Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: Current state, chemical physics-based insights and outlook
Venkata Pradeep
Indrakanti
a,
James D.
Kubicki
b and
Harold H.
Schobert
*a
aEMS Energy Institute, Department of Energy and Mineral Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: schobert@ems.psu.edu; Fax: +1 814-863-7432; Tel: +1 814-863-1337
bDepartment of Geosciences, The Earth and Environmental Systems Institute, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: kubicki@geosc.psu.edu; Fax: +1 814-863-8724; Tel: +1 814-865-3951
First published on 30th March 2009
Abstract
This article is a review of the current knowledge of the chemical physics of carbon dioxide (CO2) conversion to fuels using light energy and water (CO2 photoreduction) on titania (TiO2)-based catalysts and Ti-species in porous materials. Fairly comprehensive literature reviews of CO2 photoreduction are available already. However, this article is focused on CO2 photoreduction on Ti-based catalysts, and incorporates fundamental aspects of CO2 photoreduction, knowledge from surface science studies of TiO2 and the surface chemistry of CO2. Firstly, the current state of development of this field is briefly reviewed, followed by a description of and insights from surface state and surface site approaches. Using examples such as metal-doping of TiO2, dye-sensitization, oxygen vacancies in TiO2 and isolated-Ti centers in microporous/mesoporous materials, the utility of these approaches to understand photoinduced reactions involved in CO2activation is examined. Finally, challenges and prospects for further development of this field are presented. Enhanced understanding of the CO2 : TiO2 system, with a combination of computational and experimental studies is required to develop catalysts exhibiting higher activity towards CO2 photoreduction.
 Venkata Pradeep Indrakanti Venkata Pradeep Indrakanti | Pradeep Indrakanti will graduate with a PhD in Energy and Geo-Environmental Engineering from The Pennsylvania State University in May 2009. He has received a B.Tech. in Chemical Engineering from the National Institute of Technology, Tiruchirapalli, India. His research interests include the application of industrial ecology and green chemistry principles to clean energy technologies, carbon dioxide chemistry and semiconductor photocatalysis. He has process experience in cement and fertilizer production, and co-authors an energy blog. |
 James D. Kubicki James D. Kubicki | Kubicki is Associate Professor of Geosciences at The Pennsylvania State University and affiliated with the Earth and Environmental Systems Institute. He has received a B.S. in Geology from the California State University, Fullerton and PhD in Geochemistry from Yale University. Recent research has focused on mineral surface reactions with particular emphasis on the surface chemistry of TiO2 phases. |
 Harold H. Schobert Harold H. Schobert | Harold Schobert has been involved in coal research for over thirty years, beginning with a project on making hydrogen sulfide from coal, at Deepsea Ventures Inc. He subsequently was involved in research and development on low-rank coals for the University of North Dakota Energy Research Center and its predecessor organizations, in Grand Forks ND, where he managed the Coal Science Division. He moved to Penn State in 1986, where a major research activity has been the coal-based jet fuel program, now entering commercialization. Another research interest is carbon dioxide chemistry for mineral carbonation and CO2 utilization. |
Broader contextThe conversion of carbon dioxide (CO2) to produce fuels using light energy has the potential to reduce anthropogenic CO2 emissions, and lower the consumption of fossil fuels. Although there have been interesting developments in the photoinduced reduction of CO2 (CO2 photoreduction) on various Ti-based catalysts, a detailed discussion of the physico-chemical aspects of the light-induced activation of CO2 has been lacking. This article is a review of current knowledge of the chemical physics of CO2 photoreduction on titania (TiO2)-based catalysts and Ti-species in porous materials. Firstly, a comprehensive review of current literature on CO2 photoreduction on Ti-based materials is presented, and comparisons are made between reduction of CO2 using solar hydrogen generation and the direct photoreduction of CO2. Various advances in this field are discussed using the surface site and surface state approaches, and future research needs for improving the efficiency of carbon dioxide conversion with sunlight are outlined. |
Introduction
CO2 utilization
There is a growing need to mitigate CO2 emissions. Some of the strategies to mitigate CO2 emissions are energy conservation, carbon capture and storage and using CO2 as a raw material in chemical processes as well as enhanced oil recovery (EOR) (CO2 utilization). Catalytic reactions involving CO2 and producing value-added products are reviewed by Xiaoding and Moulijn.1 More recent reviews by Song,2 Sakakura et al.3 and Aresta and Dibenedetto4 detail additional opportunities to use CO2 as a feedstock. Reactions involving CO2 typically require energy input and/or a high energy substrate. As pointed out by Song,2 processes involving CO2 could be endergonic (or endothermic) and can still be feasible, provided the products are valuable enough. Moreover, as carbon legislation becomes imminent, a price for CO2 (either as a carbon tax, or as a CO2 offset in a cap-and-trade system) would benefit processes having a positive net CO2 balance.What is the potential for utilizing CO2 by the use of various renewable energy technologies? Current global industrial consumption of CO2 (approximately 115 million metric tons (Mt) year−1)5 is insignificant compared to anthropogenic CO2 emissions (∼26 giga tons (Gt) of CO2 year−1). However, we note that U.S. CO2 consumption for enhanced oil recovery as well as chemical synthesis (∼40 Mt year−1)6 is similar in scale to other projected means of U.S. greenhouse gas (GHG) reduction (data from a McKinsey report7), such as using solar PV electricity, wind energy and cellulosic biofuels, as shown in Fig. 1. With the caveat that most processes utilizing CO2 would need reducing power and/or a source of energy which is not reflected in the dotted lines shown in Fig. 1, we note that comparatively larger amounts of GHG mitigation could be possible if CO2 is converted into fuels. This conversion will lead to a reduction in CO2 emissions only if the underlying energy infrastructure is not based on primary fossil fuels.5
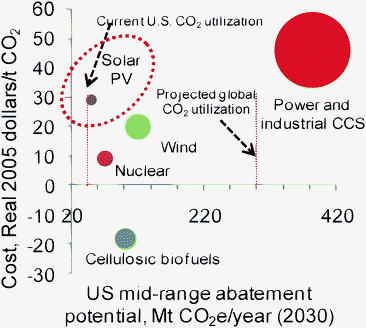 | ||
| Fig. 1 Comparison of the greenhouse gas (GHG) abatement potential (in million metric tonnes of CO2 equivalents (MtCO2e) year−1) for selected technology options. The size of the circle approximately denotes the potential for abatement; negative costs represent net-savings from reducing CO2 emissions. The data (from a McKinsey report, showing the US mid-range abatement potentials and costs) do not include a price for carbon. The estimated potential for CO2 abatement using nuclear, wind, and cellulosic biofuel technologies is of the same order of magnitude as current CO2 utilization. Aresta and Dibenedetto8 project that direct solar conversion of CO2 to fuels would mitigate 300–700 Mt CO2 year−1. | ||
For example: the quantity of U.S. CO2 emissions mitigated by solar PV technologies (50 Mt CO2e year−1 by 2030) represents approximately 50% of the CO2 currently utilized by the global chemical industry. The data shown in Fig. 1 for solar photovoltaic (PV) technologies could be considered as representing the upper limits for converting light energy to electricity. Solar fuel production could be thought of as involving solar PV electricity production, followed by generation of hydrogen or reduction of CO2 using the electrical energy. If we assume (simplistically) that the efficiency of conversion dictates the potential size of the market, using 10% efficiency for this solar-electricity-chemicals conversion results in a 5 Mt CO2 year−1 potential for the abatement of greenhouse gas emissions using solar photoreduction (of CO2 or water).† Given reasonable economies of scale, and conversion efficiencies, the costs of producing fuels from sunlight and CO2 could be offset by CO2 emission credits (under a carbon cap-and-trade framework) and the sale of the hydrocarbons produced. On the other hand, Aresta and Dibenedetto8 point that global CO2 utilization to make chemicals and fuels could reach 300–700 Mt CO2 year−1 globally in the near- to long-term. Therefore, the potential for the indirect utilization of CO2 by using renewable electricity/hydrogen would be less than the direct solar conversion of CO2 and water to chemicals or fuels.
