Organic materials for hydrogen storage applications: from physisorption on organic solids to chemisorption in organic molecules
Philippe
Makowski
a,
Arne
Thomas
a,
Pierre
Kuhn
*a and
Frédéric
Goettmann
*b
aColloid Chemistry Department, Max-Planck Institute of Colloids and Interfaces, Scientific Campus Golm, 14476, Potsdam, Germany. E-mail: pierre.kuhn@mpikg.mpg.de
bInstitut de Chimie Séparative de Marcoule, UMR 5257, ICSM site de Marcoule, BP 17171, 30207, Bagnols sur Cèze, France. E-mail: frederic.goettmann@cea.fr
First published on 18th March 2009
Abstract
Finding suitable (transportable) storage devices for hydrogen is one of the key points to really access the foreseen “hydrogen economy”. While numerous studies are devoted to finding inorganic materials enabling the storage of hydrogen with high energy densities under “mild” conditions, relatively less effort has been focused on the search for purely organic storage solutions. This review aims to focus on these pioneering works.
 Philippe Makowski Philippe Makowski | Philippe Makowski was born in Cologne (Germany) in 1981. He received his Master of Science in supramolecular and colloidal chemistry in 2006 from the Université de Versailles St Quentin in Versailles (France). He is currently pursuing a PhD degree at the Max Planck Institute of colloids and interfaces in the group of Dr Arne Thomas under the supervision of Prof. Markus Antonietti. The focus of the PhD project is the replacement or the improvement of noble metals catalysts in heterogeneous catalysis. |
 Arne Thomas Arne Thomas | Arne Thomas received his diploma in chemistry from the Philipps University Marburg and his PhD from the University of Potsdam and the Max Planck Institute of Colloids and Interfaces with Prof. Antonietti in 2003. After a postdoctoral fellowship with Professor Galen Stucky at UCSB he now heads a group at the Max Planck Institute of Colloids and Interfaces researching meso/microporous polymers and organic frameworks. |
Pierre Kuhn was born in Mulhouse (France) in 1977. He studied Chemistry at the Université Louis Pasteur in Strasbourg, where he obtained a Diplôme d'Etudes Approfondies in Transition Metal Chemistry and Molecular Engineering in 2002. After receiving his PhD from ULP in 2006 (topic: polyolefin synthesis with nickel phosphine complexes) with Prof. P. Lutz and Prof. D. Matt, he did his post-doctoral work with Prof. M. Antonietti at the Max Planck Institute for Colloids and Interfaces in Potsdam (Germany) on the synthesis of new carbon nitride based materials and their applications in catalysis. |
 Frédéric Goettmann Frédéric Goettmann | After receiving his degree from the Ecole Polytechnique in Palaiseau (France), Frédéric Goettmann prepared a PhD, at the University Pierre et Marie Curie in Paris (France), under the supervision of Dr Clément Sanchez. He then spent two years as a post-doctoral fellow at the Max Planck Institute of Colloids and Interfaces in Potsdam (Germany), in the group of Prof. M. Antonietti. He is currently holding a permanent position at the Institut of Separative Chemistry in Marcoule (France), focusing on the development of noble metal free heterogeneous catalysis. |
Broader contextHydrogen is foreseen to become a major energy carrier in a relatively close future. The shift towards a so called hydrogen economy is driven by both a shortage in fossil fuels (while it seems possible to gain “green” hydrogen from renewable sources) and tremendous progresses in the production of fuel cells (making them suitable for transportation applications). A major problem to complete this shift is to find suitable ways to store this hydrogen. Indeed, to be implemented at an industrial scale, storage devices must be light and store as much hydrogen as possible within the smallest possible volume. Moreover, the storage as well as the release temperatures must be as close as possible to ambient conditions. Achieving such goals proved to be a rather difficult task. Among the many possible systems, which were considered, we felt that devices based on organic compounds (polymers as well as molecules) were not paid enough attention. In particular, they tend to be lighter and easier to handle than their inorganic counterparts. In this short review, we thus wanted to highlight some recent advances in the field of hydrogen storage in/with organic compounds. |
1 Introduction
“The sudden recognition that an energy crisis is facing the world has loosed upon that same long-suffering world an avalanche of articles minatory, pontifical or plain gloating: heaven knows how much energy has been used in the production and absorption of these writings. Most of the prophets of doom are concerned only with present fuels and present modes of extraction and use, and very properly—given their premises—come to deeply pessimistic conclusions…” These few sentences could seem extracted from some recent edition of some British or German Newspapers, possibly under the title “Fuelling the future”.1 And actually many very good articles dealing with such questions were released in recent years.2–5 However, these words were written by the materials science correspondent of Nature in May 19736 (6 months before the first oil shock, when the crude oil barrel price reached and exceeded 45 US$, in real value). As a possible solution to over come this predicted energy crisis the author mentioned the introduction of “Hydrogen Economy”, a concept coined as early as 1972 by J. O'M Bockris.7 This concept involved the use of nuclear energy as a primary energy source and hydrogen as a universal energy carrier. In his article in Science, Prof. Bockris envisaged only three difficulties for implementing “hydrogen economy”: “i- conservatism; ii- the absence of education or training in electrochemical engineering; and iii- the public's fear of hydrogen” [sic]. One year later, in a brilliant and still pertinent contribution, Sir Alan Cottrell, identified that some basic materials science problems could hinder or at least delay the advent of the hydrogen age (the notion of “hydrogen age” having actually only been introduced in the nineties8,9). He, however, remained optimistic, saying that: “Engineering materials make the large-scale modern production of energy possible. The technical problems of the power industry are very largely problems of materials. One of the socially most important contributions that materials scientists and technologists can make is to help remove the material barriers to the provision of abundant, clean, cheap energy”.10 Well, we are in 2009 and hydrogen economy has not yet become a daily reality. But Sir Alan was right, the key point of developing hydrogen economy turned out to be a materials science problem: namely finding suitable materials for hydrogen storage.Actually, scientists have done (and still do) their best to achieve ideal hydrogen storage devices. The amount of work published on hydrogen storage has literally exploded since the early nineties (as evidenced by the number of articles found on ISI Web of Science®11 for the entry “Hydrogen storage”, as depicted in Fig. 1). Considering such a figure, one could easily get the feeling that everything has been tested and written about hydrogen storage. And, indeed, very good reviews and papers are available, which focus on given classes of materials, which are considered for storage purposes, as for example: metal hydrides12–15 and nitrides,16 organometallic complexes17 metal-organic frameworks,18–20 carbonaceous materials21–23 and zeolites.24 When considering these reviews or more general ones25,26 two main, quite surprising, figures are appearing:
(i) They are all mainly concerned with hydrogen storage for transportation application.
