Ternary cobalt spinel oxides for solar driven hydrogen production: Theory and experiment
Aron
Walsh
*a,
Kwang-Soon
Ahn
b,
Sudhakar
Shet
a,
Muhammad N.
Huda
a,
Todd G.
Deutsch
a,
Heli
Wang
a,
John A.
Turner
a,
Su-Huai
Wei
a,
Yanfa
Yan
a and
Mowafak M.
Al-Jassim
a
aNational Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: a.walsh@ucl.ac.uk
bSchool of Display and Chemical Engineering, Yeungnam University, Dae-Dong, Kyungsan 712-749, South Korea
First published on 25th March 2009
Abstract
Discovery of a chemically stable, light absorbing and low resistivity metal oxide with band edges aligned to the water redox potentials has been a goal of physical scientists for the past forty years. Despite an immense amount of effort, no solution has been uncovered. We present a combined theoretical and experimental exploration of a series of unconventional ternary cobalt spinel oxides, which offer chemical functionality through substitution on the octahedral spinel B site. First-principles predictions of the substitution of group 13 cations (Al, Ga, In) in Co3O4 to form a series of homologous CoX2O4 spinel compounds are combined with experimental synthesis and photoelectrochemical characterization. Ultimately, while tunable band gaps in the visible range can be obtained, the material performance is limited by poor carrier transport properties associated with small polaron carriers. Future design pathways for metal oxide exploration are discussed.
Broader contextThe efficient generation of hydrogen from water using visible light would provide a means to produce a chemical energy carrier providing current solar cell technologies with energy storage as well as providing a transportation fuel and a chemical feedstock. The conceptually simple photoelectrochemical process represents the most efficient pathway for accomplishing this goal. Light absorbed in a semiconductor photoelectrode produces electrons and holes which in turn generate H2 and O2, respectively. The grand challenge is discovering the right semiconductor material which can sufficiently balance cost, stability and efficiency to make the process commercially viable. In this work, we explore a series of ternary cobalt spinels oxides, where group 13 cations (Al, Ga, In) occupy the octahedral spinel sites. We demonstrate that while good optical absorption can be obtained, the transport properties are very poor owing to the localized Co 3d states, and hence the observed photoelectrochemical activity is minimal. While this rules out this class of oxide materials (combining 3dn and ns0 cations), we suggest a number of alternative routes for forming multiternary oxides, which may overcome these intrinsic limitations and provide candidate photoelectrode materials in the near-term. |
Introduction
Photoelectrochemical (PEC) decomposition of water by visible light remains the most desirable hydrogen production method for post fossil-fuel energy employment (Fig. 1).1,2 Metal oxide photoelectrodes are of particular interest due to their low cost and relatively high stability in aqueous media. Despite the good catalytic activity of materials such as TiO2,3,4 oxides are generally limited by too large band gaps, which fail to absorb a significant fraction of visible light, resulting in poor solar to hydrogen conversion efficiencies under terrestrial conditions. After almost four decades of intensive research, no material has been found to simultaneously satisfy all the criteria required for widespread PEC application:5–9 (i) low band gap (1.7–2.2 eV), (ii) low resistivity, (iii) low cost, (iv) corrosion resistant, (v) correct alignment of band edges with respect to the water redox potentials.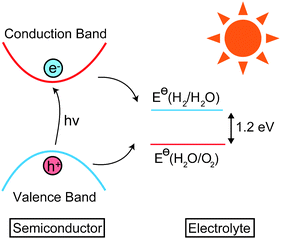 | ||
| Fig. 1 Schematic redox cycle for decomposition of water using a photoactive semiconductor. | ||
The majority of PEC oxide research has centered on trying to overcome issues associated with known photoactive materials (e.g. TiO2, Fe2O3, WO3) through doping or alloying. However, the incremental increases in efficiency presently achieved will not be enough to make PEC based hydrogen production commercially viable. We have recently been active in developing a unified approach of material design, synthesis and characterization to explore new classes of multiternary oxide semiconductor photoelectrodes. Similar to the way that the combination of several specific cations is required for multiternary copper oxides to exhibit high temperature superconductivity, it is our desire to combine multiple cations based on their individual chemical and physical properties (e.g. structural stability, catalytic activity, light absorption) to tune the material properties for enhanced oxide PEC response.
The potential of transition metal (Fe and Co) based oxide spinels has recently been highlighted via initial high-throughput experimental screening by Parkinson et al.10,11 and our subsequent theoretical analysis.12 Considering the magnitude of the band gaps alone, cobalt oxide (Co3O4) is too low (<1.7 eV) for direct PEC water decomposition. However, the spinel structure of Co3O4 contains two cation coordination environments, a four-fold tetrahedral site (A, 2+) and a six-fold octahedral site (B, 3+) giving the overall formula AB2O4. For Co3O4, the band edges are determined by a combination of the crystal field split Co 3d states on the octahedral and tetrahedral cobalt sites (Fig. 2).12–14Isovalent substitution on the spinel cations sites can therefore, in principle, be used to influence both the band edge character and the magnitude of the electronic band gaps and optical absorption.