Although CO2 utilization may not make an impact on directly reducing emissions, it may provide a means to limit the use of fossil fuels and thereby contribute indirectly. As noted by Aresta and Dibenedetto,9 a life cycle analysis (LCA) approach should be used to determine whether a given process indeed decreases fossil fuel use and the carbon intensity.
CO2 photoreduction
CO2 photoreduction refers to the conversion of CO2 to reduced C1 and C2 compounds using light-induced reactions. Since CO2 does not absorb either visible or UV radiation in the wavelengths 200–900 nm, this process requires suitable photosensitizers. Both metal complexes and semiconductors have been utilized to absorb visible/UV radiation and transfer this energy to CO2.10 Additionally, various Ti species in silicate-based micro/mesoporous materials are also active towards CO2 photoreduction. These include isolated, tetrahedral Ti species substituting framework-Si in mesoporous silicates,11,12 TiO2+ species prepared via ion-exchange in microporous aluminosilicates,13 isolated14,15 and bimetallic16Zr4+- and Ti4+-containing species grafted onto the pore surface of mesoporous materials.Whereas the semiconductor-mediated reduction of CO2 involves the generation of electron-hole pairs and their subsequent transfer to CO2 and a reductant respectively, the metal complexes undergo local charge transfer transitions leading to electron transfer to CO2. Analogously, the isolated and bimetallic Ti-centers in micro/mesoporous silicates also undergo local ligand-to-metal charge transfer (LMCT) and metal-to-metal charge transfer (MMCT) transitions respectively. Although the metal complex photocatalysts exhibit comparatively higher yields and turnovers towards CO2 photoreduction, a sacrificial electron donor (typically an amine) is required to regenerate the photocatalyst. On the other hand, a semiconductor which has its conduction and valence bands placed suitably to transfer electrons to CO2 and oxidize inexpensive reductants such as water, can be designed. Among various semiconductors, TiO2 is widely used in many photoinduced processes because of its comparatively low cost, low toxicity17 and its ability to resist photocorrosion.18 TiO2 is a wide-band gap semiconductor (Eg ∼ 3.2 eV). It occurs as three polymorphs: anatase, rutile and brookite. Among these, anatase is obtained via “soft” chemical syntheses, and transforms upon heating (to temperatures greater than 700–800 K) to the rutile phase. Surface science aspects of TiO2 have been described in detail by Diebold.19Anatase is the more photocatalytically active form of TiO2, though not for all applications. It also has a larger band gap compared to rutile. However, surface science studies of anatase single crystals have only begun in the past eight years,20–28 whereas the rutile surfaces have been studied more extensively.29–33
A semiconductor photocatalyst mediating CO2reduction and water oxidation needs to absorb light energy, generate electron hole pairs, spatially separate them, transfer them to redox active species across the interface and minimize electron hole recombination. This requires the semiconductor to have its conduction band electrons higher energy compared to the CO2reduction potential while the holes in the valence band to be able to oxidize water to O2. A single semiconductor does not usually satisfy these requirements. Moreover, current CO2 photoreduction catalysts do not perform as effectively as current solar hydrogen generation catalysts. Therefore, to develop better Ti-based CO2 photoreduction catalysts, one needs to understand interactions between CO2 on ground and excited state TiO2 surfaces or Ti-based catalyst sites.
The reduction of CO2 is an endergonic process requiring a hydrogen source and/or energy. The most abundant sources of energy and hydrogen are sunlight and water, respectively. Solar photocatalytic reduction to produce fuels using water as the hydrogen source thus has the potential to be a means to store intermittent solar energy and to recycle CO2 while decreasing the use of fossil fuels. It is not clear if the state of the TiO2 surface is restored during the photoreaction reduction of CO2 for the photoreduction to be photocatalytic.34 However, in commonality with other photoinduced surface reactions such as the photogeneration of hydrogen from water,35 we can expect that both bulk as well as local phenomena influence the reactivity. Firstly, the location of the frontier orbitals of CO2 with respect to the valence and conduction bands of TiO2 determines the feasibility and direction of charge transfer. Secondly, local structure at the surface, involving coordinatively unsaturated atoms as well as surface/bulk defects such as cationic and anionic vacancies and interstitials can also influence reactivity to CO2.
The organization of this article is as follows: initially, the current state of development in CO2 photoreduction is compared to other photosynthetic processes such as solar H2 production. Thereafter, the thermodynamics of CO2 conversion to useful chemicals is presented. Plausible initial steps of CO2activation are identified. Subsequently, the CO2 surface state energy levels and the influence of doping on the work function of TiO2 are discussed using surface state representation of the CO2 : TiO2 interface. Thereafter, the influence of local factors such as surface specificity, anion vacancies and coordination of the Ti atom are discussed using the surface site approach. Using examples such as metal-doping of TiO2, dye-sensitization, oxygen vacancies in TiO2 and isolated-Ti centers in microporous/mesoporous materials, the utility of these approaches to understand photoinduced reactions involved in CO2activation is examined. Some of the outstanding challenges for CO2 photoreduction on TiO2 are identified, and are briefly discussed. This review will focus mainly on the CO2activation at the gas/vapor–TiO2 interface (Fig. 2).
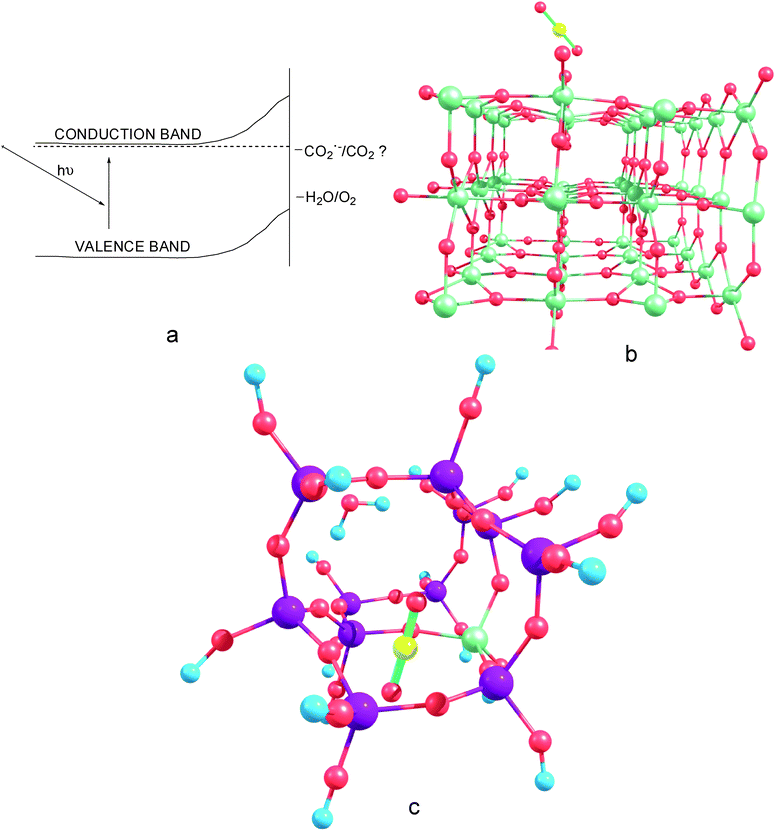 | ||
| Fig. 2 Scope of the aspects of CO2 photoreduction discussed in this review: (a): Surface state models indicating the location of the CO2−/CO2 energy levels in comparison with the conduction and valence bands of TiO2, surface site models of CO2 on Ti-based catalysts, (b): CO2 adsorption on doped/undoped TiO2 surfaces, (c): CO2 interacting with isolated model Ti sites in meso/microporous materials such as titanosilicates. | ||
Current state of CO2 photoreduction on TiO2-based catalysts