This point is somehow strange because History shows that a new technology is usually implemented in fixed devices before entering the field of transportation. The first working steam machines (Fig. 2) were, for example, designed by Savery and Newcowen between 1695 and 1712 for pumping water out of mines.27,28 Their engines were enhanced by Watt and Trevithick who introduced the rotary motion and eliminated the need of a condenser between 1782 and 1800.27,29,30 These engines were mainly used as power sources for factories and about ten more years were needed before the Salamanca, the first commercial steam locomotive was made available by Matthew Murray. Also the diesel engine was firstly designed as a small power supply for factories but took a very long time to be implemented in daily transport means. Indeed, the first working engine was designed in 1897, the first commercial successes were recorded around 1908, but the first ships powered by diesel engines appeared in the twenties, the first locomotives in the fifties.31 The current focus on mobile applications for fuel cells (and thus hydrogen storage devices) thus seems premature as long as fuel cells did not find wide spread stationary applications (for example for the supply of electricity, heat and water in remote areas). However the current fuel cell concepts for stationary (e.g. solid oxide fuel cells with high working temperatures) and mobile (e.g. polymer electrolyte membrane fuel cells) are very different, which explains why they are developed in parallel. Anyhow, transportation applications (which are more demanding with regards to H-storage) have become the common reference for people working in the area of hydrogen storage and the targets set by the US Department of Energy their Holy Grail.32
(ii) They all deal with inorganic or at least hybrid or metal organic systems.
Oil and natural gas are, besides electricity, the two most current means to transport power. They were selected because they are a naturally occurring fuel source, but also because they are easy to store or transport† and have a very high energy density. An ideal system for storing hydrogen should thus be as convenient to use and handle as oil and gas are. But oil and gas are essentially organic compounds. It is thus a slight surprise that the overwhelming majority of materials, which have been studied for hydrogen storage applications, are not purely organic.
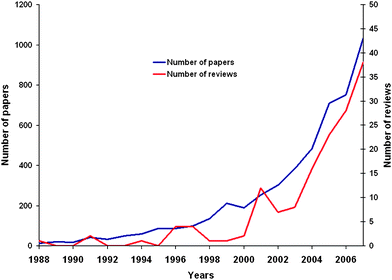 | ||
| Fig. 1 Approximate number of publications (blue line) and reviews (red line) dealing with “hydrogen storage” as obtained from ISI Web of Science®. | ||
 | ||
| Fig. 2 Timeline to show the development of steam engines, from the first pressure cooker to the first commercial locomotive. | ||
As we think that organic materials at least have as much potential for H-storage as inorganic ones, we decided to devote this short review to them. We thus will focus on purely organic materials for H-storage by physisorption (thus excluding purely carbonaceous materials as well as MOFs33) or organic molecules for H-storage by chemisorption (thus excluding hydrides; ammonia will, however, be considered here, for commodity reasons). Clarthrates, i.e. molecular cages composed mainly of carbon, are also out of the scope of this paper and were reviewed elsewhere.34
2 Physisorption of molecular hydrogen on porous organic solids
2.1 Introduction
Hydrogen storage by means of physisorption on porous materials is a straightforward approach, as tanks can be simply built up by filling a container with a given material. In addition, any porous material can be directly tested for physisorption applications, which surely partly explains why so many materials have been tested. However, the physisorption of hydrogen is suffering from major limitations. It is governed by Van der Waals interactions between hydrogen molecules and the surface of the solid.35 Due to the physical properties of hydrogen, it is adsorbed only as a monolayer at temperatures higher than its boiling point (20.4 K).35 Therefore the uptake of hydrogen for a given material is directly given by two factors:(i) Firstly, the uptake of hydrogen is related to the specific surface area of the tested solid. Thus, the density of the material has to be as low as possible, i.e. it has to be composed of light elements. As carbon is the lightest element from which porous architectures could be synthesised, it is not a surprise that in general, activated carbons are showing the best storage abilities. The specific surface area of a carbon material can however not be infinite. For example, an infinite graphene sheet was found to have a calculated surface area of 2965 m2 g−1.36 Through fragmentation of the graphene layer, this value can rise to 7745 m2 g−1 for isolated benzene rings (without hydrogen atoms).36 Considering a kinetic diameter of 0.289 nm, a hydrogen molecule would cover a surface of 2.08 × 10−20 m2. Assuming a hydrogen monolayer features a close packed arrangement, 1 gram of “infinite” graphene layers would be able to accommodate 0.09 moles of H2i.e. 18 wt%. The maximum uptakes of hydrogen reported so far are still lying well beyond this value.
(ii) The enthalpy of adsorption is the other determining parameter for hydrogen storage. Indeed, for such applications, hydrogen has to be stored at temperatures as close as possible to ambient temperature, to avoid technical problems related to low temperature tanks, especially during refilling.35 The chemical nature of the surfaces and the morphology of the pores have thus to be tuned to enable a high adsorption enthalpy.37
Studies on activated carbons showed that the adsorption enthalpy is far higher than on inorganic oxides such as silica or zeolites.37 A major drawback of activated carbons is however that these materials are composed almost of carbon, and even if their pore morphology can be slightly tuned, it remains difficult to functionalise them. The future of physisorption therefore relies on organic materials composed of well defined organic moieties. The tuning of porous materials containing well defined organic building blocks was well achieved with MOFs. These materials, even if they are presenting very interesting properties (tunability of the pore structure, chemical nature, swelling kinetic trapping) also present major drawbacks: they mostly rely on heavy atoms, which reduces their maximal specific surface area, and their structure is often chemically unstable.
For these reasons, all organic microporous materials seem to be promising candidates for the physisorption of hydrogen.37 The development of a stable microporosity being difficult in soft matter, only a few studies have emerged in the past few years dealing with such systems.38 In this part we thus will focus on the performances of the well known polystyrene (PS) derived resins, on more evolved materials, namely polymers with intrinsic microporosity (PIMs), and on the enhancement of the performances by changing the functionality of hypercrosslinked polymers (HCP).