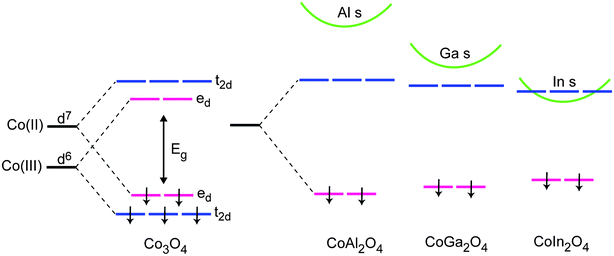 | ||
| Fig. 2 Illustrated crystal field splitting of the minority spin Co 3d states in Co3O4 and band edge electronic structure in the ternary CoX2O4 (X = Al, Ga, In) spinels. | ||
In this paper, we combine theoretical electronic structure calculations with experimental synthesis and PEC characterization to perform a comprehensive examination of how the pertinent chemical and physical properties of ternary cobalt spinels can be tailored towards those of an ideal PEC catalytic photoelectrode for solar driven hydrogen production. In particular, we explore each member of the CoX2O4 (X = Al, Ga, In) chemical series. Aluminum can be viewed as an ionic spectator in the CoAl2O4 lattice, which preserves overall charge neutrality, but contributes little to the density of states or optical absorption. However, the substitution of heavier group 13 cations helps reduce the electronic band gap and increase optical absorption through combination of the higher binding energy cation s states and reduced Co d–d crystal field splitting. Unfortunately, while we demonstrate that the band gaps can be tuned through a large range, the intrinsic charge transport properties result in poor PEC performance due to inefficient electron–hole extraction rates and high resistivity associated with small polaron carriers. Finally, approaches to overcome this behavior, and future pathways for oxide material design, are discussed.
Methods
Theoretical
Calculations were performed using density functional theory (DFT)15,16 within a plane wave basis set as implemented in the VASP17,18 code. The spinel (Oh7) unit cell was repeated under periodic boundary conditions to approximate an infinite solid.19 The Perdew–Burke–Ernzerhof (PBE)20 exchange–correlation functional was supplemented with a Coulomb U (DFT + U) to more correctly describe the correlated nature of the Co 3d states (U − J = 2 eV).12,21 The core electrons (Co, Ga:[Ar], Al:[Ne], In:[Kr], O:[He]) were treated within the projector augmented wave (PAW) method.22 A well converged 8 × 8 × 8 k-point mesh and plane wave cut-off of 500 eV were employed, with atomic forces minimized to below 1 meV Å−1. The equilibrium volumes were obtained through fitting energy–volume data to the Murnaghan equation of state.23 Cation inversion in the spinel structure was treated via the special quasi-random structure method.24,25 The optical absorption spectra were summed over all direct valence to conduction band transitions on a denser k-point mesh (12 × 12 × 12) using the all-electron WIEN2K code26 and within the random phase approximation; excitonic excitations are not considered at this level.27Experimental
Co1 + δX2 − δO4 (X = Al, Ga, In) thin films were grown on Ag-coated stainless steel plates (Ag/SS) and quartz glasses for PEC measurement and optical characterization, respectively, using an RF magnetron reactive co-sputtering system. Here Ag/SS was required as the substrate, because of the high temperature oxide growth (800 °C). Two sputter guns were used for the Co–Al–O and Co–In– O syntheses, where the Co3O4 target was fixed and the secondary target was changed to Al and In2O3 for CoAl2O4 and CoIn2O4, respectively. The Co–Ga–O films were deposited using the different amounts of Ga2O3 powder on the Co3O4 target. The distance between the sputter guns and substrate was fixed at 11 cm. The substrates were rotated during deposition for enhanced uniformity and the working pressure was 1.22 Pa. The sputtering ambient environment was mixed Ar and O2 (O2:Ar = 5:1) and the RF power of each sputter gun was varied to obtain appropriate chemical stoichiometry. All of the samples were controlled to exhibit similar film thicknesses on the order of 500 nm, as measured by stylus profilometry. Structural characterization was performed by powder X-ray diffraction (XRD) measurements, using an X-ray diffractometer (XGEN-4000, SCINTAG, Inc.) operated with a Cu Kα radiation source at 45 kV and 37 mA.PEC measurements were performed in a three-electrode cell with a flat quartz window to facilitate illumination of the photoelectrode surface. The films (active area: 0.24 cm2) were used as the working electrodes. A Pt sheet (area: 10 cm2) and an Ag/AgCl electrode (with saturated KCl solution) were used as the counter and reference electrodes, respectively. A 0.5 M NaOH basic aqueous solution (pH ∼ 13) was used as the electrolyte. The PEC response was measured with a fiber-optic illuminator (150 W tungsten-halogen lamp) processed through a UV/IR cut-off filter (cut-off wavelengths: 350 and 750 nm). Light intensity with the UV/IR filter was 80 mW cm−2 as measured by a photodiode power meter. The PEC response was then measured with respect to time under chopped light on/off illumination at constant applied potential bias. Temperature effects on the dark current, due to possible sample heating on illumination, were found to be negligible.
Results
Theoretical geometric and electronic structure
| a normal/Å | a inverse/Å | ΔE/eV | X (300 K) | ΔH/eV | E g Γ–Γ/eV | |
|---|---|---|---|---|---|---|
| Al | 8.19 | 8.16 | 0.51 | 0.00 | −17.56 | 2.61 |
| Ga | 8.46 | 8.43 | 0.23 | 0.02 | −11.20 | 1.70 |
| In | 9.06 | 9.02 | 0.11 | 0.15 | −8.05 | 0.73 |
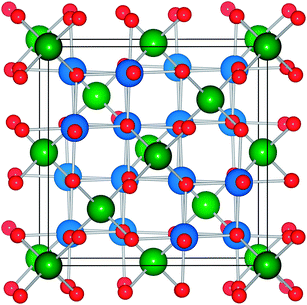 | ||
| Fig. 3 Representation of the 56 atom conventional AB2O4 spinel unit cell with 8 Td (A) and 16 Oh (B) cation sites colored green and blue, respectively. The 32 oxygen sites are colored red. | ||
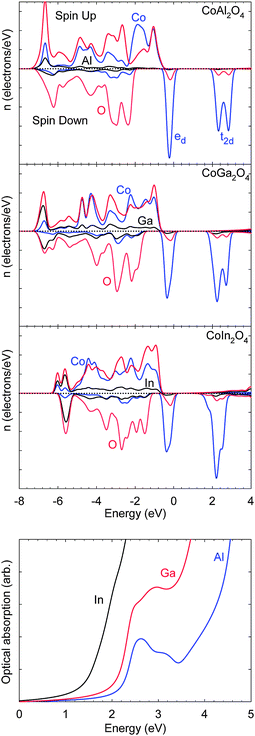 | ||
| Fig. 4 Calculated local electronic densities of states and optical absorption spectra for the CoX2O4 (X = Al, Ga, In) spinel series. The highest occupied state is set to 0 eV. | ||
Firstly, Al is a row 2 element with no occupied d bands, while both Ga and In possess filled shallow core d10 levels which lie around 10 eV below their valence s states. These d states couple with O 2p and result in increased cationic contributions to the valence band, relative to Al. Secondly, the binding energies of the Ga and In s states increase relative to Al. Both of these effects serve to decrease the fundamental electronic band gap of the ternary cobalt oxides through enhanced anion p–cation d coupling below the valence band, and a lowering of the conduction band from the influence of the group 13 cation s states. Hence, the band gap trend observed for Al2O3 (8.8 eV32), Ga2O3 (4.5 eV33) and In2O3 (2.7 eV34,35) is also maintained in the ternary cobalt oxides. Here the calculated electronic separations at the Γ point in CoX2O4 decrease from 2.61 (Al) to 1.70 (Ga) to 0.73 eV (In). While the quantitative reduction is overestimated due to the well-documented DFT over-binding of unoccupied cation s states, the qualitative trend remains valid. Increased dispersion is most visible in the lower conduction band density of states of CoIn2O4 where the conduction band minimum state is composed of majority In 5s character, which should substantially improve electron conductivity in comparison to the other ternary oxides; however, it is unclear whether the intrinsic p-type nature of Co3O4 or n-type nature of In2O3 would dominate in the ternary composite system.
The calculated optical absorption spectra are also shown for each compound in Fig. 4. A clear redshift in the onset of absorption is observed, consistent with the electronic band structure. For CoAl2O4, the weak absorption onset is associated with low intensity d → d transitions. As the spinel lattice constant increases with the heavier group 13 cations, the splitting of the filled and empty d states is reduced and the weak absorption feature moves to lower energies. The onset of strong absorption occurring from above 3 eV in CoAl2O4 arises from a combination of band to band transitions composed of p → d, d → s and p → s character. For CoGa2O4, the energy level trends discussed above, in particular the lowering of the cation s level, result in the substantial increase in optical absorption at lower photon energies. For CoIn2O4, the weak onset feature is completely lost in favour of a highly desirable sharp rise in absorption coefficient.
Experimental synthesis and characterization
The representative experimental optical data for each ternary cobalt oxide material, synthesized under a range of sputtering conditions, are shown in Fig. 5. From both the measured optical absorption coefficient and a fit to the Tauc relation for direct transitions, tailoring of the optical band gap from 1.5 to 2.25 eV can be observed. Taking into account that the separation between the H2/H2O reduction and O2/H2O oxidation potentials is 1.23 eV, and that due to the presence of unavoidable losses (e.g. component resistance, electron-hole recombination) an additional overpotential is required (raising the optimal voltage to ∼ 1.7 eV), this optical band gap range appears quite promising for PEC water-splitting application. For CoAl2O4, the measured absorption features are consistent with previous reports.36,37 The low energy absorption peak for CoAl2O4 originates from the spin allowed, parity forbidden Co4A2 → 4T1 transitions, while the drop in absorption coefficient at higher energies results from a range of low intensity spin forbidden transitions. The same general features, including the onset of strong absorption at shorter wavelengths, are observed in the calculated optical absorption spectrum (Fig. 4). The shift to longer wavelengths with lower Al sputtering power can be understood as a transition towards the inverse Co2AlO4 spinel, which is known to possess a lower band gap.12 For CoGa2O4, the strong absorption profile is redshifted to around 2.25 eV and begins to overlap with the low energy absorption feature; this overlap is even more pronounced for CoIn2O4 at high Co3O4sputtering power. At high In2O3sputtering power, the Co–In–O samples exhibit the high levels of visible transmission expected from mixed phase Co substituted In2O3, i.e. the ternary composite is not fully formed.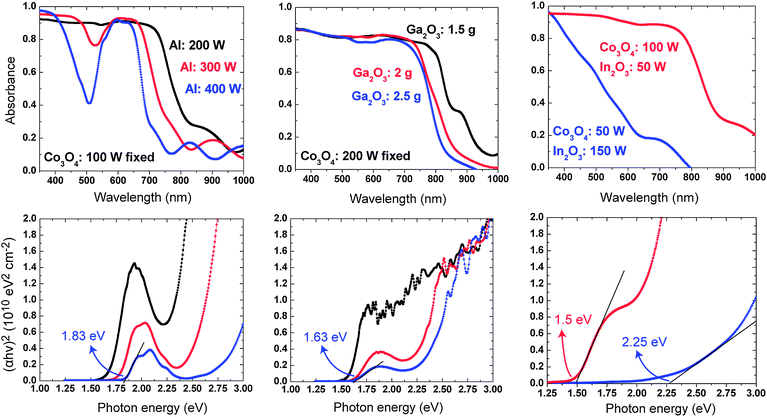 | ||
| Fig. 5 Measured optical absorption spectra (upper) and direct band gap fits (lower) of Co1 + δX2 − δO4 compounds as a function of sputtering conditions: (left) X = Al, (centre) X = Ga and (right) X = In. | ||
The time dependant PEC response of each material was investigated under chopped light illumination at a constant applied bias potential (vs. Ag/AgCl). The cathodic currents of CoAl2O4 and CoGa2O4 were found to increase on illumination, Fig. 6. This p-type PEC response suggests the presence of intrinsic hole carriers, which prior calculations have identified as cation vacancies.38 However, poor photocurrents on the order of 20 µA cm−2 are observed in both cases (currents on the order of 10 mA cm−2 will be required from commercially viable materials). It is worth noting that the PEC response of CoAl2O4 is better than that of CoGa2O4, which will be discussed in more detail shortly. Unfortunately, all synthesized CoIn2O4 films failed to exhibit any significant PEC response. Indeed, the weak PEC response varied from p-type to n-type with different samples, but no significant photocurrent was observed in either case.
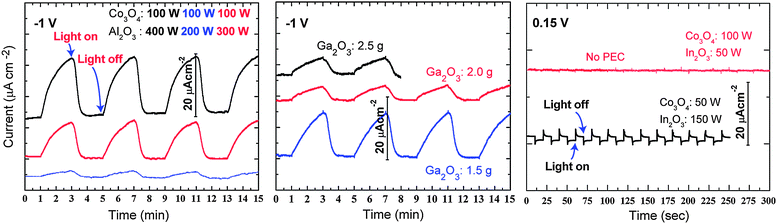 | ||
| Fig. 6 Time dependant photoelectrochemical response under light on/off illumination at constant applied voltage for (left) Al, (centre) Ga and (right) In ternary cobalt oxides. For the Al and Ga spinels, illumination induces an increase in the background cathodic current (p-type response), while for In a small increase in the anodic current is observed (n-type response). | ||
In addition to the low generated photocurrents, the second discouraging trend emerging from Fig. 6 is the long relaxation times between sample illumination. While for the majority of PEC materials, the recovery time on the removal of light is on the order of seconds or less, for these materials it is on the order of minutes. This implies poor carrier transport kinetics, originating from confined electrical carriers (heavy hole effective masses). A more detailed comparison between the relaxation times of CoAl2O4 and CoGa2O4 is shown in Fig. 7. Ideally the photocurrent decay for the chopped light PEC system will follow close to a square wave; however, here the response deviates greatly. Direct comparison of the normalized current of the Al and Ga ternaries (Fig. 7), shows much faster decay in the former, indicating better carrier transport kinetics, and hence CoAl2O4 exhibits marginally improved PEC response. One encouraging outcome is that both materials exhibited no evidence of corrosion in solution for sustained PEC testing periods.