Titania (TiO2) based photocatalysts have been used to convert CO2 to useful compounds, both in gas and aqueous phase photoreactions. The conversion of CO2 and water to simple C products such as formic acid and formaldehyde using irradiated aqueous suspensions of titania was first demonstrated by Inoue et al.53 Researchers have also used homogeneous metal complexes in solution to reduce CO2.54 A summary of recent literature on CO2 photoreduction involving isolated Ti-species as well as bulk TiO2 materials is given in Table 1 and Table 2. Some recent developments in this field have been moved towards rational photocatalyst design, the use of highly active isolated Ti-species in mesoporous and microporous materials,11,14,15,36–38,55,56 metal-doping of TiO2, development of catalysts active at longer wavelengths than can be achieved with commercially available titania, and the elucidation of reaction mechanisms through in situ spectroscopic studies.14,57 A comparison of the CO2 conversion rates from various studies is shown in Fig. 3. Although it would be more instructive to compare the conversion efficiencies and quantum yields of various TiO2-based catalysts, often such data is not readily available. The data in Fig. 3 illustrate typical rates of CO2 photoreduction, and are dependent upon many variables such as metal doping, CO2 : H2O ratios, dispersion of Ti, coordination of Ti species, light intensity, type of lamp used, etc., which are already discussed in detail in the literature (see for example, Usubharatana et al.,10 and references therein). In references (B)–(C), (F) and (G) in Fig. 3, Anpo et al. used Ti-species in microporous or mesoporous silicates as catalysts whereas the rest of the works cited used doped/undoped TiO2.| Catalyst | Reaction system used | T/K and P/Pa | CO2 : H2O mole ratio | Light source used | Notes | References |
|---|---|---|---|---|---|---|
| Pt impregnation of (a) Ti-ion-exchanged zeolites and, (b) framework-Si substituting Ti-MCM-48. | Gas–solid system. Flat-bottomed quartz cell connected to conventional vacuum system (1 × 10−4 Pa), 88 ml | 328 K, pCO2 = 734 Pa | 0.2 | 75 W high-pressure Hg lamp, λ > 280 nm | Pt addition increased selectivity towards CH4 compared to CH3OH, and quenched photoluminescence yields. | Anpo et al.,13 Yamashita et al.,36 Anpo et al.11 |
| Fluorination of Ti-FSM-16 | do | 323 K | 0.2 | 100 W high-pressure Hg lamp, λ > 250 nm | Fluorination (hydrophobicity) increased selectivity towards CH3OH over CH4. | Ikeue et al.37 |
| Ti-containing porous silica thin film photocatalyst | do | 323 K | 0.2 | 100 W high-pressure Hg lamp, λ > 250 nm | Thin film photocatalyst exhibited high quantum yield (0.28%). | Ikeue et al.38 |
| Framework-Si substituting Ti-MCM-41 | Miniature infrared vacuum cell, 3.4 ml | 298 K, P = 0.1 MPa | 61.67 | 266 nm | CO, O2 detected as products of single photon, two-electron transfer. H2O confirmed as a stoichiometric electron donor. | Lin et al.15 |
| Ti(IV)–Sn(II) bimetallic redox centers in MCM-41 | Infrared vacuum cell | — | — | Third harmonic emission of a Nd:YAG laser, 355 nm | Ti3+ centers formed under 355 nm and visible light irradiation due to Sn(II) → Ti(IV) MMCT. | Lin and Frei16 |
| Catalyst modification | Reaction system used | T, (K) and P, (Pa) | CO2 : H2O | Light source | Notes | References |
|---|---|---|---|---|---|---|
| Cu(II) impregnation of JRC-TiO-4 | Gas–solid system. Flat- bottomed quartz cell connected to conventional vacuum system (1.33 × 10−4 Pa), 60 ml | 275 K | 0.33 | 75 W high-pressure Hg lamp, λ > 290 nm | Increased selectivity towards CH3OH over CH4. | Yamashita et al.39 |
| Cu(I) doping of TiO2, sol–gel synthesis | Slurry photoreactor, 300 ml | ∼323 K, P = 0.1 MPa | — | 8 W Hg lamp, UV-C, λ = 254 nm | Increased selectivity towards CH3OH over CH4, Cu(I) active species. | Tseng et al.40 |
| Cu(I) doping of TiO2, sol–gel | Gas–solid continuous optical fiber photoreactor (OFPR), 120 optical fibers | 348 K, pCO2 = 0.1 MPa | 50 | λ = 365 nm | Cu2O clusters influenced CH3OH formation. | Wu et al.41 |
| Nanoscale Ag-coated TiO2 particles embedded in Nafion membrane | High-pressure optical cell, stacked catalyst films. 2 ml H2O added after irradiation | pCO2 = 13.7 MPa | — | 990 W Xe arc source, water filter. | Small TiO2nanoparticles (4–6 nm) embedded in ionomer. CH3OH main product after Ag-coating of TiO2. Catalyst films could be reused and were chemically stable. | Pathak et al.42 |
| Dye ([Ru(Bpy)3]2+, perylene diimide based)-sensitized, Pt-promoted, thin and thick film-TiO2 photocatalysts | Glass chamber connected to a vacuum line | 298 K, pCO2 = 0.09 MPa | 34.5 | 75 W visible daylight lamp, 150 W UV A lamp | Pt-impregnation resulted in higher CH4 yields under UV irradiation compared to sol–gel synthesized Pt/TiO2. Organic sensitizers decreased catalytic activity under UV light and resulted in CH4 evolution under visible light irradiation. | Ozcan et al.43 |
| TiO2-coated Pyrex glass pellets, H2, H2O as reductants | Packed bed, circulated photocatalytic reactor | 316 K | 1, H2 : CO2 = 90 | 4 near-UV lamps, λ = 365 nm | CH4, CO and C2H6 formed as reaction products. | Lo et al.44 |
| Cu, Fe substitution in TiO2–SiO2, sol–gel synthesis | Same as above, 216 fibers | 348 K | — | 150 W high-pressure Hg lamp, λ = 320–500 nm, concentrated sunlight, λ = 500–800 nm | Fe substitution resulted in full visible light absorption. Cu–Fe/TiO2 favors C2H2 formation, Cu–Fe/TiO2–SiO2 favoredCH4 formation (quantum yield: 0.0235%). | Nguyen et al.45,46 |
| Ruthenium dye (N3)-sensitized Cu–Fe/TiO2catalysts | Same as above, 216 ml | 348 K | — | Same as above, Isolar = 20 mW cm−2, Ilamp = 225 mW cm−2 | Dye sensitization resulted in higher CH4 yields under concentrated sunlight due to stronger absorption bands in the visible spectrum. N3 dye stable after 6 h photoreactions. | Nguyen et al.47 |
| Mixed phase TiO2 nanocomposite prepared by TiCl4 hydrolysis in the presence of anatase powder | Annular glass reactor, isopropanol hole scavenger, 1280 ml | 293–298 K | — | 450 W, medium-pressure Hg lamp, UV-visible irradiation | Anatase-rutile nanocomposite exhibited higher CH4 evolution rates compared to Degussa P-25 TiO2 and pure anatase catalysts. Effective charge separation between anatase and rutile phases postulated to explain the higher photoactivity. | Li et al.48 |
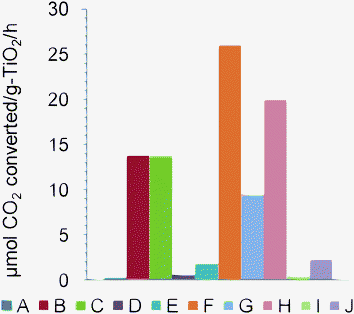 | ||
| Fig. 3 The highest specific rates of CO2 photoreduction (µmol CO2 converted g−1 TiO2 h−1) obtained using various doped and undoped Ti-based catalysts in selected recent articles. The values are presented here to illustrate typical rates of CO2 photoreduction, and are dependent upon many variables such as metal doping, CO2 : H2O ratios, dispersion of Ti, coordination of Ti species, light intensity, type of lamp used, etc., which are already discussed in detail in the literature (see for example, Usubharatana et al.10 and references therein). (A): Yamashita et al., (1994),49 (B): Anpo et al.,13 (C): Yamashita et al.,36 (D): Kaneco et al.,50 (E): Kaneco et al.,51 (F): Ikeue et al.,37 (G): Ikeue et al.,38 (H): Tseng et al.,40 (I): Wuet al.,41 (J): Nguyen et al.6,52 | ||
Comparisons of CO2 photoreduction rates to solar H2 generation
Although CO2 photoreduction has been studied since the 1970s, to the authors' knowledge, there are no studies on its performance vs. alternative approaches. Comparing the rates of CO2 photoreduction with other photosynthetic processes, such as solar hydrogen generation presents another means to assess the viability of CO2 photoreduction to limit GHG emissions. To better understand the current state of CO2 photoreduction using Ti-based catalysts, we converted the specific weights in Fig. 3 to units of micromoles CO2 converted h−1, using the catalyst weights, and titania weight percentages. Because most of the studies in Fig. 3 used electric lamps as the light source (for UV light), we illustrated the potential for improvements in CO2 photoreduction as the ratio of the amount of CO2 converted to fuels to the amount of CO2 emitted due to the consumption of electrical energy (assuming coal-fired power generation). While we realize that the ultimate goal of CO2 photoreduction would be to use sunlight to convert CO2 to fuels, we think that this ratio would be a good metric for comparing future advances in the case of UV light-irradiated systems, because quantitative values for quantum efficiency are difficult to obtain in the case of powder photocatalysts. The results (Fig. 4) indicate that current Ti-based photoinduced CO2 conversion processes emit more CO2 than that converted. As a caveat, we note that only minimal efforts were likely made to improve the light absorption in the references cited in Fig. 4. This might be one reason why the overall efficiency (kg CO2 converted kg−1 CO2 emitted by lamp) is low. However, significant improvements, both in the specific rate of CO2reduction as well as the quantum yield of the photoreaction are needed to make this process economically feasible.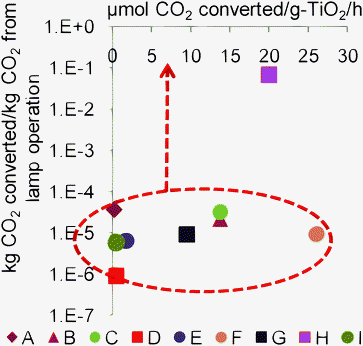 | ||
| Fig. 4 Amount of CO2 converted to reduced-C species per unit of CO2 evolved from lamp operation (assuming coal-fired power generation), from recent literature data on CO2 photoreduction using Ti-based catalysts. Current Ti-based photoinduced CO2 conversion processes emit more CO2 than they convert. Significant improvements, both in the specific rate of CO2reduction as well as the quantum yield of the photoreaction are needed to make this process feasible. A lamp power of 150 W was assumed in Wuet al. (2005).41 All references except Tseng et al.40 used lamp wattages ≥75 W. References are the same as those in Fig. 3. | ||
There are two conceptual routes to producing renewable C-containing fuels using solar energy. The first, the topic of this review, is the direct photoreduction of CO2 using water as a reductant, whereas the second route involves the photolysis of water to generate hydrogen and further reaction of this hydrogen with carbon dioxide forming C1–C2fuels. Both the efficiency of the conversion of solar energy to useful chemical energy as well as the rates of photoreduction of hydrogen/reduced C1–C2 compounds are pertinent parameters which determine economic feasibility of such processes. Assuming that the efficiency of solar energy conversion is almost the same in the two routes, the choice between direct and indirect hydrogenation of CO2 is determined by the rates of photoreduction.
Doped metal oxide photoelectrodes for solar hydrogen generation have typical hydrogen evolution rates of 400–700 µmol h−1 under visible irradiation.58 Recently, Grimes et al.59 demonstrated that nanostructured TiO2 photoanodes in photoelectrochemical cells (under voltage bias) resulted in hydrogen generation rates of ∼7000 µmol h−1 under AM 1.5 radiation. On a “sunny” day, using a single unit photoelectrocatalysis system without applied voltage bias, Ichikawa and Doi60 found a hydrogen evolution rate of 0.42 l h−1 m−2 (approximately 4 µmol H2 h−1 cm−2-irradiated photocatalyst area).
On the other hand, typical rates of CO2 photoreduction in aqueous systems containing suspended metal oxides (TiO2, BaTiO3etc.) are much lower, approximately 1 µmol h−1 C1/C2 product h−1 under UV radiation.55 Using copper-doped TiO2 and by conducting the photoreduction under a pH bias, Tseng and Wu61 obtained a slightly higher methanol formation rate of 12 µmol h−1. In gas–solid systems, using isolated Ti-sites in micro/mesoporous materials, Anpo et al.11–13 obtained CH4/CH3OH formation rates of approximately 0.05 µmol h−1. Although the intensity of the light used in these various studies is not the same, the above numbers indicate that current CO2 photoreduction catalysts do not perform well in comparison with solar-hydrogen generating catalysts, both in terms of product formation rates, and the wavelengths of light needed to sustain reactions. This is because both water oxidation as well as fuels formation from CO2 are multi-electron transfer reactions. Whereas oxygen evolution viawater oxidation is the major limiting factor in solar hydrogen production, both CO2 photoreduction as well as water oxidation have to occur simultaneously in a photoreactor using water as the reductant. In this connection, we also note that a recent design for a 100 T year−1 pilot plant producing methanol62 from CO2 involves solar photolysis of water to produce H2 and subsequent hydrogenation of CO2 using a heterogeneous catalyst and not the direct photoreduction of CO2. Additionally, Zeman and Keith propose producing carbon-neutral hydrocarbons from atmospheric CO2 and renewable hydrogen.63
Although the kinetics of CO2 photoreduction are two–three orders of magnitude slower than hydrogen photoreduction, one advantage of the direct photoreduction of CO2 compared to hydrogen evolution is the lower energy needed/electron transferred to convert CO2 to CH4 or CH3OH, Additionally, the use of carbon-based solar-derived fuels is likely to not need significant investments in infrastructure, unlike solar hydrogen generation and further reactions with CO2. The thermodynamics of CO2 photoreduction are discussed in the following section.