2.2 Physisorption on porous Hypercrosslinked PS polymers
Polystyrene derived porous polymers, also known as hypercrosslinked polymers are well known polymer networks that were first described by Staudinger and Husemann in 1935.39 They can be of two types: PS-DVB resins, where styrene is copolymerized with divinylbenzene and “Davankov” type resins, synthesized following a post-crosslinking strategy of linear polystyrene chains through Friedel–Crafts reactions. These approaches have been derivatized, and for example crosslinked resins could be obtained from the direct Friedel–Crafts reaction of poly-4-chlorostyrene. The PS-DVB resins are typically macroporous, but also show some microporosity at high DVB content. On the other hand, the Davankov type resins are typically microporous. Whereas the macroporosity originates from phase separation during the polymerization, the microporosity is related to the stiff cyclic microstructure of the polymer network. These materials are nowadays well established, several resins being commercialized. Their potential use as hydrogen storage materials was however only assessed in recent years.40In 2006, Germain et al. presented a study of several commercially available porous polymer beads.41 These, with SBET ranging from 370 to 1200 m2 g−1 showed hydrogen uptakes in the range of 0.5–1.3 wt% at 1 bar and 77 K. It should be noted that the surface area determined by nitrogen sorption did not correlate with the H2 uptake. Rather the surface area determined by hydrogen sorption was found to fit better. It was clearly identified that the hypercrosslinked polymers exhibit much higher uptakes than that of the PS-DVB resins. Therefore, efforts were turned to high surface area hypercrosslinked polymers. To further get rid of diffusion limitations in purely microporous materials, a material derived from the crosslinking of poly-4-chlorostyrene-DVB showing a bimodal micro- and mesoporosity was prepared. The surface area was not very high, and in turn the uptake value of 1.2 wt% stayed modest.
In parallel to this study, Lee et al. reported at the same time the study of another self-made hypercrosslinked polymer network from the Friedel–Crafts crosslinking of poly-4-chlorostyrene.42 The performance of this material was compared to other reference materials. Uptakes of up to 3.0 wt% were achieved at 10 bar, for an uptake of 1.3 wt% at 1 bar, in line with the results of Germain et al.41 In a further study, they investigated a large number of different hypercrosslinked polymers prepared through self polymerization of dichloroxylene, by variation of the isomers and mixtures thereof.43 Optimization led to materials with slightly increased uptakes (up to 3.7 wt% at 15 bar). As already known, their microstructure had a deep impact on hydrogen uptake. The microstructure of the network was also found to be dependant on the geometric properties of the building units.
A further study aimed at understanding the interaction of hydrogen molecules in these polymer networks on the basis of FT-IR spectroscopy.44 The measurements showed that hydrogen adsorbs perpendicular to the surface, the preferential adsorption sites being benzene rings. Thus, aromatic rings with high electronic densities were found to increase the adsorption enthalpy.
2.3 Physisorption on porous PIMs
Parallel to hypercrosslinked polymers, another type of microporous polymers was developed in recent years, namely polymers of intrinsic microporosity. PIMs are polymers featuring a packing breaking unit in otherwise stiff chains, which prevent a complete collapse of the structure of the polymers and thus preserves a given porosity without templating agents.45 One typical way for preparing such polymers is to condense catechols with tetrafluoroaryl monomers, resulting in both stiff and contorted molecular structures (Fig. 3).45 Several PIMs with various microstructures were prepared, with surface areas up to 1000 m2 g−1. Numerous studies have been carried out to test the hydrogen storage capacity of such materials, but consistently with their surface areas, their measured capacities remained low (Fig. 4).46–48 Interestingly, polymers showing no surface area by nitrogen sorption were found to have a significant uptake of hydrogen.49 This behaviour was attributed to the swelling ability of these polymers in a gas atmosphere, as demonstrated by SAXS measurements. Future works will thus aim at designing polymer networks that do not necessarily present very high surface areas, but microstructures featuring sub-nanometer cavities.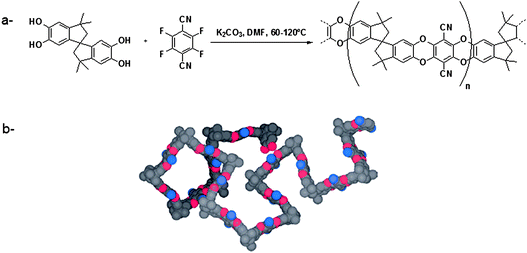 | ||
| Fig. 3 a: Principle of the synthesis of PIM-1 and b: the corresponding space filling model (adapted from ref. 45). | ||
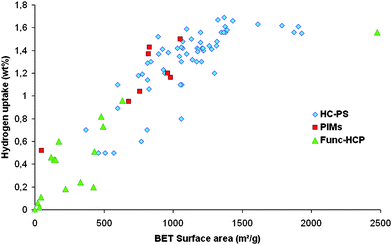 | ||
Fig. 4 Hydrogen uptake of porous organic solids as a function of their surface area (at 77 K under 0.12 Mpa).  stand for Hypercrosslinked polystyrenes (from ref. 41 and 43), stand for Hypercrosslinked polystyrenes (from ref. 41 and 43),  for polymers with intrinsic microporosity (from ref. 46 and 49), for polymers with intrinsic microporosity (from ref. 46 and 49),  for functional hypercrosslinked polymers (from ref. 51 and 58). for functional hypercrosslinked polymers (from ref. 51 and 58). | ||
2.4 Physisorption on functional HCPs
As already mentioned, the introduction of functional groups in polymer networks can provide an enhancement of the hydrogen uptake or of the free enthalpy of adsorption. A theoretical study was thus carried out to estimate the interaction enthalpy of hydrogen towards different organic or inorganic entities.50 Due to the relative difficulty to prepare microporous polymer networks, only a few examples are dealing with hydrogen sorption on functional polymer networks. The main studies are focussed on hypercrosslinked polyaniline and polyanilinium networks, resulting in the introduction of nitrogen atoms in a polyaromatic network.51–53 Again, even if promising preliminary results were obtained (reaching up to 0.96 wt% of hydrogen uptake), the storage capacity of these materials remained low due to modest surface areas. Composite materials of polyaniline were also studied.54 In line with the fact that identification of the most relevant functional groups to enhance the storage capacity is necessary, Cooper et al. carried out theoretical calculations on materials containing fluoride ions. These anions were found to present very high binding affinities for hydrogen molecules.55Finally transition metal decorated polymers were investigated, but even if these fragments can enhance binding affinities towards hydrogen, their heaviness leads to low storage capacities.56,57
2.5 Conclusions
Hydrogen storage by means of physisorption suffers from major limitations. Fig. 3 shows the hydrogen uptake of various organic solids as a function of their specific surface area at 77 K under 0.12 MPa. Two features can be observed: (i) There is no clear difference between the behaviour of polystyrene, PIMs and hypercrosslinked polymeres based materials (even if PIMs seem to perform a little better at comparable BET surface areas) (ii) Until 1200 m2 g−1 and 1.5 wt% uptake, there is a linear correlation between the surface area of the materials and their ability to store hydrogen. Above this limit a kind of plateau seems to be reached which makes the rush for higher surface areas at least questionable. However, organic materials seem very promising materials in this context, due to their high potential of structural and functional tuning, as well as good thermal and chemical stability. Actually they perform at least as well as carbonaceous materials, which also tend to store about 1.5 wt% of hydrogen around 1000 m2 g−1 BET surface area.25 Efforts still have to be done to design well defined microporous polymer architectures. The main problem is to develop materials with ultra-high surface areas. Yaghi et al. recently developed purely organic systems with features similar to MOFs, namely covalent organic frameworks (COFs). These crystalline materials, based on aromatic units linked together by boronic moieties, feature very high surface areas (up to 1600 m2 g−1).59 COFs are very promising materials for hydrogen storage and preliminary results hint towards at least 3.4 wt% of storage capacities at 55 bar and 77 K. Theoretical investigations even predict that storage as high as 19% can be achieved.60 Additionally, another group prepared polytriazine derived materials exhibiting surface areas up to 2500 m2 g−1 (Fig. 5) and featuring hydrogen uptakes of up to 1.5% (under 1 bar and at 77 K).58 Furthermore, these systems showed to be highly tuneable in term of chemical functionalities and structural parameters.61,62 The detailed investigation of their storage capability is still in progress. These examples, anyhow, show that it should be possible to push forward the maximal hydrogen storage capacities by means of physisorption on porous materials.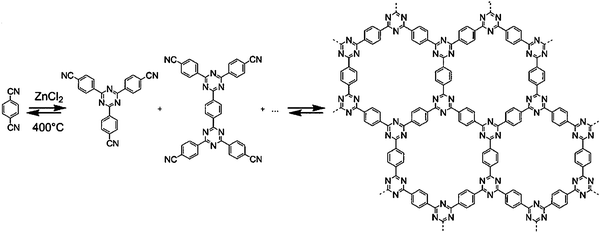 | ||
| Fig. 5 Principle of the ionothermal synthesis of porous triazine based networks (adapted from ref. 58). | ||
3 Hydrogen storage in organic molecules
As previously mentioned, the two most common fuels used to power our cars, petrol and diesel, are molecular compounds which are liquid at room temperature. This proved very convenient to allow fast refilling times for cars, low fuel losses by evaporation and long driving autonomy with relative low volumes of fuel on board.63 In order to preserve these important advantages, it would be of great interest to achieve hydrogen storage materials with similar features. Such concerns explain the current research efforts on direct liquid-feed fuel cells, in which the fuel is not hydrogen anymore, but directly an organic molecule.64 Such fuel cells, however, impose very high requirements on the anodic catalyst (especially resistance against poisoning and avoidance of side reactions) and are thus likely to require a long development time.An ideal molecular storage system for hydrogen should thus be liquid and non-hazardous, it should also store as much hydrogen as possible and release it between 80 and 150 °C (which is about the working temperature of polymer membrane fuel cells and would thus allow to avoid an increased energy consumption by additional heating), most likely in a catalytic process (in order to avoid spontaneous dehydrogenation). Another requirement would be for the dehydrogenation to be reversible so that the dehydrogenated compound could be collected and re-hydrogenated in big industrial plants.
Table 1, displays a non-exhaustive list of pairs of hydrogenated and dehydrogenated molecules, with the calculated enthalpy change associated with the corresponding dehydrogenation reaction (per released H2 molecule, all calculations being made in the gas phase) and the wt% of stored hydrogen. Water (Table 1, entry 1), being the “out gas” of hydrogen fuel cells, is obviously not a suitable means of storing hydrogen, and was only put as a reference. As hydrogen is a relatively unstable molecule (i.e. an energy rich one, and thus a good energy carrier), the hydrogenated molecules (i.e. oxidized forms of hydrogen) are always more stable than hydrogen itself. This explains why all the dehydrogenation reactions we considered are endothermic (an archetypical example being the conversion of ethane to ethylene, Table 1, entry 6, which costs 39 kcal mol−1). It is however worth noticing here that a dehydrogenation reaction splits one molecule into two or more and is thus entropically favoured. The Gibbs energy change associated with these dehydrogenation reactions is thus lower (especially at higher temperatures),65 but as it is easier to access data about ΔH°, we preferred to report these data here. Anyhow, in order to have access to dehydrogenation reactions which are thermodynamically favoured by around 100 °C (when the TΔS term of the Gibbs energy is still dominated by the ΔH° term), one has to search for dehydrogenation products, which are intrinsically stable. CO2 and N2 are typically very stable molecules. The conversion of formic acid into CO2 only costs 7.8 kcal mol−1 for example (Table 1, entry 5) while the dehydration of hydrazine (Table 1, entry 8), is almost athermic. Aromatic molecules are stable too, which explains that they have also been considered for hydrogen storage applications.
| Entry | Hydrogenated form | Dehydrogenated form | ΔH° of dehydrogenation per mol of released H2/kcal mol−1 | Weight % of stored H2 |
|---|---|---|---|---|
| a These calculations were done in the gas phase and do not take into account phase transition enthalpies. | ||||
| 1 | H2O | O2 | 68.9 | 11.1 |
| 2 | CH4 (+2 H2O) | CO2 | 13.9 | 15.4 |
| 3 | MeOH | CO | 20.7 | 25.0 |
| 4 | MeOH (+H2O) | CO2 | 9.4 | 12.0 |
| 5 | HCOOH | CO2 | 7.8 | 4.3 |
| 6 | C2H6 | C2H4 | 39.1 | 6.6 |
| 7 | NH3 | N2 | 7.4 | 20.0 |
| 8 | N2H4 | N2 | 0.2 | 12.5 |
| 9 |

|

|
19.5 | 7.1 |
| 10 |

|

|
17.5 | 3.0 |
| 11 |

|
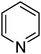
|
19.1 | 7.1 |
| 12 |

|

|
19.6 | 3.0 |
| 13 |

|
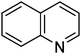
|
16.7 | 7.2 |
| 14 |

|

|
13.1 | 5.5 |
| 15 |
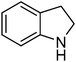
|
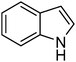
|
13.7 | 1.7 |
| 16 |

|

|
14.3 | 6.7 |
3.1 CO2 producing storage systems
With a subtitle starting with “CO2 producing…”, such systems can spontaneously appear as environmentally unsustainable. But the systems we will consider in this section are organic molecules, which can be extracted from biomass or directly produced by the hydrogenation of CO2 and are thus formally CO2 neutral (formally, because this does not take into account the fact that harvesting and/or converting these molecules into the wanted storage system is not 100% effective).| CH3OH + 1.5 O2 → CO2 + 2 H2O | (1) |
This dual application of alcohol could be a key point in the installation of an economy based on methanol. In a first step, methanol could be used alone or mixed with gasoline in ICE, allowing for the development of the infrastructure, required for its large scale production and distribution. In a second step ICE would progressively be replaced by a DMFC, which is more efficient. This two step path could also provide the time needed to overcome some major difficulties. A DMFC system was first recognized in 1922 by Müller, but research on possible applications were boosted only in the 1990's with the introduction of DuPont's Nafion® proton-exchange membrane (PEM) based fuel cells.67–69 They are now regarded as one of the most promising energy technologies in the 21st century.70 But, in practice, a DMFC suffers from lower anode electrocatalytic activity and high methanol crossover through the membrane.71 This crossover results in self-discharge of methanol, which provides additional heat instead of electricity, and reduction of the cathode voltage and efficiency due to the oxidation of methanol at the cathode.72 Methanol must be diluted in water in concentrations of approximately 10% to avoid this “chemical short circuit”.73 As a result, the fuel energy density is strongly reduced. The power density available from methanol oxidation is less important than for hydrogen oxidation resulting in an increased need for platinum at the anode. Last but not least, poisoning of the catalyst by CO, and as in proton exchange membrane fuel cells, membrane cost and life-time are limiting factors, and technological breakthroughs are needed to increase DMFC in terms of performance and costs.
Steam reforming (Table 1 entry 2) can thus appear as a valuable alternative to use alcohols as hydrogen source/storage devices. But the required temperatures for this reaction (up to 800 °C) does not allow for the development of onboard steam reforming devices for ethanol or methanol. These reaction temperatures can be lowered to around 400–500 °C in certain conditions, or with specific catalysts but this is still not sufficient to be useful in mobile applications.74–76
A last drawback of methanol is its high toxicity and water solubility, which makes it a very hazardous fuel with regards to the potential environmental impact of a car crash. Nevertheless, it has to be compared to classical fuel used every day such as gasoline which is recognized to contain carcinogenic compounds like benzene. Ingestion of 25 to 90 mL of methanol may be fatal if not treated in time (compared to 120–300 mL of gasoline), but as shown in different studies the risk for consumers is limited and in any cases not greater than those associated with the use of gasoline.66
3.2 Ammonia, ammonia-boranes and hydrazine
In the search of a perfect energy carrier, ammonia (NH3) has several advantages. The synthesis of ammonia by the Haber–Bosch process is carried out on an industrial scale with an annual production exceeding 120 million tons, and it is one of the most studied processes worldwide.97 Until now methane steam reforming yields the hydrogen needed for this process. Ammonia could thus only be considered as a CO2-free energy carrier if its production process was using “green” sources of hydrogen. It, anyhow, should be reminded that hydrogen production is necessary for all hydrogen based carriers, so the problem of “green” hydrogen is not limited to ammonia. Liquid ammonia is not flammable in air and the infrastructure for its delivery is already established.98 It can be used directly in high temperature solid oxide fuel cells (SOFC) but not for mobile applications due to the high temperature (above 200 °C). Another possibility is to decompose ammonia and to use it in low temperature fuel cells such as polymer electrolyte membrane fuel cells (PEMFC). Once again the high temperature needed to decompose ammonia can be a problem to design small devices.99 The dehydrogenation of ammonia can be catalysed by nickel on alumina, caesium promoted ruthenium100–104 or even much less expensive systems including barium, zirconium oxinitride, iron cobalt alloys or even the residual iron of commercial carbon nanotubes.105–107 Even if the hydrogen storage capacity in ammonia is high (more than 17 wt%), it is too stable to easily release H2. Furthermore NH3 suffers from its high toxicity which prevents its direct use in mobile application. Indeed, even at low concentrations it is highly toxic to aquatic animals, and for this reason it is classified as dangerous for the environment. However it is worth noticing here that ammonia has a very strong and unpleasant smell even at concentrations far below its toxicity limit. It is thus very unlikely for most people to keep staying somewhere where ammonia has reached a critical concentration.108A possibility to overcome this drawback is to confine ammonia in solid form by binding it in metal amine salts such as Mg(NH3)6Cl2 or Ca(NH3)8Cl8. The total amount of available hydrogen is thus reduced to 9.78 wt% but such salts are safer than pure ammonia (Fig. 6 and 7). More over, ammonia should be able to adsorb and desorb reversibly under mild conditions. Indeed several studies investigated the ammonia desorption temperature which can be achieved at low temperatures (between 100 and 200 °C).109–112 Ammonia can be stored and released on demand with this safe system, unfortunately ammonia decomposition is done under too harsh a conditions to be used in mobile applications. Borohydrides M+BH4− (where M is Li or Na)113 and boranes NHnBHn (with n from 1 to 4)114–116 could be a better solution. Despite a high hydrogen storage capacity in borohydrides, for example up to 19% of the total mass for LiBH4, they suffer from two major drawbacks, high decomposition temperatures and large activation barriers to rehydrogenation.
 | ||
| Fig. 6 Tablets of Mg(NH3)6Cl2 which can be handled.109 | ||

On the other hand, by simply replacing the metal cation by ammonium ion, hydrogen content increases to an impressive 24 wt%, but only a part of it is available for desorption at room temperature as seen in Table 2. Remaining hydrogen comes off at higher temperatures. Unfortunately no path for reversibly reloading NHBH has been found yet.117
| Reaction | Wt% H2 | T/°C |
|---|---|---|
| NH4BH4 → NH3BH3 + H2 | 6.1 | <25 |
| NH3BH3 → NH2BH2 + H2 | 6.5 | <120 |
| NH2BH2 → NHBH + H2 | 6.9 | ∼155 |
| NHBH → BN + H2 | 7.3 | >500 |
Hydrazine, NH2–NH2, could also have some applications in energy storage and even replace hydrogen or alcohol in direct hydrazine fuel cells (DHFC), which show a higher electric motive force.118 The use of hydrazine in fuel cells is not new, but only a few studies have been devoted to this topic,119–121 mostly because of the high toxicity of hydrazine confining its use to military or space applications. Recently, however, this idea attracted renewed interest. It turned out possible to overcome the toxicity problem, by binding hydrazine to a carbonyl group rich polymer. Release was achieved through a solvent during power generation.122 This solution, however, needs to be further developed to be completely viable.
3.3. Cycloalkanes
In the field of hydrogen storage, cycloalkanes have attracted some attention in the past two decades under the name “liquid organic hydrides” (LOH),123–127 which refers to solid hydrides such as NaBH4, LiBH4etc. The hydrogen content of cycloalkanes is comprised of between 3 wt% for tetralin and 7.1 wt% for cyclohexane. A cycloalkane based H-storage system relies on an in situ catalytic dehydrogenation coupled to a recycling hydrogenation of the corresponding aromatic compound (see Fig. 8). This method presents several advantages and is really attractive to store hydrogen. Indeed, the dehydrogenation of the storing molecules can theoretically be fully reversible, which makes it a fully recyclable CO2 free medium. In addition, the toxicity of the molecules involved in such a cycle can be equivalent to that of gasoline. However, like in the previous sections, cycloalkanes suffer from a major drawback: their high dehydrogenation temperature. Indeed, dehydrogenation of molecules including cyclohexane, methylcyclohexane, cyclohexylbenzene, bicyclohexyl,1-methyldecalin are endothermic and require between 64 to 69 kJ mole−1 of H2. Noble metal catalysts such as Pt, and bimetallic catalysts are known to be efficient in dehydrogenation of cycloalkanes. Biniwale et al. reviewed various studies concerning dehydrogenation catalysts128 and reactor systems (batch-wise, flow or spray system). Hydrogen evolution rates depend on two main parameters: the temperature of the reaction (between 200 °C and 320 °C) and the nature of the catalysts Pt/AC (active carbon), Pt/C, Ni/AC, Pt–Ir/C.129–132 Saito et al., for example, described the activity of Wilkinson's catalyst in the dehydrogenation of cyclooctane to cyclooctene at 180 °C.133,134 Liquid organic hydrides, i.e. aromatics-cycloalkanes pairs, are indeed valuable candidates for hydrogen storage, but need further development of efficient catalyst and reactors in order to reduce the dehydrogenation temperature to around 100 °C.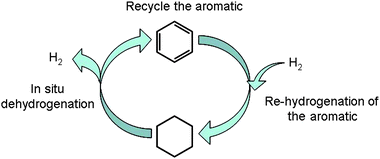
3.4 Heterocyclic compounds
As already seen, liquid organic hydrides and especially cycloalkanes suffer from high dehydrogenation reaction temperatures.135 A recent computational and experimental study, however, showed that the use of N or O heterocycles could help to substantially lower the dehydrogenation temperatures.136 The authors achieved complete dehydrogenation of 2,3-dihydro-1H-indole under mild conditions (toluene reflux), and computed theoretical thermodynamic values for dehydrogenation of bicyclic compounds. In another paper they compiled theoretical results for a large range of nitrogen substituted heterocycles.65 This work shows that a number of design factors can help to tune the dehydrogenation temperature. Five-membered rings tend to be most effective in lowering this temperature. In addition, Cui et al. recently show that various substituents also can have a positive impact on the dehydrogenation temperatures of piperidines and octahydroindoles, using Pt/C, Pd/C, and Rh/C as catalysts.137 Nevertheless, to be fully operational and used in mobile applications such as cars, these very promising hydrogen carriers need more research especially in the area of catalyst development.3.5 Conclusions
In this section we saw a broad range of solutions to the problem of hydrogen storage. These solutions are all based on the use of a liquid carrier to store hydrogen. This overcomes problems that occur with liquid or gaseous hydrogen. This ideal media should meet the following requirements: have available hydrogen content above 6 wt%, be liquid and non-volatile, non-toxic (relative to normal gasoline), have an enthalpy low enough for the dehydrogenation to be thermodynamically favoured at low temperature (below 150 °C). None of the proposed solutions achieve all of these requirements yet. Each of them has some advantages and disadvantages, and technological breakthroughs are needed in almost all cases.Conclusions
In this short overview of recent efforts to develop organic hydrogen storage systems, we aimed to underline their decisive advantages over inorganic systems: most organic materials are light, cheap and green and they allow for high hydrogen densities. Anyhow, two major drawbacks remain: storage devices relying on physisosorbtion still feature too low a hydrogen capacity and most devices in which hydrogen is chemically bonded still require too high a desorbtion temperature. These two problems must be overcome to have a chance to see organic storage systems entering an industrial phase. The most promising ways to achieve this (fluorinated polymers on the one side, nitrogen containing cycles on the other) both strongly rely on heteroelements. This could be a lesson for further studies: an ideal organic storage device for hydrogen should, indeed, have features similar to coal or gasoline (high energy densities, ease of handling, low toxicity, etc.), but contrary to the best coal and gasoline types it should contain more than just carbon and hydrogen.References
- Fuelling the Future, BBC World Service, http://www.bbc.co.uk/worldservice/specials/1349_energy/ Search PubMed.
- The Economist, The Edison of our age?, 30.11.2006 Search PubMed.
- S. Birch The Independent, Hydrogen fuel station: Is this really the fuel of the future? , 05.07.2007 Search PubMed.
- D. Bodanis, The Guardian, Let there be light, 30.04.2008 Search PubMed.
- F. Vorholz, Die Zeit, Auf dem Weg ins Solarzeitalter, 23.10.2008 Search PubMed.
- Nature, 1973, 243, 184–185 Search PubMed.
- J. O'M Bockris, Science, 1972, 176, 1323 CrossRef.
- T. N. Veziroglu, Int. J. Hydrogen Energy, 1998, 23, 1077–1078 CrossRef CAS.
- R. Britton, TCE, The coming of the hydrogen age, November 2004, pp. 28–29 Search PubMed.
- A. Cottrell, Metall. Trans., 1973, 4, 405–410 Search PubMed.
- Web of Science, http://www.thomsonreuters.com/products_services/scientific/Web_of_Science Search PubMed.
- S. I. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrog. Energy, 2007, 32, 1121–1140 CrossRef CAS.
- F. Schuth, B. Bogdanovic and M. Felderhoff, Chem. Commun., 2004, 2249–2258 RSC.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1315 CrossRef CAS.
- D. H. Gregory, Chem. Rec., 2008, 8, 229–239 CrossRef CAS.
- M. Grellier, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2613–2615 CrossRef CAS.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS.
- A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Sol. State Chem., 2005, 178, 2491–2510 CrossRef CAS.
- S. Banerjee, S. Murad and I. K. Puri, Proc. IEEE, 2006, 94, 1806–1814 CrossRef CAS.
- P. Kowalczyk, R. Holyst, M. Terrones and H. Terrones, Phys. Chem. Chem. Phys., 2007, 9, 1786–1792 RSC.
- R. Strobel, J. Garche, P. T. Moseley, L. Jorissen and G. Wolf, J. Power Sources, 2006, 159, 781–801 CrossRef.
- H. W. Langmi and G. S. McGrady, Coord. Chem. Rev., 2007, 251, 925–935 CrossRef CAS.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef.
- A. W. C. van den Berg and C. Otero Aréan, Chem. Commun., 2008, 668–681 RSC.
- K. Frenken and A. Nuvolari, The early development of the steam engine: an evolutionary interpretation using complexity theory, 2001, 2nd European Meeting on Applied Evolutionary Economics, Vienna, Austria Search PubMed.
- H. Davey, Nature, 1903, 69, 67–69 CrossRef.
- D. P. Miller, Ann. Sci., 2008, 65, 47–72 CrossRef.
- J. Tann, J. Econ. Hist., 1978, 38, 363–391 Search PubMed.
- L. Bryant, Technology and Culture, 1976, 17, 432–446 Search PubMed.
- U.S. Department of Energy, http://www.energy.gov/.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS.
- V. V. Struzhkin, B. Militzer, W. L. Mao, H. K. Mao and R. J. Hemley, Chem. Rev., 2007, 107, 4133–4151 CrossRef CAS.
- M. Felderhoff, C. Weidenthaler, R. von Helmolt and U. Eberle, Phys. Chem. Chem. Phys., 2007, 9, 2643–2653 RSC.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS.
- M. G. Nijkamp, J. Raaymakers, A. J. van Dillen and K. P. de Jong, Appl. Phys. A-Mater. Sci. Process., 2001, 72, 619–623 CrossRef CAS.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS.
- H. Staudinger and E. Husemann, Berichte Der Deutschen Chemischen Gesellschaft, 1935, 68, 1618–1634 Search PubMed.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2006, 66, 768–779 CrossRef CAS.
- J. Germain, J. Hradil, J. M. J. Frechet and F. Svec, Chem. Mat., 2006, 18, 4430–4435 CrossRef CAS.
- J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- C. D. Wood, B. Tan, A. Trewin, H. J. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mat., 2007, 19, 2034–2048 CrossRef CAS.
- G. Spoto, J. G. Vitillo, D. Cocina, A. Damin, F. Bonino and A. Zecchina, Phys. Chem. Chem. Phys., 2007, 9, 4992–4999 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- P. M. Budd, A. Butler, J. Selbie, K. Mahmood, N. B. McKeown, B. Ghanem, K. Msayib, D. Book and A. Walton, Phys. Chem. Chem. Phys., 2007, 9, 1802–1808 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem. Int. Ed., 2006, 45, 1804–1807 CrossRef.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357–1370 RSC.
- J. Germain, J. M. J. Frechet and F. Svec, J. Mater. Chem., 2007, 17, 4989–4997 RSC.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- B. Panella, L. Kossykh, U. Dettlaff-Weglikowska, M. Hirscher, G. Zerbi and S. Roth, Synth. Met., 2005, 151, 208–210 CrossRef CAS.
- M. U. Jurczyk, A. Kumar, S. Srinivasan and E. Stefanakos, Int. J. Hydrog. Energy, 2007, 32, 1010–1015 CrossRef CAS.
- A. Trewin, G. R. Darling and A. I. Cooper, New J. Chem., 2008, 32, 17–20 RSC.
- A. I. Cooper and M. Poliakoff, Chem. Commun., 2007, 2965–2967 RSC.
- H. Lee, W. I. Choi and J. Ihm, Phys. Rev. Lett., 2006, 97, 056104 CrossRef.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem. Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580 CrossRef CAS.
- P. Kuhn, A. Forget, J. Hartmann, A. Thomas and M. Antonietti, Adv. Mater., 2009, 21, 897–901 CrossRef.
- P. Kuhn, A. Forget, D. S. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS.
- T. J. Wallington, E. W. Kaiser and J. T. Farrell, Chem. Soc. Rev., 2006, 35, 335–347 RSC.
- U. B. Demirci, J. Power Sources, 2007, 169, 239–246 CrossRef CAS.
- E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 2231–2233 RSC.
- G. Olah and A. Goeppert, Beyond Oil and gas: the methanol economy, Wiley-VCH, Weinheim, 2006 Search PubMed.
- C. Lamy, S. Rousseau, E. M. Belgsir, C. Coutanceau and J. M. Léger, Electrochim. Acta, 2004, 49, 3901–3908 CrossRef CAS.
- D. Cao and S. H. Bergens, J. Power Sources, 2004, 134, 170–180 CrossRef CAS.
- J. H. Wee, J. Power Sources, 2006, 161, 1–10 CrossRef CAS.
- C. Lamy, J. M. Leger and S. Srinivasan, Direct Methanol Fuel Cells: From a Twentieth Century Electrochemist's Dream to a Twenty-First Century Emerging Technology, Kluwer Academic Publishers/Plenum Press, 2001 Search PubMed.
- W. S. Li, L. P. Tian, Q. M. Huang, H. Li, H. Y. Chen and X. P. Lian, J. Power Sources, 2002, 104, 281–288 CrossRef CAS.
- S. K. Kamarudin, W. R. W. Daud, S. L. Ho and U. A. Hasran, J. Power Sources, 2007, 163, 743–754 CrossRef CAS.
- M. Broussely and G. Archdale, J. Power Sources, 2004, 136, 386–394 CrossRef CAS.
- A. Haryanto, S. Fernando, N. Murali and S. Adhikari, Energy Fuels, 2005, 19, 2098–2106 CrossRef CAS.
- G. Ligthart, R. H. Meijer, M. P. J. Donners, J. Meuldijk, J. Vekemans and L. A. Hulshof, Tetrahedron Lett., 2003, 44, 1507–1509 CrossRef CAS.
- G. R. A. Adair and J. M. J. Williams, Tetrahedron Lett., 2005, 46, 8233–8235 CrossRef CAS.
- R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 3529–3530 CrossRef CAS.
- Y. Gao, J. K. Kuncheria, H. A. Jenkins, R. J. Puddephatt and G. P. A. Yap, J. Chem. Soc., Dalton Trans., 2000, 3212–3217 RSC.
- S. Ogo, H. Nishida, H. Hayashi, Y. Murata and S. Fukuzumi, Organometallics, 2005, 24, 4816–4823 CrossRef CAS.
- J. R. Hyde and M. Poliakoff, Chem. Commun., 2004, 1482–1483 RSC.
- E. Garcia-Verdugo, Z. M. Liu, E. Ramirez, J. Garcia-Serna, J. Fraga-Dubreuil, J. R. Hyde, P. A. Hamley and M. Poliakoff, Green Chem., 2006, 8, 359–364 RSC.
- R. Williams, R. S. Crandall and A. Bloom, Appl. Phys. Lett., 1978, 33, 381–383 CrossRef.
- R. S. Coffey, Chem. Commun., 1967, 923–924 Search PubMed.
- T. Yoshida, Y. Ueda and S. Otsuka, J. Am. Chem. Soc., 1978, 100, 3941–3942 CrossRef CAS.
- S. H. Strauss, K. H. Whitmire and D. F. Shriver, J. Organomet. Chem., 1979, 174, C59–C62 CrossRef CAS.
- R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 3529–3530 CrossRef CAS.
- C. Wakai, K. Yoshida, Y. Tsujino, N. Matubayasi and M. Nakahara, Chem. Lett., 2004, 33, 572–573 CrossRef CAS.
- H. A. Gasteiger, N. Markovic, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1994, 98, 617–625 CrossRef CAS.
- F. J. Scott, C. Roth and D. E. Ramaker, J. Phys. Chem. C, 2007, 111, 11403–11413 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic chemistry, Wiley-VCH, Weinheim, 1997 Search PubMed.
- Y. Inoue, H. Izumida, Y. Sasaki and H. Hashimoto, Chem. Lett., 1976, 863–864 CAS.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 3927–3941 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS.
- W. Leitner, Angew. Chem. Int., Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef.
- P. J. Feibelman, L. A. Glenn, P. Stripling, G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2005, 58, 13–15 CrossRef.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal. A, 2004, 277, 1–9 CrossRef CAS.
- W. Raróg-Pilecka, D. Szmigiel, Z. Kowalczyk, S. Jodzis and J. Zielinski, J. Catal., 2003, 218, 465–469 CrossRef CAS.
- S.-F. Yin, B.-Q. Xu, C.-F. Ng and C.-T. Au, Appl. Catal. B, 2004, 48, 237–241 CrossRef CAS.
- S. J. Wang, S. F. Yin, L. Li, B. Q. Xu, C. F. Ng and C. T. Au, Appl. Catal. B, 2004, 52, 287–299 CrossRef CAS.
- G. Papapolymerou and V. Bontozoglou, J. Mol. Catal. A, 1997, 120, 165–171 CrossRef CAS.
- T. V. Choudhary, C. Sivadinarayana, A. Klinghoffer, D. W. Goodman and J. J. S. a. T. H. F. E. Iglesia, in Studies in Surface Science and Catalysis, Elsevier, 2001, pp. 197–202 Search PubMed.
- H. Soerijanto, C. Rodel, U. Wild, M. Lerch, R. Schomacker, R. Schlogl and T. Ressler, 2007, 250, pp. 19–24.
- J. Zhang, M. Comotti, F. Schuth, R. Schlogl and D. S. Su, Chem. Commun., 2007, 1916–1918 RSC.
- J. Zhang, J. O. Muller, W. Q. Zheng, D. Wang, D. S. Su and R. Schlogl, Nano Lett., 2008, 8, 2738–2743 CrossRef CAS.
- The authors would like to acknowledge Prof. Dr. F. Schüth for this pertinent remark.
- C. H. Christensen, T. Johannessen, R. Z. Sørensen and J. K. Nørskov, Catal. Today, 2006, 111, 140–144 CrossRef CAS.
- J. S. Hummelshoj, R. Z. Sorensen, M. Y. Kustova, T. Johannessen, J. K. Norskov and C. H. Christensen, J. Am. Chem. Soc., 2006, 128, 16–17 CrossRef CAS.
- H. S. Jacobsen, H. A. Hansen, J. W. Andreasen, Q. Shi, A. Andreasen, R. Feidenhans'l, M. M. Nielsen, K. Ståhl and T. Vegge, Chem. Phys. Lett., 2007, 441, 255–260 CrossRef CAS.
- C. H. Christensen, R. Z. Sorensen, T. Johannessen, U. J. Quaade, K. Honkala, T. D. Elmoe, R. Kohler and J. K. Norskov, J. Mater. Chem., 2005, 15, 4106–4108 RSC.
- B. Bogdanovic, M. Felderhoff, A. Pommerin, T. Schuth and N. Spielkamp, Adv. Mater., 2006, 18, 1198 CrossRef.
- A. Gutowska, L. Y. Li, Y. S. Shin, C. M. M. Wang, X. H. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS.
- M. H. Matus, K. D. Anderson, D. M. Camaioni, S. T. Autrey and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 4411–4421 CrossRef CAS.
- C. W. Yoon and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 13992–13993 CrossRef CAS.
- G. W. Crabtree and M. S. Dresselhaus, MRS Bull., 2008, 33, 421–428 CAS.
- W. X. Yin, Z. P. Li, J. K. Zhu and H. Y. Qin, J. Power Sources, 2008, 182, 520–523 CrossRef CAS.
- A. J. Bard, Anal. Chem., 1963, 35, 1602–1607 CrossRef CAS.
- K. Yamada, K. Yasuda, N. Fujiwara, Z. Siroma, H. Tanaka, Y. Miyazaki and T. Kobayashi, Electrochem. Commun., 2003, 5, 892–896 CrossRef CAS.
- L. D. Burke and K. J. O'Dwyer, Electrochim. Acta, 1989, 34, 1659–1664 CrossRef CAS.
- K. Asazawa, K. Yamada, H. Tanaka, A. Oka, M. Taniguchi and T. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 8024–8027 CrossRef CAS.
- G. Cacciola, N. Giordano and G. Restuccia, Int. J. Hydrogen Energy, 1984, 9, 411–419 CrossRef CAS.
- S. Yolcular and O. Olgun, Energy Sources Part A-Recovery Util. Environ. Eff., 2008, 30, 309–315 Search PubMed.
- D. E. Schwarz, T. M. Cameron, P. J. Hay, B. L. Scott, W. Tumas and D. L. Thorn, Chem. Commun., 2005, 5919–5921 RSC.
- A. Touzani, D. Klvana and G. Bélanger, Int. J. Hydrogen Energy, 1984, 9, 929–936 CrossRef CAS.
- D. Klvana, A. Touzani, J. Chaouki and G. Bélanger, Int. J. Hydrogen Energy, 1991, 16, 55–60 CrossRef CAS.
- R. B. Biniwale, S. Rayalu, S. Devotta and M. Ichikawa, Int. J. Hydrogen Energy, 2008, 33, 360–365 CrossRef CAS.
- S. Hodoshima, H. Arai and Y. Saito, Int. J. Hydrogen Energy, 2003, 28, 197–204 CrossRef CAS.
- R. Biniwale, N. Kariya and M. Ichikawa, Catal. Lett., 2005, 105, 83–87 CrossRef CAS.
- I. Kobayashi, K. Yamamoto and H. Kameyama, Chem. Eng. Sci., 1999, 54, 1319–1323 CrossRef CAS.
- N. Kariya, A. Fukuoka, T. Utagawa, M. Sakuramoto, Y. Goto and M. Ichikawa, Appl. Catal. A, 2003, 247, 247–259 CrossRef CAS.
- T. Fujii and Y. Saito, J. Chem. Soc., Chem. Commun., 1990, 757 RSC.
- K. Yukawa, T. Fujii and Y. Saito, J. Chem. Soc., Chem. Commun., 1991, 1548 RSC.
- G. P. Pez, A. R. Scott, A. C. Cooper, H. Cheng, L. D. Bagzis and J. B. Appleby, US, 2006.
- A. Moores, M. Poyatos, Y. Luo and R. H. Crabtree, New J. Chem., 2006, 30, 1675–1678 RSC.
- Y. Cui, S. Kwok, A. Bucholtz, B. Davis, R. A. Whitney and P. G. Jessop, New J. Chem., 2008, 32, 1027–1037 RSC.
- R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
Footnote |
| † It is worth noticing here, that handling and transporting natural gas over large distances (especially over seas) is more awkward; see for example ref. 138. |
| This journal is © The Royal Society of Chemistry 2009 |