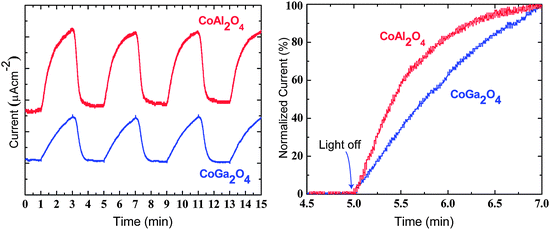 | ||
| Fig. 7 Photocurrent decay comparison between the CoAl2O4 and CoGa2O4 samples with the same bias voltage (−1 V). | ||
Based on our initial electronic structure analyses, it was anticipated that the Co–In spinel may possess both the lowest band gap and highest n-type conductivity (through the presence of the In 5s conduction states). However, while the synthesized In based ternary oxides did not exhibit any significant photocurrent on illumination, the n-type PEC response time was much shorter, indicating the beneficial influence of the delocalized In 5s orbitals in the lower conduction band. To explore the origin of the performance failure in more detail, we first measured the dark currents without illumination, as shown in Fig. 8. Even for small applied potentials, the dark current is large, indicating that the film is not an intrinsic semiconductor, but is in fact closer to a semi-metallic state. The XRD curves are also shown in Fig. 8. The measurements clearly show that the deposited films are not pure CoIn2O4 but undergo significant phase segregation into Co3O4 and In2O3 (this has been confirmed by transmission electron microscopy analysis). Taking into account that In2O3 itself exhibits degenerate electron conduction behavior as an n-type transparent conducting oxide,39–41 the presence of In2O3 in the film will contribute to both the inferior PEC response and high dark current levels. Within the limitations of our co-sputtering system, we could not succeed in synthesizing a homogeneous CoIn2O4 film.
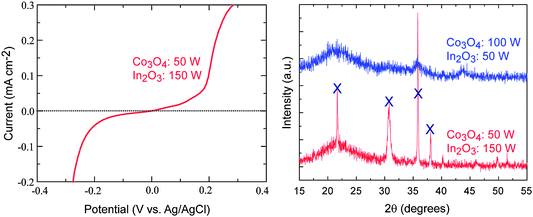 | ||
| Fig. 8 Measured dark current–voltage curve and powder X-ray diffraction (XRD) data for synthesized Co–In systems. The crosses in the XRD spectrum correspond to reflections associated with bixbyite In2O3. | ||
Discussion
The absence of efficient water decomposition
While the prediction of tunable electronic band gaps based on density functional theory calculations was realized experimentally, the ternary cobalt samples all exhibit poor PEC performance, which can be related to the inherent conductivity mechanisms in transition metal oxides. From examination of intrinsic point defect formation in CoAl2O4, we previously identified doubly ionized cobalt vacancies as the likely source of p-type conductivity38 (eqn (1), in Kröger–Vink notation42), which is in agreement with early experimental observations.43| CoCo → V//Co + 2h˙ + Co(s) | (1) |
Within the realm of traditional semiconductor defect theory, the generated electron holes (h˙) are considered to behave as free carriers moving through delocalized bands. However, for conductivity in localized states (such as partially filled 3d levels), this is not the case. From a chemistry perspective, each hole formally corresponds to a net cobalt oxidation (i.e. h˙ ≡ Co˙Co), which introduces a local lattice distortion through the contraction in the Co–O interatomic distances associated with the loss of an electron. The accessibility of the higher oxidation state (Co 3+) explains why these spinels favour p-type conductivity (electron deficiency). Mobility of this so-called small polaron will have an associated activation energy corresponding to the hopping of the electronic defect and associated structural distortion (eqn (2)). From a physics perspective, such polaron mediated conductivity will dominate when the electronic band width (a measure of electron delocalization in reciprocal space) of the conductive state is less than the polarization energy gained by inducing a local lattice distortion.44 In other words, it is an effect typical of materials with heavy carrier effective masses.45
| Co˙Co + CoCo + CoCo ⇌ CoCo + Co˙Co + CoCo ⇌ CoCo + CoCo + Co˙Co | (2) |
The presence of localized carriers is detrimental for any potential semiconductor photoelectrode. The introduction of an Arrhenius-like activation energy decreases the carrier mobility by several orders of magnitude at room temperature. Furthermore, trapping of photoexcited carriers at such sites will result in an increased probability for electron–hole recombination, as well as the excessive carrier relaxation times observed in our PEC measurements for the ternary spinels.
From the reported electrical properties of single crystal CoAl2O4, the conductivity is indeed characterized by an exponential rise with temperature, combined with an activation barrier on the order of 1.8 eV.46 This results in carrier mobilities far below 1 cm2V−1 s−1 and highly resistive behavior at room temperature. For Co3O4, manipulation of the growth conditions has been shown to increase the conductivity by several orders of magnitude;47 however, such an effect has not been reported in the corresponding ternary cobalt oxide compounds. The principal reason why the conductivity of the ternary spinels is diminished with respect to binary cobalt oxide, is that as the Co concentration is diluted, the Co–Co separations are increased, inducing lower hopping probabilities. Indeed, the average Co–Co separations increase by 33% from Co3O4 to CoIn2O4, which, assuming a 1/r2 dependence, would result in an 89% decrease in polaron conductivity. It is worth noting that the related class of ternary ZnX2O4 spinels, where X = Co, Rh, Ir offer much improved p-type conductivities.48 This is due to the presence of Co on the edge sharing spinel octahedra, which significantly lowers the barrier for polaron mobility. However, for this case, the octahedral crystal field induces a low spin Co (t2g6eg0) configuration, with optical band gaps greater than 2.3 eV, and hence they have been gaining interest for application as p-type transparent conductors.49
One potential avenue to overcome the limitations of the CoX2O4 spinels is to make the materials grossly non-stoichiometric so that the cation vacancy defect levels begin to overlap, inducing a transition to band conductivity. However, such a highly defective system would not be optimal for PEC application. The only other alternatives are to further alloy with cations that can provide more dispersive character at the top of the valence band to facilitate rapid hole transport, or to employ nanoparticle or ultra thin films, where the bottleneck provided by low carrier mobility would be reduced. In particular, the application of suspended nanoparticles in a slurry phase PEC reactor, where the particle sizes are on the same order as the carrier diffusion length is a possibility for overcoming such poor transport behavior.
Moving forward
Our combined experimental and theoretical exploration has demonstrated how the fundamental properties of multicomponent oxides can be manipulated by the combination of appropriate cations. However, we have also demonstrated that serious issues exist with the inclusion of transition metal cations with localized 3d levels situated at the band edges. Indeed this is also the main limitation of Fe2O3, where the good band gap and visible light absorption are offset by inefficient photogenerated carrier separation and lackluster carrier mobility.50 This experience can be employed as a future selection criterion in the choice of optimal cations.51For high levels of conductivity in oxides, the dispersive conduction bands of ns0 cations (Zn 2+, In 3+, Sn 4+) are highly desirable,39,52,53 and it has been well demonstrated in the literature that nd0 cations (e.g. Ti 4+, V 5+, W 6+) can act as beneficial catalytic redox centers.54–57 Unfortunately, both cations tend to produce oxides with band gaps outside the visible photon range. Furthermore, it is undesirable to further lower the conduction band energy, as this will require a significant bias voltage to drive forward the hydrogen reduction reaction. It is therefore required to increase the valence band energy, which can be achieved through cation functionalization by the inclusion of cations with occupied low binding energy d or s states such as Cu/Ag (nd10) or Bi/Pb (ns2). To ensure stability in solution, it may be beneficial to also include cations which form strong metal–oxygen bonds to provide a stable structural framework in the multiternary systems, e.g.Al, Zr, Si.
By following these intuitive design principles, we can suggest a number of potential routes for improving oxide PEC performance. An oxide class of immense current interest are the ABO2 delafossite oxides;58–60 where A = Cu or Ag, and B can be any trivalent cation. While the prototype delafossite CuAlO2 first gathered interest as a p-type transparent conductor,61 replacement of Al with alternative cations such as Cr62,63 can reduce the otherwise large optical band gaps and simultaneously improve the conductivity.64 This homologous series is unique in maintaining a Cu 1+oxidation state; Cu containing spinels tend to induce the formation of the less desirable magnetic 2+d9 configuration. Similar to the ternary Co spinels presented in our current work, it should be possible to tune the band gap of the group 13 Cu delafossites through formation of the Cu(Al1 − x − yGaxIny)O2 alloy. Due to the reduced symmetry of the resulting ordered or disordered alloys, the weak band edge optical transitions associated with typical ternary delafossites58 could be overcome.
Alternatively, post-transition metal containing oxides such as BiVO4 have also been gathering recent attention.65–69 Occupied cation s states serve both to raise the valence band energy through the addition of cation s–anion p coupling70,71 and induce lighter hole masses than typical oxides, e.g. good p-type mobilities and conductivity have been reported in SnO.72 Many mineral structures combining these cations exist (e.g.ilmenite,73 rosiaite74 and trirutile75 based compounds) and there are certainly many more complex configurations that remain to be discovered and explored. In particular, we would refer readers to recent work on bismuth technetates,76 ternary Sn, Pb, Bi and Sb oxides,77 and Sn based niobates and tantalates.78 This serves to emphasize that there are many viable lines for future research beyond the PEC oxide standards such as TiO2.
Conclusions
We have presented a combination of theoretical electronic structure calculations with experimental synthesis and characterization of a series of ternary cobalt spinel oxides. While these materials combine excellent stability in solution and good visible light absorption properties, their performance as photoelectrochemical catalysts is limited by the poor transport properties induced by small polaron mobility. This is an enhancement of the same effect that limits the performance of Fe2O3 based photocatalysts. While incremental improvements in charge carrier extraction efficiencies may be possible through the alteration and tuning of growth conditions and crystal morphology, it is unlikely that the limitations of late 3d transition metal oxides will be overcome to provide the performance required for commercial hydrogen production. We have suggested a number of potential future pathways for reducing the band gaps of metal oxides, without sacrificing electron transport performance.Acknowledgements
We would like to thank B. A. Parkinson and M. Woodhouse for useful discussions in relation to their high throughput screening results. This work is supported by the U.S. Department of Energy (DOE) under contract no. DE-AC36-08GO28308. Computing resources of the National Energy Research Scientific Computing Center were employed, which is supported by DOE under Contract No. DE-AC02-05CH11231.References
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Science, 2005, 308, 1901 CrossRef CAS.
- J. A. Turner, Science, 1999, 285, 687 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- A. Fujishima, K. Kohayakawa and K. Honda, J. Electrochem. Soc., 1975, 122, 1487 CAS.
- B. D. Alexander, P. J. Kulesza, I. Rutkowska, R. Solarska and J. Augustynski, J. Mater. Chem., 2008, 18, 2298 RSC.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991 CrossRef CAS.
- N. S. Lewis, Nature, 2001, 414, 589 CrossRef CAS.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- M. Woodhouse, G. S. Herman and B. A. Parkinson, Chem. Mater., 2005, 17, 4318 CrossRef CAS.
- M. Woodhouse and B. A. Parkinson, Chem. Mater., 2008, 20, 2495 CrossRef CAS.
- A. Walsh, S.-H. Wei, Y. Yan, M. M. Al-Jassim, J. A. Turner, M. Woodhouse and B. A. Parkinson, Phys. Rev. B, 2007, 76, 165119 CrossRef.
- K. M. E. Miedzinska, B. R. Hollebone and J. G. Cook, J. Phys. Chem. Solids, 1987, 48, 649–656 CrossRef CAS.
- Y. Jugnet and T. M. Duc, J. Phys. Chem. Solids, 1979, 40, 29 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B, 1998, 57, 1505 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953 CrossRef.
- F. D. Murnaghan, Proc. Nat. Acad. Sci. U. S. A., 1944, 30, 244.
- S.-H. Wei and S. B. Zhang, Phys. Rev. B, 2001, 63, 045112 CrossRef.
- S.-H. Wei, L. G. Ferreira, J. E. Bernard and A. Zunger, Phys. Rev. B, 1990, 42, 9622 CrossRef CAS.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka and J. Luitz, WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties, Techn. Universitat Wien, Austria, 2001 Search PubMed.
- C. Ambrosch-Draxl and J. O. Sofo, Comput. Phys. Commun., 2006, 175, 1 CrossRef CAS.
- N. Tristan, J. Hemberger, A. Krimmel, H. A. K. von Nidda, V. Tsurkan and A. Loidl, Phys. Rev. B, 2005, 72, 174404 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751 CrossRef.
- H. Furuhashi, M. Inagaki and S. Naka, J. Inorg. Nucl. Chem., 1973, 35, 3009 CrossRef CAS.
- A. Nakatsuka, Y. Ikeda, N. Nakayama and T. Mizota, Acta Crystallogr., Sect. E, 2006, 62, i109 CrossRef.
- T. Perevalov, A. Shaposhnikov, V. Gritsenko, H. Wong, J. Han and C. Kim, JETP Lett., 2007, 85, 165 CrossRef CAS.
- G. Schmitz, P. Gassmann and R. Franchy, J. Appl. Phys., 1998, 83, 2533 CrossRef CAS.
- A. Walsh, J. L. F. Da Silva, S.-H. Wei, C. Korber, A. Klein, L. F. J. Piper, A. DeMasi, K. E. Smith, G. Panaccione, P. Torelli, D. J. Payne, A. Bourlange and R. G. Egdell, Phys. Rev. Lett., 2008, 100, 167402 CrossRef.
- A. Bourlange, D. J. Payne, R. G. Egdell, J. S. Foord, P. P. Edwards, M. O. Jones, A. Schertel, P. J. Dobson and J. L. Hutchison, Appl. Phys. Lett., 2008, 92, 092117 CrossRef.
- U. Lavrenčič Štangar, B. Orel and M. Krajnc, J. Sol–Gel Sci. Technol., 2003, 26, 771–775 CrossRef CAS.
- M. Zayat and D. Levy, Chem. Mater., 2000, 12, 2763 CrossRef CAS.
- A. Walsh, Y. Yan, M. M. Al-Jassim and S.-H. Wei, J. Phys. Chem. C, 2008, 112, 12044 CrossRef CAS.
- A. Walsh, J. L. F. Da Silva and S.-H. Wei, Phys. Rev. B, 2008, 78, 075211 CrossRef.
- C. G. Granqvist and A. Hultaker, Thin Solid Films, 2002, 411, 1 CrossRef CAS.
- P. D. C. King, T. D. Veal, D. J. Payne, A. Bourlange, R. G. Egdell and C. F. McConville, Phys. Rev. Lett., 2008, 101, 116808 CrossRef CAS.
- D. M. Smyth, The Defect Chemistry of Metal Oxides, Oxford University Press, Oxford, 2000 Search PubMed.
- C. Greskovich and H. Schmalzried, J. Phys. Chem. Solids, 1970, 31, 639 CrossRef CAS.
- P. A. Cox, The Electronic Structure and Chemistry of Solids, Oxford University Press, New York, 1987 Search PubMed.
- N. F. Mott and R. Peierls, Proc. Phys. Soc., London, 1937, 49, 72 CrossRef.
- N. G. Matveeva and A. I. Shelykh, Phys. Status Solidi B, 1972, 50, 83–86 CrossRef CAS.
- F. Tronel, L. Guerlou-Demourgues, M. Menetrier, L. Croguennec, L. Goubault, P. Bernard and C. Delmas, Chem. Mater., 2006, 18, 5840 CrossRef CAS.
- M. Dekkers, G. Rijnders and D. H. A. Blank, Appl. Phys. Lett., 2007, 90, 021903 CrossRef.
- H. Hosono, Thin Solid Films, 2007, 515, 6000–6014 CrossRef CAS.
- A. Kleiman-Shwarsctein, Y.-S. Hu, A. J. Forman, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2008, 112, 15900 CrossRef CAS.
- M. N. Huda, A. Walsh, T. G. Deutsch, Y. Yan, S.-H. Wei, J. A. Turner and M. M. Al-Jassim, unpublished.
- A. Janotti and C. G. Van de Walle, Phys. Rev. B, 2007, 76, 165202 CrossRef.
- K. G. Godinho, A. Walsh and G. W. Watson, J. Phys. Chem. C, 2009, 113, 439 CrossRef CAS.
- F. A. Grant, Rev. Mod. Phys., 1959, 31, 646 CrossRef CAS.
- M. N. Huda, Y. Yan, C. Y. Moon, S.-H. Wei and M. M. Al-Jassim, Phys. Rev. B, 2008, 77, 195102 CrossRef.
- S. K. Deb, Sol. Energy Mater. Sol. Cells, 2008, 92, 245 CrossRef CAS.
- B. J. Morgan and G. W. Watson, Surf. Sci., 2007, 601, 5034 CrossRef CAS.
- X. Nie, S.-H. Wei and S. B. Zhang, Phys. Rev. Lett., 2002, 88, 066405 CrossRef.
- L.-J. Shi, Z.-J. Fang and J. Li, J. Appl. Phys., 2008, 104, 073527 CrossRef.
- W. C. Sheets, E. S. Stampler, M. I. Bertoni, M. Sasaki, T. J. Marks, T. O. Mason and K. R. Poeppelmeier, Inorg. Chem., 2008, 47, 2696 CrossRef CAS.
- H. Kawazoe, M. Yasukawa, H. Hyodo, M. Kurita, H. Yanagi and H. Hosono, Nature, 1997, 389, 939 CrossRef CAS.
- D. O. Scanlon, A. Walsh, B. J. Morgan, G. W. Watson, D. J. Payne and R. G. Egdell, Phys. Rev. B, 2009, 79, 035101 CrossRef.
- T. Arnold, D. J. Payne, A. Bourlange, J. P. Hu, R. G. Egdell, L. F. J. Piper, L. Colakerol, A. De Masi, P.-A. Glans, T. Learmonth, K. E. Smith, J. Guo, D. O. Scanlon, A. Walsh, B. J. Morgan and G. W. Watson, Phys. Rev. B, 2009, 79, 075102 CrossRef.
- R. Nagarajan, A. D. Draeseke, A. W. Sleight and J. Tate, J. Appl. Phys., 2001, 89, 8022 CrossRef CAS.
- G. Li, D. Zhang and J. C. Yu, Chem. Mater., 2008, 20, 3983 CrossRef CAS.
- I. C. Vinke, J. Diepgrond, B. A. Boukamp, K. J. de Vries and A. J. Burggraaf, Solid State Ionics, 1992, 57, 83 CrossRef CAS.
- K. Sayama, A. Nomura, Z. Zou, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2003, 2908 RSC.
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S.-H. Wei, Chem. Mater., 2009, 21, 547 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- D. J. Payne, R. G. Egdell, A. Walsh, G. W. Watson, J. Guo, P. A. Glans, T. Learmonth and K. E. Smith, Phys. Rev. Lett., 2006, 96, 157403 CrossRef CAS.
- A. Walsh and G. W. Watson, J. Solid State Chem., 2005, 178, 1422–1428 CrossRef CAS.
- Y. Ogo, H. Hiramatsu, K. Nomura, H. Yanagi, T. Kamiya, M. Hirano and H. Hosono, Appl. Phys. Lett., 2008, 93, 032113 CrossRef.
- T. F. W. Barth and E. Posnjak, Z. Kristallogr. Kristallgeom. Kristallphys. Kristallchem., 1934, 88, 265 Search PubMed.
- A. Magneli, Ark. Kem., 1941, 15, 1 Search PubMed.
- A. Bystroem, B. Hoek and B. Mason, Ark. Kem., 1942, 15, 1 Search PubMed.
- E. E. Rodriguez, F. Poineau, A. Llobet, K. Czerwinski, R. Seshadri and A. K. Cheetham, Inorg. Chem., 2008, 47, 6281 CrossRef CAS.
- H. Mizoguchi and P. M. Woodward, Chem. Mater., 2004, 16, 5233 CrossRef CAS.
- Y. Hosogi, Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, Chem. Mater., 2008, 20, 1299 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |