Bio-oxygenates and the peroxide number: a safety issue alert
Viktória Fábosa, Gabriella Koczóa, Hasan Mehdia, László Bodab and István T. Horváth*a
aInstitute of Chemistry, Eötvös University, Pázmány Péter Sétány 1/A, H-1117, Budapest, Hungary. E-mail: istvan.t.horvath@att.net
bMOL Plc, DS Development, Százhalombatta, Hungary
First published on 14th April 2009
Broader ContextThe conversion of biomass to carbon based chemicals is a rapidly growing field in sustainable development. Since monomers, oligomers, and polymers of carbohydrates are key components of biomass, various conversion schemes involve step-by-step removal of oxygen atoms leading to the formation of bio-oxygenates, generally with fewer and fewer oxygen atoms. As laboratory and commercial applications of new biomass-based chemicals increase daily, their safe production, storage, transportation and use must also become part of the development process. One of the most dangerous transformations of organic compounds under air is the formation of organic peroxides, these can in some circumstances cause violent explosions on either a laboratory or industrial scale. Also, peroxide reactions can initiate various other chemical reactions and thus the formation of impurities that may alter product performance. We suggest that anyone working with new bio-oxygenates should check peroxide formation as soon as possible to ensure their safe use. In addition, synthesis procedures leading to these oxygenates should be carefully analyzed to see if peroxide-forming chemical(s) can form as side product(s) resulting in unexpected but serious risk for the user. |
The conversion of biomass to carbon based chemicals is a rapidly growing field in sustainable development.1 Since monomers, oligomers, and polymers of carbohydrates are key components of biomass,2 the various conversion schemes involve step-by-step removal of oxygen atoms leading to the formation of bio-oxygenates generally with less and less numbers of oxygen atoms.3 It was recently suggested4 that γ-valerolactone (GVL) can be considered as a sustainable liquid for the production of energy and carbon based chemicals,5 and some of the key processes of a GVL-economy were demonstrated.6 For example, the conversion of C6-carbohydrates (C6H12O6), preferentially obtained from non-edible sources,7 to C5-hydrocarbons (C5H12) proceeds through oxygenates with different numbers of carbon, hydrogen, and oxygen atoms, as depicted in Scheme 1.
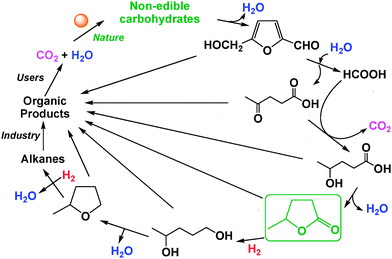 | ||
| Scheme 1 C6H6O3 (5-hydroxymethyl-furfural, HMF) → C5H8O3 (levulinic acid) + C1H2O2 (formic acid)→C5H10O3 (4-hydroxy-valeric acid)→C5H8O2 (GVL) → C5H10O2 (4-hydroxy-pentanol) → C5H10O1, (2-methyltetrahydrofuran, 2-Me-THF).6 | ||
In general, the products of carbohydrate-based biomass conversion can be multifunctional compounds containing, in addition to typical C,H-functional group(s), various combinations of hydroxy, ether, aldehyde, carbonate or acid groups, and finally mono-functional alcohols, ethers, aldehydes, carbonates, and acids. Further functionalization of these molecules will result in even greater structural diversity and so physical and chemical characteristics. As the daily use in laboratory and commercial applications of these old and new molecules increase, their safe production, storage, transportation and use must also become part of the development process.
The formation of organic peroxides is one of the most dangerous transformations of organic compounds under air that can cause accidents during production, storage, transportation and use.8–11 In addition, the formation of peroxides is not just a safety issue; they can lead to the initiation of various chemical reactions and thus the formation of impurities that could also alter product performance. Structurally, organic peroxides are derivatives of H2O2 having one or both hydrogen atoms replaced by an organic moiety. The relatively weak peroxide bonds (–O–O–) readily undergo spontaneous or catalytic bond cleavage leading sometimes violent explosions on a laboratory or industrial scale. It should be noted that some organic peroxides are extremely sensitive to shock, heat, spark, friction, impact, and ultraviolet light.
While most chemists and chemical engineers are well aware of the safety issues concerning the use of organic peroxides even on a small scale, they are generally less informed about the structures of the potentially peroxide-forming chemicals. The rate of the formation of peroxides via insertion of an oxygen molecule to a C,H- or a C,C-bond under aerobic conditions strongly depends on the strengths of the C,H- or C,C-bonds, these are significantly effected by their local environment. For example, the peroxizability of a C,H-bond in an isopropyl group attached to an oxygen atom is so high, that diisopropyl ether readily forms explosive levels of peroxides without concentration. Consequently, it is considered as a Group A solvent, forming peroxides at explosive levels without concentration (e.g. without evaporation by standing in an open flask or distillation under air or vacuum).9 Other Group A compounds are divinyl acetylene, vinylidene chloride, liquid butadiene, chloroprene and tetrafluoroethylene. Group A compounds should be tested for peroxide formation before use or discarded after 3 months. Most solvents require concentration to have serious peroxide risks and are classified Group B compounds. This group includes: acetaldehyde, benzyl alcohol, dioxanes, chlorofluoroethylene, isopropylbenzene, cyclohexene, cyclopentene, decahydronaphthalene, diacetylene, dicyclopentadiene, diglyme, diethyl ether, ethylene glycol ether acetates, furan, methyl acetylene, methyl-isobutyl ketone, tetrahydrofuran (THF), tetrahydronaphthalene, vinyl ethers and secondary alcohols. Group B compounds should be tested for peroxide formation before distillation, evaporation, and used or discarded after 1 year. It should be mentioned, that some chemicals belonging to Group C (chlorobutadiene, vinyl acetate, styrene, tetrafluoroethylene, vinyl chloride, vinylidene chloride, liquid butadiene and chloroprene) and could become hazardous by peroxide initiated autopolymerization. Group C chemicals should be tested for peroxide formation before distillation, evaporation, and use or discarded after 6 months. Since the structural diversity of peroxide forming chemicals are great, the peroxidizability of new biomass-based chemicals especially the large number of oxygenates, should be considered as key chemicals and safety properly investigated by the inventors as part of the discovery process.
We have recently demonstrated that gamma-valerolactone (GVL) can be considered as a sustainable liquid for the production of energy and carbon-based chemicals.5,6 It was observed during the first test of GVL as an additive to 95 octane gasoline that the peroxide number12 of the mixed fuels increased from 3.7 to 11.1 by increasing the GVL content from 3% to 9%, respectively (Table 1).
| AN-95 gasoline | AN-95 + 3% GVL | AN-95 + 6% GVL | AN-95 + 9% GVL | |
|---|---|---|---|---|
| Peroxide number/mg kg−1 | < 0.15 | 3.7 | 8.5 | 11.1 |
Since we could not find any literature data for the formation of peroxides from GVL directly and only one paper reported the conversion of levulinic acid to gamma-hydroperoxo-gamma-valerolactone by hydrogen peroxide,13 we measured the peroxide number of the 98% pure commercial GVL we used. To our surprise, the peroxide number was 400 ppm, which is well above the 100 ppm limit considered safe without the presence of a stabilizer. In order to establish that the high peroxide number was due to peroxides formed from an impurity and not from GVL, we prepared a peroxide-free GVL by treating it with FeSO4 in water. Indeed, no peroxide formation was observed for GVL after one month at 60 °C. This result suggested that the high peroxide number of the commercial GVL was due to a peroxidizable impurity in GVL. Since the commercial GVL was most likely produced by heterogeneous hydrogenation of levulinic acid, we have assumed that the heterogeneous catalyst is also capable of reducing the GVL to 1,4-pentan-diol, which then readily loses water to form 2-methyl-tetrahydrofuran (2-Me-THF). The latter is considered a fuel additive14,15 as well as a solvent, and was shown to readily form peroxides, especially when it was intensively mixed under air.16 Indeed, a systematic investigation of the peroxizability of several oxygenates (Table 2) has revealed that peroxide formation could be a serious safety issue for THF, 2-Me-THF, and 2,5-diMe-furan. MTBE and furan show much slower peroxide formation, which is of course why MTBE could be used safely as an additive in gasoline for years.
| Material | Time (h) | Peroxide number (ppm) |
|---|---|---|
| a nd: not detectable; nm: not measured. | ||
| Tetrahydrofuran (THF) | 0 | nd |
| 41 | 4 | |
| 163 | nm | |
| 330 | 11 | |
| 378 | 12 | |
| 476 | 160 | |
| 546 | 376 | |
| 2-Methyl-tetrahydrofuran (2-Me-THF) | 0 | nd |
| 74 | 46 | |
| 118 | 111 | |
| 220 | 430 | |
| 288 | 716 | |
| 2,5-Dimethylfuran | 0 | nd |
| 48 | 180 | |
| 146 | 422 | |
| 313 | 856 | |
| 476 | 1493 | |
| 674 | 1583 | |
| Furan | 0 | nd |
| 27 | 1 | |
| 193 | 8 | |
| 338 | 9 | |
| Methyl-t-butyl-ether (MTBE) | 0 | nd |
| 48 | 3 | |
| 117 | 4 | |
| 183 | 6 | |
| 663 | 9 | |
In summary, our results suggest that chemists and chemical engineers working with new oxygenates should check for peroxide formation as soon as possible to ensure their safe use. In addition, the synthesis procedures to these oxygenates should be carefully analyzed to see if peroxide-forming chemical(s) could form as side product(s) resulting in unexpected but serious risk for the user.
Experimental
Peroxide number measurement12
Required solutions: (a) an acetic acid solution was prepared by the addition of 4 ml cc. HCl to 996 ml glacial acetic acid; (b) a 0.005 N Na2S2O3 solution was obtained by dissolving 1.25 g Na2S2O3 in distilled 1000 ml water; (c) a starch solution was prepared by adding 3 g of starch to 500 mL distilled water, which was boiled until the starch was dissolved completely. 10 g potassium hydroxide was added and stirred vigorously for 10 min. It was kept at room temperature for 2 h and then 6 mL of glacial acetic acid was added. The pH = 4 was set by 20% hydrogen chloride (approximately 5 mL).In a typical experiment, 2 g of the sample was placed in a 100 ml glass flask and 8 ml of the acetic acid solution and a freshly prepared solution of 0.96 g KI in 1 ml of distilled water was added. The flask was closed, mixed vigorously for 21 s and kept at room temperature for 5 min. After the addition of 40 ml distilled water the solution was titrated with 0.005 N Na2S2O3. When the color changed from orange to light yellow 2 ml of the starch solution was added resulting in a blue color. The titration was continued with 0.005 N Na2S2O3 until the disappearance of the blue color. A blank was titrated just prior to each set of measurements. The peroxide number was calculated as:
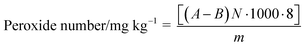
Synthesis of peroxide free oxygenates
In the case of the commercial samples containing stabilizers (THF, 2-Me-THF, furan) purification was performed only by distillation. In all other cases we prepared peroxide-free samples by treatment with FeSO4 solution. In the case of GVL, 500 ml GVL and a solution of 45 g FeSO4 in 150 ml distilled water were placed in a separatory funnel, which was vigorously shaken six times. The aqueous phase was extracted with 75 ml ethyl acetate three times and the combined organic phase was dried over Na2SO4. The ethyl acetate was removed under reduced pressure, and the residual pale-yellow liquid was distilled under vacuum to give a clear, colorless liquid. In the cases of MTBE and 2,5-diMe-furan, 300 ml of a sample was treated with 30 g FeSO4 in 100 mL distilled water. After separation of the organic phase, it was dried over Na2SO4 and the residual liquid was distilled under N2 at ambient pressure. In all cases the peroxide number was zero after distillation. The samples were kept at room temperature for weeks.Acknowledgements
This work was funded by the Hungarian National Scientific Research Fund (T047207).References
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS.
- F. W. Lichtenthaler, Carbohydrates as Organic Raw Materials, VCH, Weinheim, 1991 Search PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 Search PubMed.
- I. T. Horváth, 10th Annual Green Chemistry & Engineering Conference, Washington, DC, July 26–30, 2006, abstr. no. 27 Search PubMed.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49 CrossRef CAS.
- I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169 CrossRef CAS.
- D. E. Clark, Chem. Health Saf., 2001, 12 Search PubMed.
- H. L. Jackson, W. B. McCormack, C. S. Rondestvedt, K. C. Smetz and I. E. Viele, J. Chem. Educ., 1970, 47, A175 CrossRef CAS.
- J. Bailey, D. Blair, L. Boada-Clista, D. Marsick, D. Quigley, F. Simmons and H. Whyte, Chem. Health Saf., 2004, 11(5), 14 Search PubMed.
- J. Bailey, D. Blair, L. Boada-Clista, D. Marsick, D. Quigley, F. Simmons and H. Whyte, Chem. Health Saf., 2004, 11(5), 17 Search PubMed.
- ASTM D 3703–99 http://www.astm.org/Standards/D3703.htm.
- R. C. P. Cubbon and C. Hewlett, J. Chem. Soc. C, 1968, 2986 RSC.
- S. F. Paul, US Pat. 5 697 987, 1996 Search PubMed.
- DOE, Alternative Fuel Transportation Program; P-series fuels (Proposed Rules), Fed. Reg., 1998, 63, 40202 Search PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |