficiency of TE devices and to develop scalable TE materials and devices using those elements that are abundant on Earth and environmentally benign.
In the near-term (2–5 years), the recent progress in improving the materials ZT needs to be accelerated by comprehensive strategies combining the know-how in suppressing the lattice thermal conductivity with both proven and new ideas for increasing the power factor, while at the same time suppressing the electronic thermal conductivity. In addition, there is an urgent need to demonstrate efficient TE devices and systems based on recently reported nano-related and other advanced materials with improved ZT. Advances already made in the laboratory need to be scaled up and commercialized.
For the mid-term (5–10 years), many remaining problems in the design, assembly, and reliability of TE devices will need to be addressed. Issues such as stress caused by thermal expansion mismatch, bonding layers with high thermal and electrical conductivity, passivating TE surfaces and interfaces with regard to sublimation and corrosion require renewed attention with the implementation of new high-ZT materials. The long-term stability of nano-based TE devices needs to be investigated and addressed.
The long-term goal (>10 years) is to scale up the synthesis of bulk and thin film nanostructured TE materials with ZT > 2, with a focus on constituents, such as Si, silicides, oxides, etc., that are available globally at the level necessary for a major worldwide implementation of TE.
Thermal insulation and thermal management
Heating and cooling account for a large fraction of energy consumption for residential homes, commercial buildings, transportation, and industrial processes. Significant energy savings can potentially be achieved by a revolution in thermal insulation and thermal management technologies. Breakthroughs in this direction will be enabled by the nanoscale design of materials with controllable thermal transport properties that surpass existing limits.192Thermal transport in a solid is dominated by electrons in metals and phonons in insulators and intrinsic semiconductors. In semimetals and small-bandgap semiconductors, thermal conductivity consists of non-negligible contributions from phonons, electrons, holes, and the bi-polar contribution. The thermal conductivity due to electrons or phonons is proportional to the specific heat, group velocity, and scattering mean free path of the carriers. The room-temperature thermal conductivity of dense solids often falls between the upper limit of about 3000 W m−1 K found in high quality single-crystal diamonds193 with strong sp3 bonds (allowing a high phonon group velocity) and few defects (hence long mean free paths on the order of microns), and the lower limit of about 1 W m−1 K value or slightly lower in amorphous materials with a short mean free path approaching the atomic spacing. Porous materials commonly used as thermal insulation in buildings as well as polymers and aerogels can achieve lower thermal conductivity than the amorphous limit in a dense solid, and in some cases can approach the thermal conductivity of air of 0.026 W m−1 K.194 However, these materials often cannot endure the temperature or stress encountered in many industrial applications, such as TE devices and thermal protection coatings used for gas turbine blades, which require dense solids with low thermal conductivity, ideally much lower than the amorphous limit.
Nanoscale design of materials can lead to new approaches to breaking the existing limits in the thermal conductivity by manipulating the transport of heat carriers. One recent method for reducing the thermal conductivity is to incorporate a large density of interfaces that effectively scatter the heat carriers. Using this approach, thermal conductivity values approaching or lower than the amorphous limit have been reported in thin film superlattices,195 layered films,196,197 nanocomposites,59 and nanowires,198,199 all of which are dense solids. The large thermal conductivity suppression has been attributed to the numerous interfaces that scatter a wide spectrum of phonons effectively and the resulting thermal boundary resistances, although it has also been suggested that phonon interference is responsible for a thermal conductivity minimum observed in thin film superlattices.200,201 For the case of disordered, layered films,196 the cross-plane thermal conductivity approaches that of air (Fig. 9). In addition, the ultralow cross-plane thermal conductivity is accompanied by a highly anisotropic thermal transport with a much higher in-plane thermal conductivity.202 Hence, it is thought that phonons are scattered by the boundaries between adjacent layers into predominantly in-plane modes, resulting in the ultra-low cross-plane thermal conductivity.
 | ||
Fig. 9 (a) High resolution transmission electron micrograph (HRTEM) of a 65 nm disordered, layered WSe2 film from Ref. 203. Reprinted with permission from MRS. (b) Summary of the measured thermal conductivities of the WSe2 films as a function of the measurement temperature on a log-log scale. Each curve is labeled by the film thickness. Data for a bulk single crystal are included for comparison. The ion-irradiated sample (irrad) was subjected to a 1 MeV Kr+ ion dose of 3 × 1015 cm−2. The line ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) is the calculated minimum thermal conductivity for amorphous WSe2 films in the cross-plane direction from Ref. 196. Reprinted with permission from AAAS. is the calculated minimum thermal conductivity for amorphous WSe2 films in the cross-plane direction from Ref. 196. Reprinted with permission from AAAS. | ||
As the phonon thermal conductivity is significantly reduced in nanostructured materials, heat transfer by thermal radiation in the materials becomes important. For porous materials with an ultralow thermal conductivity, it is essential to suppress the thermal radiation contribution to the overall thermal transport.194 Here, photonic crystals provide a possible approach to reduce the thermal radiation transfer in the material, by overlapping the thermal emission spectrum with forbidden energy gaps in the photonic crystal dispersion relations.
One example particularly relevant to energy conservation is the development of smart windows based on electrochromic materials, such as tungsten oxide, which can be reversibly switched between a highly disordered, opaque form and a crystalline, transparent structure via a simple reaction of the oxide with ion impurities and electrons at the application of an electric field.204 Similar electric field-tuned optical properties can be realized in materials based on suspended particles and liquid crystals,205 where the alignment of the micro/nano constituents of the materials is altered by the electric field. Such smart windows allow users to control the amount of light and heat passing through the window throughout the day, providing a feasible method to reduce energy consumption for heating, cooling, and lighting. Here, a better understanding of the nanoscale processes governing the structural transformation and its effects on the electronic structure and photon transport is the key for developing smart window materials with a much improved optical property range and reduced electrical power use.206
To capitalize on these advances in controlling the thermal properties through nanoscale design and to make a significant impact on global thermal energy consumption, scientific breakthroughs and technological advances need to be made in a number of areas, as summarized below.
In the near-term (2–5 years), a better understanding of many unresolved fundamental questions regarding thermal transport in nanomaterials will need to be developed. This includes uncovering the physical origins and identifying the means to control the ultralow thermal conductivity in the disordered, layered films196 and the minimum thermal conductivity in superlattice films.200,201 Reliable theoretical models and new experimental techniques will be needed to investigate thermal transport at the nanoscale and to probe intriguing nanoscale transport phenomena, including phonon transport and electron–phonon coupling at interfaces, phonon interference in superlattices, phonon localization, electronic thermal transport, bi-polar contributions to thermal transport, spin waves and magnons that can be additional carriers of heat in solids.207
For the mid-term (5–10 years), low-cost materials with a thermal conductivity comparable to or perhaps even higher than that of diamond remain to be explored. The intrinsic thermal conductivity values of defect-free CNTs208 and of graphene along the plane can be comparable to that of diamond. However, the problem of large contact resistances to such nanostructures209 needs to be addressed in order to enable large-scale applications of these nanostructures with high intrinsic thermal conductivity. On the other hand, the equivalent thermal conductivity of advanced heat pipes can be made to exceed that of diamond,210 and can potentially be increased further with the use of nanostructured materials of high thermal conductivity and large surface area for heat transfer enhancement and capillary fluid pumping in heat pipe devices, which enable a transfer of significant heat load over large distances using only passive means with no moving parts.
The long-term goal (>10 years) is to develop materials with tunable or directional conduction, convection, and radiation properties similar to what is presently being investigated in smart windows but with a much larger tunable range so as to allow the effective use and manipulation of thermal energy. Achieving this goal relies on the development of low-cost, large-scale synthesis methods in order to scale up the production of nanostructured materials and to enable their use in thermal insulation and thermal management applications at a scale relevant to the global energy and environmental challenge.
Solid-state lighting
Solid-state lighting (SSL) based on light-emitting diode (LED) technology has the potential to reduce energy consumption for lighting by 33% by the year 2025, thereby circumventing the need to build 41 GW power plants in the United States alone.211 As ∼22% of electrical power generated in the U.S. is applied to general illumination, the energy savings potential of this technology represents a significant contribution to addressing the global energy challenge. Emerging legislation,212 that addresses both the inefficiency of incandescent lighting (∼6% of the electrical input power is transformed to visible light) and the mercury disposal problem of compact fluorescent technology, is creating an opening for the early introduction of SSL. Although SSL products based on LEDs are already on the market, and organic LEDs (OLEDs) are close to market introduction,213 many scientific, engineering and manufacturing challenges remain. To realize the potential of SSL, the DOE has outlined a roadmap culminating in 2025 with lamps that achieve 50% wallplug efficiency for sunlight quality light (color temperature and color rendering comparable to sunlight) at a cost that is competitive with established technologies.214Among the scientific challenges that impede progress toward efficient LED lighting are the “green gap” and “droop.” The green gap refers to the lack of a high efficiency LED source in the yellow-green portion of the spectrum, near 560 nm (Fig. 10). At the blue and red ends of the spectrum—470 nm LEDs based on (In,Ga)N and 625 nm LEDs based on (Al,Ga,In)P—luminous efficiencies of commercial, high-volume LEDs are in the range of 25–30%,214 and the best devices exceed 50%. Extending (Al,Ga,In)P towards the yellow-green is complicated by the lack of direct bandgap alloys in the arsenide–phosphide system with energy gaps above ∼2.3 eV. Extending (In,Ga)N towards the yellow-green is impeded by the large lattice mismatch strain between GaN and InN (10.8% in the basal plane). The large mismatch severely limits the thickness of (In,Ga)N that can be grown without roughening, islanding, phase separation, or misfit dislocation introduction. Hence, the active region of a GaN-based LED is a series of quantum wells, each of which is limited in thickness to a few nanometers.216 The point group of the wurzite crystal structure permits both pyroelectricity (a spontaneous dipole moment along the c-axis) and piezoelectricity (a change in the dipole moment induced by coherency stress). The net effect is a large internal electric field in the quantum well, giving rise to a quantum-confined Stark effect that spatially separates the electron and hole wave functions, thereby impeding radiative recombination. These coupled effects result in a rapidly decreasing internal quantum efficiency as the emission wavelength is increased beyond approximately 500 nm, yielding wallplug efficiencies well below 10% for wavelengths greater than 560 nm (Fig. 10).
 | ||
| Fig. 10 Energy conversion efficiency and luminous efficacy of LED lamps as a function of peak emission wavelength (after ref. 215). Efficient monochromatic LEDs have been achieved in the blue-to-green and amber-to-red portions of the visible spectrum with nitride and phosphide III–V heterostructure diodes, respectively. To achieve white LEDs with acceptable color quality and 50% wallplug efficiency, it will be necessary to fill the “green gap” with efficient yellow-green emitters. The wallplug efficiencies of today's compact fluorescent (CFL) and incandescent (W) technologies are indicated on the figure. | ||
Concomitant with the reduction in internal quantum efficiency as the wavelength is increased is the emergence of the loss in efficiency with increasing current density. This problem, known as “droop,” has plagued the development of green LEDs for lighting applications. Droop is not currently understood. Several theories have been proposed. Among the most prominent is the effect of the small recombination volume (necessitated by the large lattice mismatch) in enhancing the probability of nonradiative Auger processes, proportional to the cube of the excess carrier density, over radiative recombination, which is proportional to the square of the excess carrier density.217 An alternative explanation is based on carrier leakage through the quantum wells.218 Although no consensus has emerged, it is likely that more than one of the proposed explanations have roles in the observed phenomenon. For example, filling of the available quantum confined states in the (In,Ga)N wells or quantum dots will reduce the confinement of excitons bound by bandgap fluctuations, thereby enhancing the probability of non-radiative recombination at threading dislocations.
Many approaches to address the droop and green gap problems have been proposed. They include switching to semipolar or nonpolar orientations of GaN, employing nanostructures to elastically relax a portion of the mismatch strain, and moving toward bulk GaN substrates to minimize the density of threading dislocations. The current practical solution in “warm” (i.e., color temperatures comparable to that of sunlight) white LED lamps is to use phosphors to downconvert blue photons into yellow-green photons to fill in the green gap. The Stokes shift of the phosphor, however, puts an upper limit on the efficiency of this process, as this energy is converted to heat.
Although the primary focus of solid-state lighting research has been on the more mature inorganic semiconductor LED technology, OLED technology is also advancing rapidly. OLEDs offer the potential for low-cost, flexible, spatially-distributed light sources. OLEDs combine organic phosphors emitting at several visible wavelengths to generate warm white light. The luminous efficacy of the best warm white OLEDs is currently 45 lm W−1,219 substantially below the 2015 DOE target of 100 lm W−1. The stability of the blue phosphor is a major concern as it limits the lifetime of today's OLED devices.213 Combining organic and inorganic constituents appears to be an interesting current research direction.
For solid-state lighting to reach its full potential for energy savings, the following nanoscale scientific and engineering challenges should be addressed:
Environmental aspects of energy
The problems of energy (generation, storage, and conservation) and the environment (preservation and stewardship) are intimately linked, both in terms of technology development and by virtue of their global reach. Both are truly worldwide concerns, and the solution of the energy problem not only depends on the local environment (e.g., winds, solar, tides), but also will affect the environment on a global scale. Although we argue for urgent and massive investments into rapid development of a diverse portfolio of renewable energy technologies aiming at a 100% carbon-dioxide-neutral sustainable energy future, the path to this future will inevitably involve utilization of carbon-based energy sources for a significant period of time. Considering the cumulative nature of the effect of CO2 emissions on the environment, development and deployment of carbon dioxide capture and storage (CCS) technologies may not be just optional, but required for a sustainable transition from the present fossil fuel dominated energy mix to carbon-free energy, while avoiding an environmental disaster in the interim. Apart from the environmental protection aspect of carbon dioxide capture, which initially is focused on CO2 sequestration, advanced carbon dioxide capture technologies may potentially become an important enabling component of the reagent supply chain for renewable carbon-based fuels by providing a concentrated source of CO2 for fuel synthesis reactions (e.g., photocatalytic production of solar fuels or via photosynthetic routes, such as algae cultivation).220 To this end, we briefly touch upon the role of nanotechnology in developing energy-efficient and scalable techniques for carbon dioxide capture.CCS will initially focus on capture of CO2 from large point sources, such as coal-fired, electricity generating power plants. Both carbon dioxide capture and carbon sequestration are currently technically feasible, but the costs of deploying CCS on a wide scale have prevented significant adoption of the technology. Of the components of CCS, it is the capture technology that has been consistently shown to bear the largest cost, with the sequestration of the CO2 playing a less significant, but still important role in the overall cost of the technology. As such, advances in nanotechnology are needed to help reduce the cost of carbon dioxide capture.
Carbon dioxide capture technologies of two main types are needed, (i) technologies for the capture of CO2 from existing coal-fired power plants and (ii) technologies for capture from new coal plants specifically designed to allow for easier carbon dioxide capture. In the first category, CO2 must be captured from a flue gas stream with a low CO2 partial pressure and significant water content. Existing, mature technologies based on CO2 absorption in liquid amine solutions provide a benchmark cost for carbon dioxide capture, with the US National Energy Technology Laboratory (NETL) suggesting that implementation will result in a parasitic consumption of 38% of total plant energy output leading to more than a doubling of the cost of the electricity produced by the plant.5 New types of solvents such as ionic liquids, which have been shown to be effective for the absorption of both CO2 and SO2 and have the advantage of being non-volatile, are being investigated for their potential use in carbon dioxide capture.221,222 Adsorption technologies, where the CO2 is captured by a solid sorbent rather than a liquid solution, hold great promise for alternative, lower cost capture technologies. Specifically, nanoengineered solids that capture CO2 efficiently and can be easily regenerated are needed. As an example, solid materials functionalized with amine sites that chemisorb CO2 have been engineered to have large capacities for CO2.223,224 Alternatively, functionalized ionic liquids can be coated onto nanoporus materials and utilized for CO2 capture.225 Advances in the ability to design the sorbent materials at the nanoscale will enable the atomic or molecular scale engineering of the CO2 binding properties (e.g., enthalpy of adsorption) as well as the critical sorbent transport properties (e.g., gas diffusion coefficients) that directly determine the efficacy of the adsorbents. In parallel, new regenerable sorbents for the capture of NOx and SO2 are needed, and it is anticipated that such materials may be developed by nanoscale design combining theory and experiment.
New power plant designs, for example, integrated gasification combined cycle (IGCC) and oxy–combustion plants, that are more amenable to low cost CCS, are under development. These designs offer different constraints on the CO2 capture problem. In IGCC, CO2 is captured at high temperatures from a H2/CO2 mixture. Dense metal membranes are one attractive route to capturing CO2 from this mixture, although challenges remain to develop membrane materials that are resistant to the chemical impurities ubiquitous in coal gasification.226 Controlling the structure of metal films on nanometer length scales, either by modifying the surface properties of films or by using amorphous structures rather than crystalline structures, has the potential for making step changes in the performance of these membranes.227
Thus, advances in nanoscale materials design are needed to allow expanded use of fossil fuels with CCS while slowing the rate of increase of emissions of CO2 in the short term, and while simultaneous work on nanoscale design of materials for solar and thermal energy management is carried out, seeking long-term energy solutions. In particular, the future efforts should focus on:
In the near-term (2–5 years), conventional amine-based absorption/stripping processes will be the first to be deployed for post-combustion CO2 capture. Therefore, immediate efforts should be directed at developing new liquid absorbent systems with superior performance properties that can serve as “drop in” replacements for amines. These systems would be designed using computational and nanoengineering techniques to have optimal capacity and regeneration characteristics, thereby lowering parasitic energy losses and costs. These processes will still be costly and inefficient, however, and therefore broad research efforts should be initiated to search for new, breakthrough technologies that can “leapfrog” these absorption processes.
In the mid-term (5–10 years), the goal will be to develop revolutionary new materials designed specifically for large-scale CO2 separation. New materials, such as nanoengineered porous materials and membranes, as well as “hybrid” materials such as chemically functionalized solids, will enable separation processes that approach high thermodynamic efficiency without the large thermal regeneration loads required by chemical solvents.
In the long-term (>10 years), conventional coal-fired power plants will gradually be replaced with new technologies utilizing gasification technologies. Materials will need to be developed that are optimized for H2/CO2 separation under high pressures, high temperatures, and in the presence of contaminants. These technologies will be relevant for gasification of feedstocks varying from coal to municipal waste and biomass, and therefore the design of materials and processes to optimize systems performance in diverse settings will be vital.
Summary and outlook
In this article, we critically assess the challenges faced by the research community in meeting the demands for sustainable energy in the near-, mid-, and long-term time frames. The projected need is to globally deliver power up to 25–30 TW by 2050 and 45–50 TW by 2100, with >50% of it coming from renewable sources. Providing the world with a sustainable energy supply without damage to the global environment poses a truly grand challenge to the world, crossing national boundaries and demanding massive scale societal investment in the search of the sustainable energy generation, storage, and distribution infrastructure of the future. This societal investment should have multiple dimensions: current industrial practices should become much more “energy-aware” and fully embrace energy conservation technologies and policies; development of largely CO2-emission-free transportation should be encouraged, especially in dense urban communities; and pubic awareness of future energy challenges must be raised including the need for policy changes to respond to these challenges. However, first and foremost, the magnitude of the challenge and urgency to achieve the desired goal within the designated timeframe demand rapid acceleration of scientific progress in energy-related disciplines and technologies. Such a result can only be realized through a combination of significant investments in energy research and development along with advanced education and training of a large contingent of scientists and engineers, working on the most pressing scientific and engineering challenges in the field of energy. Further, considering the relatively short time (∼40 to 90 years) we have to double-to-triple the present global energy supply, as well as to make a dramatic change in the distribution of the energy mix (from primarily fossil fuels to largely renewable sources), we argue that only revolutionary change in how we produce energy (convert it from the primary source to its useful form) and how we manage energy (store, distribute and conserve) will allow us to meet the demand goals within the pressing time schedule.We focus our attention on a critically important subset of renewable technologies concerned with solar, thermal, and electrochemical energy conversion, storage, and conservation. On the basis of this discussion we highlight the need for a step-wise advance in energy science and technology within the next decades, and we emphasize the importance of nanoscience and nanotechnology to enable such advances to occur. The search for new materials compositions and nanostructures with intrinsically superior transport and storage properties should be at the focus of the basic science efforts, towards evaluating the broadest range of chemistries using powerful combinatorial screening tools. Through this nanoscience-enabled search, the champion(s) class of materials/structures will likely emerge, materials which are either naturally abundant and readily accessible or should serve as a proxy for the development of affordable synthetic materials possessing similar properties, thus forming an elementary building block for the design of energy conversion and storage devices and systems. These accelerated advances in nanoscale design of materials should proceed hand-in-hand with the development of tools and methodologies for economically sound, scalable manufacturing and integration practices. This grand challenge calls for joint work between scientists and engineers across the spectrum of disciplines which, in this paper, we broadly refer to as the “nanoscale design for renewable energy revolution”.
By examining the specifics of the underlying science and the challenges to be addressed, we identified several cross-cutting themes of nanoscale design that are critical to the development of the next generation of solar, thermal, and electrochemical energy conversion, storage, and conservation technologies, expressed in terms of the manipulation and control of basic energy carriers (i.e., photons, excitons, electrons/holes, phonons, and molecules/ions), emphasizing the importance of interactions that occur on the nanoscale. These unifying themes, in which “nano” enables marrying diverse functionalities, suggest priority directions for present investment in nanoscale research that will lead to the greatest and broadest future impact across the entire spectrum of nanotechnology-enabled energy conversion, storage, and conservation technologies.
• Multifunctional and selective active interfaces
Engineering the interfaces of nanostructures is of the outmost importance to achieve the desired carrier transport characteristics and efficient coupling between different energy conversion processes with minimal dissipative losses. Selective interfaces with spectrally tunable properties to enable wavelength-selective filtering of energy carriers, and with chemical functionality to promote preferential adsorption/selective separation of chemical species will enable efficient utilization of the entire frequency spectrum of the primary energy source (be it solar or thermal) and efficient solar/thermal-to-electro/chemical energy conversion and storage.• Scale coupling for energy conversion and storage
Energy storage occurs at the nanoscale, primarily in the energy bonding one atom to another and, on this fundamental level, it cannot be separated from energy conversion and transport. Thus, matching the time scales (relaxation times) of the relevant energy carriers participating in energy conversion processes (i.e., photons, electrons, phonons) by managing and/or engineering their characteristic length scales (mean free paths and wavelength) and propagation velocities is an essential prerequisite to sound nanoscale design of energy conversion and storage devices.• Catalyst design for fast, high density conversion and storage
Design of catalytically active materials on the nanoscale offers an opportunity to dramatically lower the activation energy for catalytic reactions (e.g., photocatalytic production of renewable fuels and electrochemical conversion and storage devices) that enable an exponential increase in the rates of energy conversion and storage. Catalytic activity is an essential driver to achieve a Moore's-law-like increase in the supply of energy in technologically usable form and at low cost. Development of active catalysts with minimal materials usage and the use of cheap, abundant materials for catalysis are central to address the issue of availability/limited materials resources.• Device/process/material design for multifunctionality
Multifunctional processes and devices with an intimate coupling of energy conversion and storage, although fundamentally more complex and difficult to engineer at the nanoscale, have the greatest potential for major impact. Synthesis of biofuels and photocatalytic production of solar fuels are examples of such processes, which not only convert and store solar energy into practically useful, high energy density substances, but also consume greenhouse pollutant CO2 as a co-reagent of the reaction. An ability to fine-tune the properties of materials (e.g., via domain-selective doping) and to precisely control their structural features (e.g., catalyst active site and supporting structure) are potentially promising avenues for overcoming limitations on performance that appear to be fundamental barriers when material/device design is done on a conventional (macro) scale.• Hybrid materials for energy efficient separation
Energy efficient approaches to selective chemical species separation at high throughput using nanoengineered membranes and porous sorbents, in particular targeting carbon dioxide capture and hydrogen purification, are fundamental to viability of a host of energy storage and conversion technologies, from fuel cells and hydrogen storage to biofuels and solar fuels synthesis and processing for power generation. In particular, hybrid materials, such as chemically-functionalized nanostructured solids are promising for large-scale CO2 separation for its capture, with an energy efficiency approaching the thermodynamic limit.• Optimized substrates and supporting structures
The critical importance of a substrate for high efficiency and practical viability of nanoscale energy conversion should not be overlooked. The substrate is defined here in its broadest sense, including a support structure for catalysts (e.g., a meso/nano-scopic zeolite matrix hosting dispersed nanoparticles of the catalyst for photocatalysis or for electrodes in fuel cells, batteries and capacitors); a high surface area, gas-permeable backbone for an active sorbent material (e.g., CO2 capture); a substrate for solid-state-lighting devices (e.g., sapphire for white LED lamps); among others. The substrates not only must be cheap, manufacturable, but must first and foremost have the desired physico-chemical properties which often play an important functional role in device/process performance. Ultimately, the quest is for cheap meso/nano-structured substrates, such as plastic, polymers and paper-like materials that could be modified on the nanoscale to attain the desired structural and physical characteristics for the application in hand.• Exploitation of liquid/gas properties in a confined state
Engineering (tuning) liquid/gas properties of materials by nanoscale confinement is a relatively unexplored venue that can lead to breakthroughs in performance for a number of critical energy-related applications. In particular, this could result in conceptually new designs of thermal insulation materials aiming to simultaneously minimize convective, conductive and radiative heat losses, of smart windows with directional and wavelength dependent optical properties, and improved efficiency of electrochemical storage devices (batteries and capacitors). Furthermore, conventional bulk-solution-based absorption/stripping processes for post-combustion CO2 capture may be also impacted, resulting in the development of new types of liquid absorption systems with superior performance as compared to currently used amine-based CO2 capture.• Manufacturing for affordable nanostructures and devices
In addition to opening up new, untapped functionalities and enhancing the existing operating modes of conventional processes/devices/systems, nanotechnology offers a unique possibility to realize these dramatic improvements in practice on a large scale and at much lower cost. This realization of nanoscale improvement relative to the macroscale is owing to the possibility of minimizing the amount of expensive (“active”) materials that is used, which in the limit is just a single atomic layer deposited on a surface of a cheap substrate. An appealing feature of such a nanotechnology-enabled approach is that a dramatic decrease in cost often occurs not only without sacrificing, but actually with improving performance (in many cases, for example solar cells, where thinner structures result in improved performance by minimizing negative bulk effects). However, this promise could only be realized if advanced manufacturing technologies are developed for making nanostructures at high throughput and low cost. Techniques such as Atomic Layer Deposition (ALD) are very promising if they could only be made less expensive with greatly increased throughput; while also the creative adaptation of low-cost (“low-tech”) conventional manufacturing tools and processes (e.g., ball-milling has been recently used for making low-cost thermoelectric nanocomposite materials with record-setting performance numbers) for making nanomaterials is an interesting avenue that needs to be explored to utilize an already available manufacturing base and investments in such systems.• Adaptive and flexible design approach for managing complexity
To deliver on its promise of revolutionary improvement in performance, the next generation of energy conversion and storage devices needs to be designed with nano-enabled functionality in mind, thus resulting in an effective top-down approach to nanoscale design that could fully utilize the unique features and capabilities offered by nanoscale materials and structures. Nanoscale control is one side of the problem, and complexity is the other. Both need to be managed to achieve the desired performance improvement for the final processes and products which are used on the large (macro) scale. Development of nanomaterials/nanostructures that are flexible and adaptable (i.e., featuring high functionality at the nanoscale and tolerating disorder on the mesoscale) provides a route to scalable and economical manufacturing.Predicting the future is a risky task and highly speculative. Nevertheless, we believe that placing the research needs and state-of-the-art in context, as this paper has attempted to do, will prove to be useful in informing and stimulating activities that could produce such breakthroughs. Hence, the mid-term and long-term goals and unifying themes suggested in this article must be considered as a strategic process, where the mid-term and long-term goals are evolutionary and must be periodically revisited and re-calibrated based on near-term successes and failures, so that the resulting recommendations will provide scientifically sound drivers and a roadmap for focusing future research activities on the most promising approaches.
Finally, of no lesser importance is the need for educating and training a workforce that is capable to lead the revolution to a sustainable energy system. Of particular concern is the education of research personnel and engineering practitioners who are knowledgeable in the latest advances in nanoscience and nanotechnology, and are focused on and committed to energy-relevant applications. This skill set includes not only expansion of energy-specific educational programs for talented and motivated youth in colleges and universities at the undergraduate and graduate levels, but also the professional development and re-training of experienced researchers at the post-doctoral level at the leading centers of energy research. Ultimately, it is our responsibility to invest wisely and with urgency in clean technologies that offer promise in helping to solve the energy challenge and to implement the solution. The benefits of success for present and future generations will be incalculable, enabling sustainable advances in the quality of life for the entire world population without sacrificing the vitality of the environment.
Acknowledgements
Financial support for the workshop was provided by the US National Science Foundation, the Journal of Energy & Environmental Science, and the General Catalysts Partners Venture Fund. The authors thank Dr Gene Dresselhaus for his contribution to the preparation of this manuscript.References
- M. I. Hoffert, K. Caldeira, A. K. Jain, E. F. Haites, L. D. D. Harvey, S. D. Potter, M. E. Schlesinger, S. H. Schneider, R. G. Watts, T. M. L. Wigley and D. J. Wuebbles, Nature, 1998, 395, 881–884 CrossRef CAS.
- IPCC 2007 Summary for Policymakers, in Climate Change 2007: Impacts, Adaptation and Vulnerability. Contribution of Working Group II to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change (Ed. M. L. Parry, J. P. Palutikof, P. J. van der Linden and C. E. Hanson), Cambridge University Press, Cambridge, UK, 2007, 7–22 Search PubMed.
- J. Hansen, M. Sato, P. Kharecha, D. Beerling, R. Berner, V. Masson-Delmotte, M. Pagani, M. Raymo, D. L. Royer and J. C. Zachos, Open Atmos. Sci. J., 2008, 2, 217–231 Search PubMed.
- Avoiding Dangerous Climate Change, Exeter, UK, 2005, http://www.stabilisation2005.com Search PubMed.
- K. S. Fisher, C. Beitler, C. Rueter, K. Searcy, G. Rochelle, M. Jassim and J. D. Figueroa, Integrating MEA Regeneration with CO2 Compression and Peaking to Reduce CO2 Capture Costs, DOE/NETL, May 2–5, 2005 Search PubMed.
- N. S. Lewis, G. Crabtree, A. J. Nozik, M. R. Wasielewski and P. Alivisatos, Basic Research Needs for Solar Energy Utilization, DOE, 2006 Search PubMed.
- M. A. Green, K. Emery, D. L. King, Y. Hishikawa and W. Warta, Prog. Photovoltaics: Res. Appl., 2007, 15, 35–40 Search PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CAS.
- M. A. Green, Prog. Photovoltaics, 2001, 9, 123–135 CAS.
- A. J. Nozik, Physica E, 2002, 14, 115–120 CrossRef CAS.
- D. Y. Song, E. C. Cho, G. Conibeer, C. Flynn, Y. D. Huang and M. A. Green, Sol. Energy Mater. Sol. Cells, 2008, 92, 474–481 CrossRef CAS.
- R. D. Schaller, M. Sykora, J. M. Pietryga and V. I. Klimov, Nano Lett., 2006, 6, 424–429 CrossRef CAS.
- R. D. Schaller and V. I. Klimov, Phys. Rev. Lett., 2004, 92, 186601–186604 CrossRef CAS.
- M. C. Beard, K. P. Knutsen, P. Yu, J. M. Luther, Q. Song, W. K. Metzger, R. J. Ellingson and A. J. Nozik, Nano Lett., 2007, 7, 2506–2512 CrossRef CAS.
- R. J. Ellingson, M. C. Beard, J. C. Johnson, P. R. Yu, O. I. Micic, A. J. Nozik, A. Shabaev and A. L. Efros, Nano Lett., 2005, 5, 865–871 CrossRef CAS.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510–074518 CrossRef.
- D. Timmerman, I. Izeddin, P. Stallinga, I. N. Yassievich and T. Gregorkiewicz, Nat. Photon., 2008, 2, 105–109 CrossRef CAS.
- R. T. Ross and A. J. Nozik, J. Appl. Phys., 1982, 53, 3813–3818 CrossRef CAS.
- M. Neges, K. Schwarzburg and F. Willig, Sol. Energy Mater. Sol. Cells, 2006, 90, 2107–2128 CrossRef CAS.
- J. B. Baxter and E. S. Aydil, Appl. Phys. Lett., 2005, 86, 053114–053117 CrossRef.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. D. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS.
- C. Levy-Clement, R. Tena-Zaera, M. A. Ryan, A. Katty and G. Hodes, Adv. Mater., 2005, 17, 1512–1515 CrossRef CAS.
- B. M. Kayes, H. A. Atwater and N. S. Lewis, J. Appl. Phys., 2005, 97, 114302 CrossRef.
- B. O'Regan and M. Gratzel, 1991, Nature, 353, pp. 737–740 Search PubMed.
- M. Gratzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef.
- H. J. Snaith and L. Schmidt-Mende, Adv. Mater., 2007, 19, 3187–3200 CrossRef CAS.
- J. Bernreuter, Bild Wiss.., 2007, 84–87 Search PubMed.
- D. V. Talapin and C. B. Murray, Science, 2005, 310, 86–89 CrossRef CAS.
- A. N. Goldstein, C. M. Echer and A. P. Alivisatos, Science, 1992, 256, 1425–1427 CrossRef CAS.
- F. G. Shi, J. Mater. Res., 1994, 9, 1307–1312 CrossRef CAS.
- K. K. Nanda, Eur. J. Phys., 1998, 19, 471–472 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- D. L. Damm and A. G. Fedorov, Energy Convers. Manage., 2008, 49, 1674–1683 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- A. Ogden, J. L. Gole and A. G. Fedorov, J. Nanoelectron. Optoelectron., 2007, 2, 269–277 Search PubMed.
- A. Kudo, H. Kato and I. Tsuji, Chem. Lett., 2004, 33, 1534–1539 CrossRef CAS.
- Y. B. L. A. C. S. S. C. B. J. L. G. X. Chen, Adv. Funct. Mater., 2005, 15, 41–49 CrossRef CAS.
- J. L. Gole, J. D. Stout, C. Burda, Y. Lou and X. Chen, J. Phys. Chem. B, 2004, 108, 1230–1240 CrossRef CAS.
- L. Xiao, J. Zhang, Y. Cong, B. Tian, F. Chen and M. Anpo, Catal. Lett., 2006, 111, 207–211 CrossRef CAS.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- H. Fu, L. Zhang, S. Zhang, Y. Zhu and J. Zhao, J. Phys. Chem. B, 2006, 110, 3061–3065 CrossRef CAS.
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2005, 109, 8920–8927 CrossRef CAS.
- H. Habazaki, M. Yamasaki, B. P. Zhang, A. Kawashima, S. Kohno, T. Takai and K. Hashimoto, Appl. Catal., A: Gen., 1998, 172, 131–140 CrossRef CAS.
- A. Steinfeld and R. Palumbo, Encycl. Phys. Sci. Technol., 2001, 15, 237–256 Search PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS.
- R. Nakamura and H. Frei, J. Am. Chem. Soc., 2006, 128, 10668–10669 CrossRef CAS.
- H. X. Han and H. Frei, J. Phys. Chem. C, 2008, 112, 8391–8399 CrossRef CAS.
- A. Martí and A. Luque, Next Generation Photovoltaics: High Efficiency Through Full Spectrum Utilization, Institute of Physics, Bristol, UK, 2003 Search PubMed.
- P. Wurfel and W. Ruppel, IEEE Trans. Electron. Dev., 1980, 27, 745–750 CrossRef.
- M. Telkes, J. Appl. Phys., 1954, 25, 765–777 CrossRef CAS.
- H. J. Goldsmid Applications of Thermoelectricity, Wiley, London, New York, Methuen, 1960 Search PubMed.
- H. Scherrer and S. Scherrer, CRC Handbook of Thermoelectrics, 1995, p. 211–237 Search PubMed.
- T. Caillat, J. P. Fleurial, G. N. Snyder, A. Zoltan, D. Zoltan and A. Borshchevsky, IEEE, 18th International Conference on Thermoelectrics, Piscataway, NJ, 1999, pp. 473–476 Search PubMed.
- T. Kajikawa and T. Onishi, Proceedings of the 26th International Conference on Thermoelectrics (ICT 2007), 2007 Search PubMed.
- T. C. Harman, P. J. Taylor, M. P. Walsh and B. E. LaForge, Science, 2002, 297, 2229–2232 CrossRef CAS.
- K. F. Hsu, S. Loo, F. Guo, W. Chen, J. S. Dyck, C. Uher, T. Hogan, E. K. Polychroniadis and M. G. Kanatzidis, Science, 2004, 303, 818 CrossRef CAS.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS.
- B. Bitnar, W. Durisch, J. C. Mayor, H. Sigg and H. R. Tschudi, Sol. Energy Mater. Sol. Cells, 2002, 73, 221–234 CrossRef CAS.
- R. E. Nelson, Proceedings of the 1st NREL Conference on Thermophotovoltaic Generation of Electricity, 1995 Search PubMed.
- A. Heinzel, V. Boerner, A. Gombert, B. Bläsi, V. Wittwer and J. Luther, J. Mod. Opt., 2000, 47, 2399–2419 CAS.
- P. M. Martin, L. C. Olsen, J. W. Johnston and D. M. Depoy, Appl. Opt., 2002, 41, 6702–6707 CrossRef CAS.
- J. G. Fleming, S. Y. Lin, I. El-Kady, R. Biswas and K. M. Ho, Nature, 2002, 417, 52–55 CrossRef CAS.
- W. Vielstich, A. Lamm and H. A. Gasteiger, Handbook of Fuel Cells: Fundamentals, Technology, and Applications, Wiley, Chichester, England, 2003 Search PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS.
- A. Roy, M. A. Hickner, X. Yu, Y. X. Li, T. E. Glass and J. E. McGrath, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2226–2239 CrossRef CAS.
- Y. Z. Fu, A. Manthiram and M. D. Guiver, Electrochem. Commun., 2006, 8, 1386–1390 CrossRef CAS.
- K. T. Adjemian, S. J. Lee, S. Srinivasan, J. Benziger and A. B. Bocarsly, J. Electrochem. Soc., 2002, 149, A256–A261 CrossRef CAS.
- D. J. Berger, Science, 1999, 286, 49–49 CrossRef CAS.
- M. L. Anderson, R. M. Stroud and D. R. Rolison, Nano Lett., 2002, 2, 235–240 CrossRef.
- V. Raghuveer, A. Manthiram and A. J. Bard, J. Phys. Chem. B, 2005, 109, 22909–22912 CrossRef CAS.
- P. J. Ferreira, G. J. la O, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef.
- K. T. Lee and A. Manthiram, Chem. Mater., 2006, 18, 1621–1626 CrossRef CAS.
- Y. H. Huang, R. I. Dass, Z. L. Xing and J. B. Goodenough, Science, 2006, 312, 254–257 CrossRef CAS.
- C. Laberty-Robert, J. W. Long, K. A. Pettigrew, R. M. Stroud and D. R. Rolison, Adv. Mater., 2007, 19, 1734–1739 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS.
- P. B. Weisz, V. J. Frilette, R. W. Maatman and E. B. Mower, J. Catal., 1962, 1, 307–312 CrossRef CAS.
- P. B. Weisz and J. N. Miale, J. Catal., 1965, 4, 527–529 CrossRef CAS.
- A. Corma, M. S. Grande, V. GonzalezAlfaro and A. V. Orchilles, J. Catal., 1996, 159, 375–382 CrossRef CAS.
- T. A. Milne, R. J. Evans and N. Abatzoglou, Biomass Gasifier Tars: Their Nature, Formation and Conversion, National Renewable Energy Laboratory, Golden, CO, 1998, http://www.osti.gov/bridge Search PubMed.
- E. G. Baker, L. K. Mudge and M. D. Brown, Ind. Eng. Chem. Res., 1987, 26, 1335–1339 CrossRef CAS.
- K. Tomishige, M. Asadullah and K. Kunimori, Catal. Today, 2004, 89, 389–403 CrossRef CAS.
- R. L. Bain, D. C. Dayton, D. L. Carpenter, S. R. Czernik, C. J. Feik, R. J. French, K. A. Magrini-Bair and S. D. Phillips, Ind. Eng. Chem. Res., 2005, 44, 7945–7956 CrossRef CAS.
- V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115–2117 CrossRef CAS.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Bioresour. Technol., 2005, 96, 1959–1966 CrossRef CAS.
- A. Amerasekera, W. Abeelen, L. Roozendal, M. Hannemann and P. Schofield, IEEE Trans. Electron. Dev., 1992, 39, 430–436 CrossRef.
- Q. Chen and Z. J. Zhang, Appl. Phys. Lett., 1998, 73, 3156–3158 CrossRef CAS.
- C. S. Gill, B. A. Price and C. W. Jones, J. Catal., 2007, 251, 145–152 CrossRef CAS.
- N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209–2212 CrossRef.
- J. S. Lettow, Y. J. Han, P. Schmidt-Winkel, P. D. Yang, D. Y. Zhao, G. D. Stucky and J. Y. Ying, Langmuir, 2000, 16, 8291–8295 CrossRef CAS.
- A. Imhof and D. J. Pine, Nature, 1997, 389, 948–951 CrossRef CAS.
- A. V. Bridgwater and G. V. C. Peacocke, Renewable Sustainable Energy Rev., 2000, 4, 1–73 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- J. M. Moffatt and R. P. Overend, Biomass, 1985, 7, 99–123 CrossRef.
- A. V. Bridgwater, Appl. Catal., A: Gen., 1994, 116, 5–47 CrossRef CAS.
- P. T. Williams and N. Nugranad, Energy, 2000, 25, 493–513 CrossRef CAS.
- M. Olazar, R. Aguado, J. Bilbao and A. Barona, AICHE J., 2000, 46, 1025–1033 CrossRef CAS.
- T. R. Carlson, T. R. Vispute and G. W. Huber, Chemsuschem, 2008, 1, 397–400 CrossRef CAS.
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- Q. G. Yan, P. T. Doan, H. Toghiani, A. C. Gujar and M. G. White, J. Phys. Chem. C, 2008, 112, 11847–11858 CrossRef CAS.
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- J. M. H. Lo and T. Ziegler, J. Phys. Chem. C, 2008, 112, 13681–13691 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- E. Furimsky, Appl. Catal., A: Gen., 2000, 199, 147–190 CrossRef CAS.
- R. K. Sharma and N. N. Bakhshi, Can. J. Chem. Eng., 1991, 69, 1071–1081 CAS.
- S. Vitolo, M. Seggiani, P. Frediani, G. Ambrosini and L. Politi, Fuel, 1999, 78, 1147–1159 CrossRef CAS.
- C. Baerlocher and L. B. McCuskerDatabase of Zeolite Structures, http://www.iza-structure.org/databases/ Search PubMed.
- O. M. Yaghi, Nat. Mater., 2007, 6, 92–93 CrossRef CAS.
- M. Nyman, F. Bonhomme, T. M. Alam, M. A. Rodriguez, B. R. Cherry, J. L. Krumhansl, T. M. Nenoff and A. M. Sattler, Science, 2002, 297, 996–998 CrossRef CAS.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS.
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- D. K. Ross, Vacuum, 2006, 80, 1084–1089 CrossRef CAS.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef.
- M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef CAS.
- C. Liu, Y. Y. Fan, M. Liu, H. T. Cong, H. M. Cheng and M. S. Dresselhaus, Science, 1999, 286, 1127–1129 CrossRef CAS.
- R. T. Yang, Carbon, 2000, 38, 623–626 CrossRef CAS.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuettel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- S. V. Alapati, J. K. Johnson and D. S. Sholl, Phys. Chem. Chem. Phys., 2007, 9, 1438–1452 RSC.
- C. Sachs, A. Pundt, R. Kirchheim, M. Winter, M. T. Reetz and D. Fritsch, Phys. Rev. B, 2001, 64, 075408 CrossRef.
- J. J. Vajo and G. L. Olson, Scr. Mater., 2007, 56, 829–834 CrossRef CAS.
- K. Aguey-Zinsou and J. Ares-Fernandez, Chem. Mater., 2008, 20, 376–378 CrossRef CAS.
- R. Kadono, K. Shimomura, K. H. Satoh, S. Takeshita, A. Koda, K. Nishiyama, E. Akiba, R. M. Ayabe, M. Kuba and C. M. Jensen, Phys. Rev. Lett., 2008, 100, 026401 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS.
- J. S. Zhang, T. S. Fisher, P. V. Ramachandran, J. P. Gore and I. Mudawar, J. Heat Transfer, 2005, 127, 1391–1399 CrossRef CAS.
- V. Bérubé, G. Radtk, M. Dresselhaus and G. Chen, Int. J. Energy Res., 2007, 31, 637–663 CrossRef CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1316 CrossRef CAS.
- H. Smithson, C. A. Marianetti, D. Morgan, A. Van der Ven, A. Predith and G. Ceder, Phys. Rev. B, 2002, 66, 144107 CrossRef.
- R. Prasher, Phys. Rev. B, 2006, 74, 165413 CrossRef.
- W. Zhang, N. Mingo and T. Fisher, Phys. Rev. B, 2007, 76, 195429 CrossRef.
- M. J. Varady, L. McLeod, J. M. Meacham, F. L. Degertekin and A. G. Fedorov, J. Micromech. Microeng., 2007, 17, S257–S264 CrossRef CAS.
- C.-J. Winter, Int. J. Hydrogen Energy, 2006, 31, 1623–1631 CrossRef CAS.
- G. W. Scheffler, J. DeVaal, G. Kissel, J. Schneider, M. Veenstra, N. Kinoshita, G. Nicols and H. Fukumoto, Proceedings of SAE World Congress & Exposition, Detroit, MI, 2008 Search PubMed.
- S. Aceves, G. Berry, J. Martinezfrias and F. Espinosaloza, Int. J. Hydrogen Energy, 2006, 31, 2274–2283 CrossRef.
- J. Hu, J. Chen, S. Sundararaman, K. Chandrashekhara and W. Chernicoff, Int. J. Hydrogen Energy, 2008, 33, 2738–2746 CrossRef CAS.
- D. R. Rolison, J. W. Wang, J. C. Lytle, A. E. Fischer, C. P. Rhodes, T. M. McEvoy, M. E. Bourg and A. M. Lubers, Chem. Soc. Rev., 2009, 38 Search PubMed in press.
- R. J. Gummow, A. Dekock and M. M. Thackeray, Solid State Ionics, 1994, 69, 59–67 CrossRef CAS.
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2007, 154, A792–A797 CrossRef CAS.
- J. M. Han, S. T. Myung and Y. K. Sun, J. Electrochem. Soc., 2006, 153, A1290–A1295 CrossRef CAS.
- A. Nyten, S. Kamali, L. Haggstrom, T. Gustafsson and J. O. Thomas, J. Mater. Chem., 2006, 16, 2266–2272 RSC.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123–128 CrossRef CAS.
- B. Ellis, W. H. Kan, W. R. M. Makahnouk and L. F. Nazar, J. Mater. Chem., 2007, 17, 3248–3254 RSC.
- G. G. Amatucci and N. Pereira, J. Fluorine Chem., 2007, 128, 243–262 CrossRef CAS.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, Chem. Rev., 2004, 104, 4463–4492 CrossRef CAS.
- Y. Wu and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A221–A224 CrossRef CAS.
- J. W. Long, C. P. Rhodes, A. L. Young and D. R. Rolison, Nano Lett., 2003, 3, 1155–1161 CrossRef CAS.
- P. E. Tang, J. S. Sakamoto, E. Baudrin and B. Dunn, Proceedings of 7th International Symposium on Aerogels, Alexandria, VA, 2003 Search PubMed.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 Search PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- T. Zhang, L. J. Fu, J. Gao, L. C. Yang, Y. P. Wu and H. Q. Wu, Pure Appl. Chem., 2006, 78, 1889–1896 CrossRef CAS.
- P. H. L. Notten, F. Roozeboom, R. A. H. Niessen and L. Baggetto, Adv. Mater., 2007, 19, 4564–4567 CrossRef CAS.
- T. Ogasawara, A. Debart, M. Holzapfel, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281–286 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Plenum Press, New York, 1999 Search PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- A. V. Murugan and K. Vijayamohanan, Nanomaterials Chemistry, Wiley-VCH GmbH & Co., Weinheim, 2007, p. 219 Search PubMed.
- P. Simon and A. Burke, Interface, 2008, 17, 38–43 Search PubMed.
- K. Naoi and P. Simon, Interface, 2008, 17, 34–37 Search PubMed.
- J. P. Zheng and T. R. Jow, J. Electrochem. Soc., 1995, 142, L6–L8 CAS.
- W. Dmowski, T. Egami, K. E. Swider-Lyons, C. T. Love and D. R. Rolison, J. Phys. Chem. B, 2002, 106, 12677–12683 CrossRef CAS.
- T. Shinomiya, V. Gupta and N. Miura, Electrochim. Acta, 2005, 51, 4412–4419.
- Y. U. Jeong and A. Manthiram, J. Electrochem. Soc., 2002, 149, A1419–A1422 CrossRef CAS.
- Y. U. Jeong and A. Manthiram, Electrochem. Solid-State Lett., 2000, 3, 205–208 CAS.
- Y. U. Jeong and A. Manthiram, J. Electrochem. Soc., 2001, 148, A189–A193 CrossRef CAS.
- D. R. Rolison and B. Dunn, J. Mater. Chem., 2001, 11, 963–980 RSC.
- D. R. Rolison, J. W. Long, J. C. Lytle, A. E. Fischer, C. P. Rhodes, T. M. McEvoy, M. E. Bourg and A. M. Lubers, Chem. Soc. Rev., 2009, 38, 226–252 RSC.
- W. Dong, D. R. Rolison and B. Dunn, Electrochem. Solid-State Lett., 2000, 3, 457–459 CrossRef CAS.
- H. J. Goldsmid, Thermoelectric Refrigeration, Plenum Press, New York, 1964 Search PubMed.
- D. Kraemer, L. Hu, A. Muto, X. Chen, G. Chen and M. Chiesa, Appl. Phys. Lett., 2008, 92, 3.
- L. E. Bell, Science, 2008, 321, 1457–1461 CrossRef CAS.
- T. Kajikawa and T. Onishi, Proceedings of the 26th International Conference on Thermoelectrics (ICT 2007), Jeju Island, South Korea, 2007 Search PubMed.
- G. D. Mahan and J. O. Sofo, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7436–7439 CrossRef CAS.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B, 1993, 47, 12727–12731 CrossRef CAS.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B, 1993, 47, 16631–16634 CrossRef CAS.
- J. P. Heremans, V. Jovovic, E. S. Toberer, A. Saramat, K. Kurosaki, A. Charoenphakdee, S. Yamanaka and G. J. Snyder, Science, 2008, 321, 554–557 CrossRef CAS.
- D. Vashaee and A. Shakouri, Phys. Rev. Lett., 2004, 92, 106103 CrossRef.
- J. M. O. Zide, D. Vashaee, Z. X. Bian, G. Zeng, J. E. Bowers, A. Shakouri and A. C. Gossard, Phys. Rev. B, 2006, 74, 205335 CrossRef.
- R. Venkatasubramanian, E. Siivola, T. Colpitts and B. O'Quinn, Nature, 2001, 413, 597–602 CrossRef CAS.
- K. F. Hsu, S. Loo, F. Guo, W. Chen, J. S. Dyck, C. Uher, T. Hogan, E. K. Polychroniadis and M. G. Kanatzidis, Science, 2004, 303, 818–821 CrossRef CAS.
- B. Poudel, Q. Hao, Y. Ma, Y. C. Lan, A. Minnich, B. Yu, X. Yan, D. Z. Wang, A. Muto, D. Vashaee, X. Y. Chen, J. M. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS.
- G. S. Nolas, D. T. Morelli and T. M. Tritt, Annu. Rev. Mater. Sci., 1999, 29, 89–116 CrossRef CAS.
- W. Kim, J. Zide, A. Gossard, D. Klenov, S. Stemmer, A. Shakouri and A. Majumdar, Phys. Rev. Lett., 2006, 96, 045901 CrossRef.
- S. V. Faleev and F. Leonard, Phys. Rev. B, 2008, 77, 214304 CrossRef.
- M. S. Dresselhaus, G. Chen, M. Y. Tang, R. G. Yang, H. Lee, D. Z. Wang, Z. F. Ren, J. P. Fleurial and P. Gogna, Adv. Mater., 2007, 19, 1043–1053 CrossRef CAS.
- H. J. Goldsmid, Proc. Phys. Soc., 1958, 72, 17–26 CrossRef CAS.
- W. Kim, R. Wang and A. Majumdar, Nano Today, 2007, 2, 40–47 CrossRef.
- G. A. Slack, J. Appl. Phys., 1964, 35, 3460–3466 CrossRef CAS.
- L. W. Hrubesh and R. W. Pekala, J. Mater. Res., 1994, 9, 731–738 CrossRef CAS.
- G. Chen, Phonon Heat Conduction in Low-Dimensional Structures,, in Semiconductors and Semimetals, Recent Trends in Thermoelectric Materials Research III (Ed. T. A. p. Tritt, San Diego), Academic Press, San Diego, 2001 Search PubMed.
- C. Chiritescu, D. G. Cahill, N. Nguyen, D. Johnson, A. Bodapati, P. Keblinski and P. Zschack, Science, 2007, 315, 351–353 CrossRef CAS.
- R. M. Costescu, D. G. Cahill, F. H. Fabreguette, Z. A. Sechrist and S. M. George, Science, 2004, 303, 989–990 CrossRef CAS.
- A. I. Hochbaum, R. K. Chen, R. D. Delgado, W. J. Liang, E. C. Garnett, M. Najarian, A. Majumdar and P. D. Yang, Nature, 2008, 451, 163–165 CrossRef CAS.
- A. I. Boukai, Y. Bunimovich, J. Tahir-Kheli, J. K. Yu, W. A. Goddard and J. R. Heath, Nature, 2008, 451, 168–171 CrossRef CAS.
- Y. F. Chen, D. Y. Li, J. R. Lukes, Z. H. Ni and M. H. Chen, Phys. Rev. B, 2005, 72, 174302 CrossRef.
- R. Venkatasubramanian, Phys. Rev. B, 2000, 61, 3091–3097 CrossRef CAS.
- A. Mavrokefalos, N. T. Nguyen, M. T. Pettes, D. C. Johnson and L. Shi, Appl. Phys. Lett., 2007, 91, 171912 CrossRef.
- S. W. Kim, J. M. Zuo, N. T. Nguyen, D. C. Johnson and D. G. Cahill, J. Mater. Res., 2008, 23, 1064–1067 CrossRef CAS.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2000, 60, 201–262 CrossRef CAS.
- C. M. Lampert, Proceedings of International Symposium on Solar Energy Materials, Cancun, Mexico, 1996 Search PubMed.
- Department of Energy Basic Energy Sciences Advisory Committee, Basic Research Needs to Assure a Secure Energy Future, 2002 Search PubMed.
- T. Lorenz, M. Hofmann, M. Gruninger, A. Freimurth, G. S. Uhrig, M. Dumm and M. Dressel, Nature, 2002, 418, 614–617 CrossRef CAS.
- P. Kim, L. Shi, A. Majumdar and P. L. McEuen, Phys. Rev. Lett., 2001, 87, 215502 CrossRef CAS.
- S. T. Huxtable, D. G. Cahill, S. Shenogin, L. P. Xue, R. Ozisik, P. Barone, M. Usrey, M. S. Strano, G. Siddons, M. Shim and P. Keblinski, Nat. Mater., 2003, 2, 731–734 CrossRef CAS.
- A. Faghri, Heat Pipe Science and Technology, Taylor & Francis, Washington, DC, 1995 Search PubMed.
- Energy Savings Potential of Solid-State Lighting in General Illumination Applications, 2003, available at http://www.netl.doe.gov/ssl/publications-ssltechreports.htm Search PubMed.
- EU Ministers seek 2010 Bulb Ban, 2008, available at http://www.compoundsemiconductor.net Search PubMed.
- J. Broderick, LD + A Mag., 2007 Search PubMed.
- Department of Energy, Solid-State Lighting Research and Development: Multi-Year Program Plan FY′09-FY′14, 2008, available at http://www.netl.doe.gov/ssl/publications-techroadmaps.htm Search PubMed.
- M. Peter, Compd. Semiconduct., 2008 Search PubMed.
- D. Holec, P. Costa, M. J. Kappers and C. J. Humphreys, J. Cryst. Growth, 2007, 303, 314 CrossRef CAS.
- Y. C. Shen, G. O. Mueller, S. Watanabe, N. F. Gardner, A. Munkholm and M. R. Krames, Appl. Phys. Lett., 2007, 91, 141101 CrossRef.
- M. H. Kim, M. F. Schubert, Q. Dai, J. K. Kim, E. F. Schubert, J. Piprek and Y. Park, Appl. Phys. Lett., 2007, 91, 183507 CrossRef.
- Department of Energy, Efficacy of 45 lm/W Achieved in White OLED, available at http://www.netl.doe.gov/ssl/highlights_UDC07.htm) Search PubMed.
- D. L. Damm and A. G. Fedorov, Energy Convers. Manage., 2008, 49, 1674–1683 CrossRef CAS.
- J. L. Anthony, S. Aki, E. J. Maginn and J. F. Brennecke, Int. J. Environ. Technol. Manage., 2004, 4, 105–115 Search PubMed.
- J. L. Anderson, J. K. Dixon, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2006, 110, 15059–15062 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446–458 CrossRef CAS.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS.
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS.
- P. Kamakoti, B. D. Morreale, M. V. Ciocco, B. H. Howard, R. P. Killmeyer, A. V. Cugini and D. S. Sholl, Science, 2005, 307, 569–573 CrossRef CAS.
- S. Hao and D. S. Sholl, Energy Environ. Sci., 2008, 1, 175–183 RSC.
| This journal is © The Royal Society of Chemistry 2009 |