Thermodynamics and initial steps of CO2activation and further conversion
“Activation” refers to the transformation of a relatively inert species (such as CO2, CH4) to more reactive forms using some form of energy input and/or an activating agent. The activating agent may or may not be regenerated after the process. Because of its significance in steam reforming and methanol production processes, CO2activation on the surfaces of metals such as Pt, Fe, Nietc. has been studied in detail, both through in situ experiments on well defined surfaces64 as well as by computational modeling (see for example, Wang et al.,65 and references therein). Metals belonging to the platinum-group are however either expensive or will be in limited supply in the future. Nevertheless, metals play an important role in industrial catalytic processes where CO2 is either a reactant or a product. For example: Ni-based supported catalysts are used in CO2reforming/steam reforming of methane. The similarities between homogeneous metal complexes of CO2 and heterogeneous metal–CO2 surface species have been noted by Gibson in a review.66 As an example, a study by Wovchko and Yates demonstrated the activation of CO2 by a metal complex (RhI(CO)2) immobilized on an alumina support.57 Similar studies should lead to the rational design of photocatalysts mediating CO2reduction. The surface chemistry of CO2 on metals and oxide surfaces was reviewed by Freund and Roberts.67CO2 is one of the most stable compounds of carbon. Gaseous CO2 is linear and does not have a dipole moment. However, the oxygen atoms, each have a lone pair of electrons and can donate these electrons to surface Lewis acid centers. The carbon atom could also gain electrons from Lewis base centers such as oxide ions, forming carbonate-like species. Additionally, the (C–O) π electrons can also participate in reactions with electron centers. Therefore, reactions of CO2 involving Lewis acid and base centers and the π orbitals constitute the initial steps of its activation. The simplest reduction products that could be produced from CO2reduction are CO and HCOO−. One, two, four, six and eight electron reduction potentials (vs.NHE) for CO2reduction and H2O oxidation at pH 7 and 25 °C assuming unit activities for all gaseous and aqueous species are given in Scheme 1.7,8
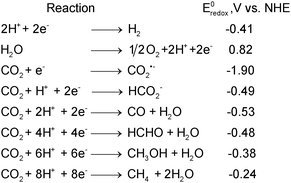 | ||
| Scheme 1 One, two, six and eight electron reduction potentials (vs.NHE) of some reactions involved in CO2 photoreduction at pH 7 and unit activity. | ||
From Scheme 1 it is clear that CO2 photoreduction is not a single-step reaction. Additionally, single electron transfer to CO2 is highly endergonic, because of the negative adiabatic electron affinity of CO2.67 CO2activation involves the formation of a negatively charged CO2δ˙− species.67,68 The first plausible step in CO2 photoreduction on TiO2 surfaces is therefore its activation to form a CO2δ˙− species. CO2˙−(g) is a 23 electron radical anion. It is significantly distorted from the linear D∞h geometry due to the repulsion among the two lone electron pairs on the oxygen atoms and the unpaired electron on the carbon atom. Therefore, the lower the O–C–O bond angle, the higher the charge of the CO2δ˙− moiety. CO2δ˙− can be detected via infra red spectroscopy69 as well as electron paramagnetic resonance (EPR) spectroscopy.70
The initial step in the photocatalytic reduction of CO2 is the generation of electron-hole pairs upon absorption of photons of energy greater than or equal to the band gap of the photocatalyst. The time scale of this electron-hole recombination is two to three orders of magnitude faster than other electron transfer processes. Therefore, any process which inhibits electron-hole recombination would greatly increase the efficiency and improve the rates of CO2 photoreduction. The kinetics of CO2 photoreduction are also dependent upon many other factors such as incident light intensity, fraction of the incident light absorbed by the photocatalyst, the specific surface area of the photocatalyst absorbing the light, etc. In this article, we examined four such factors: (a) the use of titania-supported noble metals, (b) the use of isolated Ti-tetrahedral centers in micro/mesoporous materials, (c) the use of dye-sensitized TiO2 to shift the absorption spectrum to the visible region and (d) the role of oxygen vacancies in electron trapping and activating CO2
Charge transfer from excited TiO2 surface to CO2 is influenced by both bulk and local interactions. Two different approaches for understanding electron transfer to CO2 are using surface sites and surface states. A surface site is a collection of atoms on the surface which is reactive in some manner. It may be a coordinatively under-saturated metal atom, oxygen vacancy or a combination of various other surface features resulting in an orbital having unusual electron affinity. A surface state is a localized energy level at the surface. The location of the surface state with respect to the Fermi energy (EF) of the solid determines its occupation. The surface site and surface state approaches to CO2 photoreduction on TiO2 will be described in detail in the following section.
Surface state description of CO2activation on TiO2
The main questions addressed by a surface state approach are:1. Is electron transfer from the TiO2 conduction band to CO2 feasible upon UV illumination?
2. How do surface states (created, for example: by metal doping) affect the reactivity towards CO2 photoreduction?
A knowledge of surface state energy levels for the CO2/CO2˙− couple at the TiO2 surface is required to determine the feasibility of electron transfer to CO2 from the conduction band of TiO2 A methodology to determine the energy levels (of the surface states) at the solid–gas interface has been proposed by Morrison.71 Accordingly, for the case of a non-interacting redox couple on a solid surface, the location of the standard redox potential in the absolute vacuum scale (AVS) units with respect to the edges of the conduction and valence bands of the solid at the point-of-zero-charge (pzc) determines the potential for electron transfer. Because this article concerns the gas–solid photoreduction of CO2, the relevant states in question are those at the TiO2–CO2 interface. However, it is helpful to compare the energy required to reduce CO2 in gas–liquid systems to that in gas–solid systems. The location of the energy levels associated with the CO2/CO2˙−redox couple in various media (gaseous, aqueous solutions) are shown in comparison to the location of the conduction band edge of TiO2 at pzc in Fig. 5. The standard reduction potential of the CO2/CO2˙−redox couple vs. the standard hydrogen electrode (SHE) is ∼1.9 V.72 On the absolute vacuum scale (AVS), this potential is 2.6 V, which corresponds to a redox state with an energy level at −2.6 eV. The main contributions to this energy are the electron affinity of CO2 (−0.6 eV) and the solvation of CO2˙− in water (−3.2 eV). The high free energy of solvation indicates that solvent reorganization energy will be significant. Therefore, a fluctuating energy level mechanism is needed to describe energy levels of CO2 and CO2˙−. Nevertheless, without any specific interaction with the catalyst/electrode surface, it is clear from Fig. 5 that the reduction of CO2 on TiO2 surfaces in aqueous solutions is unlikely because the energy level associated with the CO2/CO2˙−redox couple is higher in energy compared to the conduction band of TiO2.
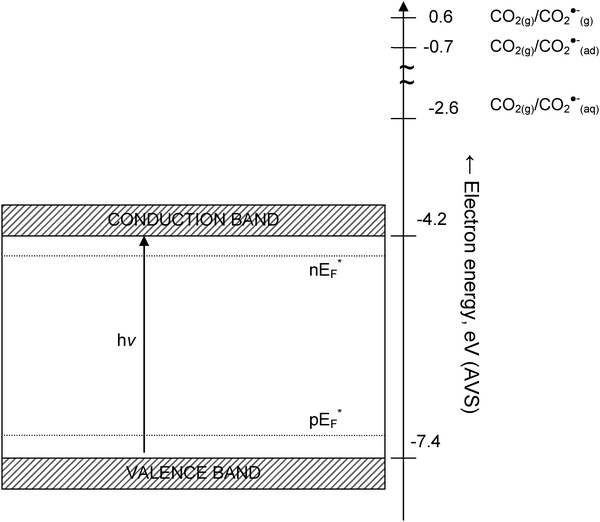 | ||
| Fig. 5 Location of the conduction band of TiO2 (rutile) particles at the point-of-zero charge (pzc) with respect to the standard energy states associated with the CO2/CO2˙− couple. Photoinduced electron transfer from stoichiometric TiO2 to CO2 is not possible because the surface state is not lower than the Fermi energy (EF) of the electrons in photoexcited TiO2. The bands are shown at the flat band condition. The exact locations of the Fermi energy levels for electrons and holes (nEF* and pEF*) are not to scale. | ||
Compared to the solid–liquid interface, measurement of band edges and surface states at the solid–gas interface is relatively more difficult. The exact location of the CO2˙−(surface)/CO2(g) surface state on TiO2 is currently not known. One way to estimate this value is using the an approach analogous to that used by Koppenol.72 CO2˙− adsorption on metal oxide surfaces can be conceptually thought of as the capture of an electron by CO2 molecule in vacuum/gas-phase forming the anion radical, followed by its adsorption on the metal oxide surface. (In practice, CO2 adsorbed on a hydrated/hydroxylated metal oxide surface captures a trapped electron either from the defect sites or trapping sites.)
In Scheme 2, EA is the adiabatic electron affinity of gaseous CO2 (in kJ mol−1) and ΔGad0 is the heat of chemisorption of CO2˙− on the surface (in kJ mol−1). F is the Faraday's constant (the charge of a mole of electrons). The overall free energy change of the reaction is ΔG0 = [ΔGad0(CO2˙−) − EA]. In the absolute vacuum scale (AVS), the energy level associated with such a conversion will be E0vac,energy = ΔG0/F (eV). In other words, the effect of interactions of CO2 with the surface is to decrease the energy required for CO2reduction.
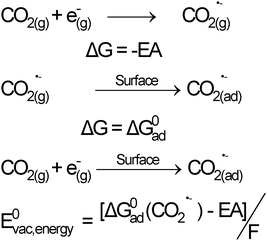 | ||
| Scheme 2 Thermodynamic cycle for the formation of CO2˙− at the TiO2 : CO2 interface. The surface state energy level of CO2 can be expressed as a function of its free energy of interaction with the surface and the electron affinity of gaseous CO2. | ||
Typical values for the heat of chemisorption of CO2˙− on metal surfaces (Ni) are of the order of ∼50 of kJ mol−1.65 If the heats of chemisorption CO2˙− on TiO2 surfaces were approximately 100 kJ mol−1 CO2, the surface state associated with the CO2/CO2˙− couple will be lowered approximately 1.3 eV from its gas-phase value, resulting in values of Eenergy,AVS0(CO2(g)/CO2˙−(ad)) of −0.7 eV, which is much more negative than that in aqueous solution. This simplistic analysis is not fully accurate, because the heat of chemisorption (arising out of local interactions of CO2˙− with the TiO2 surface) is comparable to the gas-phase electron affinity of CO2. Moreover, the electrostatic contribution (due to the Madelung potential of TiO2) is ignored. To understand this further, one needs to study the exact bonding between the CO2/CO2˙− couple and the surface. CO2 is plausibly bonded to the TiO2 surface differently compared to CO2˙−. Therefore, the so called “Frank–Condon splitting” of surface state energy levels also needs to be taken into account, which requires some description of the interaction of CO2˙− with the surface atoms. One example of this is a recent study by Bonapasta et al.73 where the energy levels of neutral and electron-attached states of O2 adsorbed on (100) anatase TiO2 surface were calculated using a surface molecule description of adsorbates in combination with periodic density functional theory calculations.
Experimentally, such surface states may be identified by spectroscopic studies of CO2 adsorbed on well-defined rutile TiO2 surfaces. Using ultraviolet photoemission spectroscopy (UPS) and metastable impact electron spectroscopy (MIES), Krischok et al.74 found that CO2 interacted with rutile TiO2 single crystals weakly, forming linearly adsorbed species. This is also supported by the results of an ab initio periodic study of CO2 adsorption on rutile.75 CO2 therefore does not form a surface state upon linear adsorption (involving Lewis acid–base interactions) in the ground-electronic state (without irradiation). Irradiation with light of energy greater than the band gap creates electron-hole pairs in TiO2, with the electrons populating the conduction band. Using the previous analysis, the energy of conduction band electrons in TiO2 is likely to be less than that required for CO2reduction to CO2˙δ−.
In conclusion, the above analysis indicates that the major factors controlling the surface state energy levels for CO2˙−(ad) are the electron affinity of CO2 (in the gas phase) and the local interaction of CO2/CO2˙−redox couple with the TiO2 surface. It is clear that a description of the energy levels of CO2/CO2˙− couple on TiO2 surfaces requires a combination of local and collective approaches. Therefore, we describe the surface site model of CO2activation on TiO2 in the next section.
Surface site description of CO2activation on TiO2
In recent years, knowledge of surface photocatalytic processes has progressed from a surface state approach to a more detailed understanding of specific surface adsorbate structures which are involved in the charge transfer. An example of this shift has been noted by Nowotny et al.35 Compared to the understanding of the initial steps of H2O oxidation on TiO2 surface, not much is known about the initial steps of CO2activation during the photocatalytic reduction. As noted in the earlier section, to better understand the nature of this charge transfer, one also needs to understand local chemistry at surface sites where activated species are formed. CO2 can be adsorbed as a carbonate and a linear species on TiO2 surfaces. Various ground-state adsorbate configurations of CO2 on TiO2 surfaces have been studied.75–78 CO2 acts as a net donor of electrons when it adsorbs with the oxygen end on Ti Lewis acid sites, and a net acceptor of electrons when the electrophilic C atom interacts with surface electron centers or Lewis basic sites (surface O atoms).67 A limiting case of the electrophilic interaction between the C atom of CO2 and the surface oxygen atoms is the formation of surface carbonate and bicarbonate species. On the other hand, if CO2 does not interact strongly with TiO2 in the ground state, the LUMO of adsorbed CO2 may not be lowered enough to ensure electron transfer from the TiO2 surface. The primary questions addressed by a surface site approach are:1. Upon illumination of the surface, how does electron transfer from the TiO2 conduction band to CO2 occur?
2. What specific sites and specific CO2 configurations on the TiO2 surface promote this electron transfer? Can these sites be engineered?
The mechanisms proposed for CO2 photoreduction over TiO2 or Ti-based catalysts assume that CO2 gains electrons from the conduction band of TiO2.10 This indicates a strong interaction between CO2 and the TiO2 surface. Photoexcitation of TiO2 produces electron-hole (Ti3+–O−) centers in TiO2. In the next step, these Ti3+ centers are proposed to interact with CO2 forming CO2δ˙− species adsorbed on the surface. These carbon dioxide radical anions then undergo further reactions to form C radicals and CHxOy end products. Electron transfer from Ti3+ surface electron centers to CO2 requires the presence of suitable unoccupied molecular orbitals on CO2. Electronic interaction between the surface and CO2 is necessary for CO2 to gain electrons. Freund and Roberts67 indicate that the lowest unoccupied molecular orbital (LUMO) of CO2 decreases with the O–C–O bond angle. This variation of the LUMO of CO2 with the O–C–O bond angle is shown in Fig. 6. One role of the surface might be to interact with CO2 reducing the bond angle. In the limiting case, this corresponds to the formation of carbonate-like species on the surface. This principle is not limited to CO2activation on irradiated metal oxides alone. Similar interactions of CO2 with the electrocatalyst are supposed to decrease the overpotential for CO2reduction79 during electrochemical reduction of CO2.
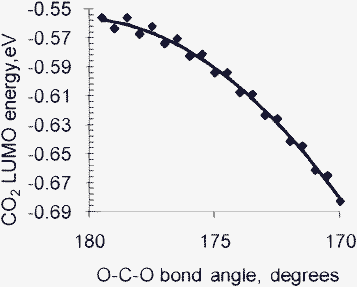 | ||
| Fig. 6 The variation in the energy of the LUMO of gaseous CO2 with the O–C–O bond angle, calculated using constrained quantum chemical calculations at the B3LYP/6 − 31 + G(d) level of theory. Decreasing the O–C–O bond angle (via surface interactions) may facilitate charge transfer to CO2 by lowering its LUMO. | ||
Techniques such as Fourier transform infra red (FT-IR) spectroscopy have been used to study CO2 adsorbed on surface electron centers (Ti3+) created due to oxygen vacancies.76 CO2 adsorption on reduced TiO2 surfaces generated IR signals corresponding to CO2δ˙− species.
In the following section, we will examine the use of these two (surface state, surface site) approaches to understand photoinduced reactivity of Ti-based catalysts towards CO2
Utility of surface state and surface site approaches in understanding the elementary reactions involved in CO2 photoreduction on Ti-based catalysts
Effects of alkali and noble metal doping on CO2 photoreduction
Some approaches to make the thermodynamics of CO2reduction on TiO2 more favorable are the use of promoters such as potassium (K) or through impregnation with noble metals. Alkali promotion of CO2activation on TiO2 has been well studied.33 The added alkali atom donates an electron to CO2, forming CO2˙− species. This reacts with the surface oxygen to form a surface carbonate species. Such promoters are stoichiometrically consumed upon charge transfer to CO2 because they usually cannot be regenerated (photo)catalytically in an environment containing water vapor and oxygen. On the other hand, the modification of TiO2 surface with metals such as Pt, Rhetc. may affect both the thermodynamics as well as kinetics of CO2activation and further conversion.In a study of photoinduced CO2 dissociation on titania-supported noble metals, it was found that the activity towards CO dissociation correlated with the work function of the polycrystalline metal for titania-supported Pt, Rh and Ir/TiO2.78 The following mechanism was proposed to account for photoinduced CO2activation on titania-supported noble metal catalysts: Chemisorption of CO2 on polycrystalline metal islands results in a bent CO2δ˙− species formed by back donation between the metal d-orbitals and the (C–O) π* orbitals. This electron transfer results in the “depopulation” of the metal surface state. Photogenerated electrons generated upon band gap illumination would be transferred from the conduction band of TiO2 to the metal island (because the work function of these metals is higher than that of reduced TiO2). Therefore, illumination of TiO2-supported noble metal catalysts in a CO2 atmosphere increased the intensity of signals corresponding to CO2δ˙− species. We note that such activation reactions require a concomitant oxidation reaction to be photocatalytic. Additionally, the study by Raskó78 demonstrates that an understanding of both surface states as well as surface sites is needed to understand CO2activation on such TiO2-supported metal photocatalysts. The case of Rh/TiO2 is especially interesting, from a catalytic viewpoint if not from a practical perspective, rhodium being one of the most expensive metals in the world currently.
Recently, CO oxidation on gold clusters supported on TiO2 has been studied extensively, both by experimental80 and modeling81,82 studies. We note that CO oxidation also involves the formation of a CO2δ˙− intermediate. Therefore, a good CO oxidation catalyst may also promote CO2activation. Most studies have been performed on metallic clusters of gold on TiO2. In these cases, density functional theory (DFT) calculations have shown that the binding strength of low-coordinated Au clusters with gaseous CO is a crucial parameter in the oxidation of CO.83 Moreover, promotion effects may also contribute to the observed low temperature oxidation of CO to CO2 in the presence of gaseous oxygen. In contrast, a Mars–van Krevelen mechanism was proposed by Chrétien and Metiu84 to account for the activity of gold doped TiO2, where the gold atom is positively charged and substitutes a five coordinate Ti atom on the rutile (110) surface.
In addition to activating CO2, noble metal impregnation may also decrease the photogenerated electron-hole recombination rates, leading to higher product yields. Using femtosecond time resolved measurements of the intensity dependence of electron hole recombination in TiO2 nanoclusters, Colombo and Bowman85 found that more than half of the conduction band electrons underwent recombination within 20 picoseconds (ps). The remaining electrons were trapped at surface sites resulting in a longer lived species. Therefore, from an overall system perspective, the greatest gain in efficiency in CO2 photoreduction may result by slowing down the rates of electron-hole recombination. Using surface photovoltage spectroscopy (SPS) measurements on Pd and Pd/RuO2 doped TiO2, Wang et al.86 showed that the rates of CO2 photoreduction to formate in aqueous solutions correlated with the surface photovoltage measured in air. Two pertinent conclusions could be drawn from their study. Firstly, surface modification of TiO2 with noble metals could lead to the formation of surface states, increasing the photogenerated electron-hole pair lifetimes. Secondly, the fact that photovoltages observed in air correlated with the yields in aqueous solutions indicates that electron-hole pair separation appears to be the primary control over the reactivity.
Visible light performance of dye-sensitized TiO2catalysts mediating CO2 photoreduction
Another means to decrease electron-hole recombination rates and use higher wavelengths of the solar spectrum is the dye-sensitization of TiO2. Closely related to this is the dye sensitized solar cell (DSSC), (see McConnell87 and references therein for details about operation and comparisons). Briefly, the DSSC is a solar electric device where the light absorption and charge transport are physically separated. The organometallic dye attached to porous TiO2 absorbs sunlight, and injects electrons into the conduction band of TiO2, which flow through an external circuit, creating electricity. The oxidized dye is reduced by a mediator which in turn, is reduced at the metallic cathode of the solar cell. This concept has been applied to the photosensitization of TiO2 anodes in photoelectrochemical (PEC) cells. A brief description of the operation of a dye-sensitized photoanode is provided by Bak et al.88 However, in the case of dye-sensitized TiO2 used for photocatalytic CO2reduction applications,43,47 it is less clear how the oxidized dye would be regenerated when the photoelectrochemical cell is short-circuited and no defined pathway for electron transport exists (RERITE). Sacrificial photooxidation of the dye or other reagents probably occurs unless the oxidized state of dye has a surface state level (Dye(surf)+/Dye(surf)) lower than that for water oxidation (O2(surf)/H2O(surf)).Nevertheless, Nguyen et al.47 found substantial improvements in the photoactivity of (Cu,Fe)-TiO2 photocatalysts towards CO2 photoreduction in the presence of water and concentrated sunlight in a optical fiber photoreactor (OFPR). The higher activity was attributed to full visible light absorption, whereas TiO2 only absorbs a fraction of the radiation above 400 nm. Only CH4 was produced under sunlight, and the photocatalyst was reported to be stable for 6 h. Whereas dye-sensitization of TiO2 enables the use of visible light, we note that future studies should consider 13CO2-isotope testing to ensure that the reduced C-products are due to CO2reduction, and not the sacrificial oxidation of the dye.
To summarize, both metal doping as well as dye-sensitization reduce the rates of electron-hole recombination, but the mechanisms involved are different. In the case of metal-doped TiO2, the electrons are transferred from the conduction band of TiO2 to the metal, whereas on a dye-sensitized photocatalyst, the dye injects electrons into the conduction band of TiO2.
Isolated Ti-species acting as active sites for CO2 photoreduction in micro/mesoporous materials
As discussed in Table 1, isolated tetrahedral Ti+4 species in zeolites or mesoporous sieves were used as catalysts by Anpo and coworkers13,36,37,38 as well as Frei et al.14,15 The location of the Ti atoms in mesoporous silica depend on the synthetic technique used.89 Framework Ti-substituted MCM-41 is prepared when the Ti precursors are included in the MCM-41 synthesis gel (Lin et al.15). On the other hand, Ti-grafted MCM-41 is prepared by post-synthesis grafting (ex: Lin and Frei16). In such Ti-grafted MCM-41, the Ti atoms are located at the surface of the silicate wall and are not a part of the framework. They are therefore more accessible to reactants. However, this might also make them more susceptible to leaching in aqueous solutions. Moreover, depending on the titanium content, a variety of configurations for Ti species are possible.90Using in situX-ray fluorescence spectroscopy (XAFS) studies to probe the local environment around an isolated Ti atom, Yamashita et al.56 studied the effects of the hydrophobic–hydrophilic properties of Ti-β zeolites in the photocatalytic reduction of CO2 with H2O. Adsorption of CO2 did not change the XANES spectra of Ti-β zeolite catalysts. On the other hand, the addition of water increased the coordination number of Ti atom from 4 to 6. Therefore, in the ground state, H2O interacted more strongly with Ti-β zeolites compared to CO2. In situphotoluminescence (PL) spectroscopy measurements on irradiated Ti-β zeolites indicated that the lifetime of the charge transfer excited state decreased in the presence of CO2 and/or H2O molecules. Therefore, while CO2 interacted with the excited state of the Ti-β zeolite, water interacted with the Ti-centers in both excited and ground electronic states. Additionally, using in situFT-IR spectroscopy to study the initial steps of CO2 photoreduction with methanol as a sacrificial oxidant in a Ti silicalite molecular sieve, Ulagappan and Frei91 recorded the formation of formic acid as the primary electron transfer product. They proposed that the initial step in this reaction involved electron transfer from photoexcited LMCT excited state of the catalyst to CO2 concurrent with methanol oxidation to form CO2− and CH3OH+ species which react further to produce formic acid and formaldehyde.
Although the above isolated tetrahedral Ti-centers in zeolites/mesoporous materials are active towards CO2 photoreduction, the absorption of these materials lies in the deep-UV (UV-C) region of the electromagnetic spectrum. To activate CO2 using visible light, Frei's research group demonstrated metal–metal charge transfer (MMCT), involving two different metal ions grafted in a mesoporous material.16 Examples include Ti–O–Cu(I) and Ti–O–Sn(II) as well as Zr–O–Cu(I) grafted in Mobile crystalline material (MCM)-41 molecular sieve. Irradiation of the binuclear Zr–O–Cu(I)/MCM-41 catalyst under a CO2 atmosphere resulted in the growth of IR signals corresponding to CO and H2O.92 Only Zr–O–Cu(I) complexes showed CO formation. CO2 dissociation was not observed on Ti–O–Cu(I) complexes. Lin and Frei attributed this to the higher reducing power of the Zr3+ transient ion. Similar studies using various other redox couples (ex: Ti4+/Fe2+, Ti4+/Mn2+) could reveal visible light-absorbing catalysts active towards CO2 photoreduction.
Although the primary charge transfer mechanism in isolated-Ti species is localized, overall reactivity is also affected by the bulk properties. For example, Yamashita et al.36 attributed higher C1 photoproduct yields using Ti-MCM-48 compared to Ti-MCM-41 or TS-1 to the large pore size and the three-dimensional channel structure of the MCM-48 catalyst. Additionally, electrostatic fields in the zeolite/mesporous material could stabilize the CO2˙− radical ion and thereby affect the product yield and selectivities.93
Effect of oxygen vacancies created by cation doping or thermal treatment of TiO2 on the activity towards CO2
As mentioned earlier, CO2 does not interact with stoichiometric rutile TiO2 surfaces. The band gaps of rutile and anatase are not widely different; therefore, this might also be expected to apply to anatase TiO2. Defects such as oxygen vacancies control most of the chemistry at many metal oxide surfaces. These defects could be created either by doping TiO2 with other anions or cations or by thermal treatments of stoichiometric TiO2.One example of this defect creation due to thermal treatments is the work of Yamashita et al.,49 who studied the photoreduction of CO2 with H2O on rutile TiO2 (100) and TiO2 (110) single crystals. Structure-dependent reactivity was observed, with the TiO2 (100) surface being more active towards CO2 photoreduction compared to the TiO2 (110) surface. Yamashita et al.49 explained this by stating that the Ti : O atomic ratio on the (100) surface was greater than the (110) surface. A pretreatment procedure involving degassing at 725 K, followed by a 4 h reoxidation in O2 at 725 K, followed by subsequent catalyst degassing at 725 K for 4 h at ∼1 × 10−6 Torr was used in the study. We note that this pretreatment procedure might create oxygen vacancies on the TiO2 surface. For example: Thompson et al.94 note that surface oxygen vacancies could be created with annealing temperatures as low as 600 K with rutile TiO2 under ultra high vacuum (UHV) conditions. Additionally, stoichiometric reactions between CO2 and water on oxygen-deficient rutile surfaces to yield reduced C1–C2 products are well known in the literature.95 In such cases, in situ surface characterization of the catalyst under light irradiation would provide information on the oxidation state of the surface, because the surface-Ti4+ and Ti3+ can be differentiated using XPS 2p core-level spectra.96
In addition to creating oxygen vacancies, transition metal doping of TiO2 also may create specific surface structures affecting the CO2 photoreduction activity. Tseng et al.40 and Wu and Lin97 found that Cu(I) doped TiO2 promoted the formation of CH3OH compared to undoped TiO2. Pathak et al.42 also observed selectivity for CH3OH formation over HCOOH using Ag-coated nanoscale TiO2 immobilized in Nafion ionomer membranes. Apart from influencing the electron-hole recombination rates (as noted in the discussion on surface states) and affecting product specificity and yields, transition metal doping of TiO2 also results in red-shifting of the absorption spectrum of TiO2. This apparent bandgap-narrowing is attributed to transitions from localized surface states above the conduction band of TiO2 and not due to bulk band-gap modification.98
Discussion
One of the major factors limiting CO2 photoreduction is the electron-hole recombination in semiconducting materials under band-gap illumination. The use of isolated Ti-species partly resolves this issue, but most Ti–O LMCT transitions occur at UV-C wavelengths (∼270 nm). Both local as well as bulk factors influence the activity of catalysts mediating CO2 photoreduction. For example: cation doping of TiO2 not only creates oxygen vacancies, but also creates localized intra-bandgap states which contribute to charge transfer transitions by absorbing light wavelengths below the bandgap.Both collective and local approaches are required to understand such light-induced electron transfer reactions. The collective, surface state approach indicates that pure TiO2 is likely to be a poor photocatalyst for CO2activation. On the other hand, localized surface sites such as oxygen vacancies have been implicated in CO2activation.
Using the surface state and surface site approaches, we have shown that both bulk and local variables affect the photoreduction of CO2. Fundamental understanding of the initial steps of CO2activation on TiO2 surfaces and a detailed knowledge of how these bulk and local phenomena affect photoreactivity is required to design better catalysts.
Challenges for CO2 photoreduction on Ti-based catalysts
As discussed in the earlier sections, significant improvements in the quantum yields and selectivities of Ti-based catalysts is required to bring CO2 photoreduction using these materials closer to commercialization. Materials not based on Ti have been used to mediate CO2 photoreduction. For example, metal chalcogenides (CdS, CdSe, ZnS) and other metal oxides (ZnO, MgO) have also been used as photocatalysts (see Usubharatana et al.10 and references therein). In most cases, the improvements in efficiency are incremental at best. On the other hand, higher quantum efficiencies are possible with homogeneous metal complexes, but a sacrificial reagent is often required to regenerate the photosensitizer.54 Breakthroughs in related fields such as solar-electric conversion, solar production of hydrogen using water and catalytic CO2activation will enable the further development of this field.Essentially, the main challenges for CO2 photoreduction using Ti-based catalysts involve finding suitable photosensitizers that can absorb visible light to produce electron-hole pairs and transfer the electrons to CO2 with minimal losses. Additionally, the photosensitizer has to be regenerated with a relatively inexpensive reductant such as water. Choi et al.99 noted that interfacial electron/hole transfer involves charged pair generation, followed “trapping” of the charged particles, their subsequent release and migration to the surface prior to the transfer. Electron-hole recombination occurs parallel to the interfacial charge transfer, at all stages of this process. As pointed out in a report on solar energy utilization,100 potential breakthroughs in CO2 photoreduction can result from significantly increasing the lifetime of the charge-separated state, designing materials which can work under visible light, catalyzing proton-coupled multi-electron transfer to CO2. This requires a combination of experimental and computational studies
Conclusions
In this article we provided a brief review of and insights into the mechanistic aspects of CO2 photoreduction using TiO2-based metal oxides or Ti-species in micro/mesoporous materials. CO2 utilization has a significant role in mitigating CO2 emissions, especially in comparison to other means of CO2 abatement. The conversion of CO2 to fuels using solar energy is one of the “grand challenges” for a variety of scientific disciplines. Significantly increased specific rates (∼10 s of millimoles CO2 converted g−1 TiO2 h−1) under visible light irradiation are required to make this process economically feasible. Paradigm shifts in TiO2 photocatalysis such as the use of bimetallic redox species in mesoporous materials, novel reactor design configurations (OFPR), the use of catalysts absorbing visible radiation, decreasing electron-hole recombination rates, surface modifications via doping or other chemical treatments, will continue to be required to drive innovation in this field.Acknowledgements
VPI thanks Penn State Institutes of Energy and the Environment (PSIEE) for financial support. Computational support was in part provided by the National Science Foundation under grant no. CHE-0431328 for the Center for Environmental Kinetics Analysis (CEKA) at The Pennsylvania State University. VPI also thanks Dr Mercedes Maroto-Valer for her comments on a section of this review.References
- X. Xu and J. A. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef.
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- M. Mazzotti, J. C. Abanades, R. Allam, K. S. Lackner, F. Meunier, E. Rubin, J. C. Sanchez, K. Yogo and R. Zevenhoven, in IPCC 2005: IPCC Special Report on Carbon dioxide Capture and Storage. Prepared by Working Group III of the Intergovernmental Panel on Climate Change., ed. B. Metz, O. Davidson, H. C. de Coninck, M. Loos and L. A. Meyer, Cambridge University Press, Cambridge, United Kingdom and New York, USA, 2005, p. 333 Search PubMed.
- H. J. Herzog and E. M. Drake, Annu. Rev. Energy Environ., 1996, 21, 145–166 CrossRef.
- McKinsey, Reducing U.S. Greenhouse Gas Emissions: How Much at What Cost?http://www.mckinsey.com/clientservice/ccsi/pdf/US_ghg_final_report.pdf, accessed 11/24/2008 Search PubMed.
- M. Aresta and A. Dibenedetto, Catal. Today, 2004, 98, 455 CrossRef CAS.
- M. Aresta, E. Quaranta and I. Tommasi, Photochem. Convers. Storage Sol. Energy, Proc. Int. Conf., 8th, 1991, 517–550 Search PubMed.
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558–2568 CrossRef CAS.
- M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, S. G. Zhang, Y. Ichihashi, D. R. Park, Y. Suzuki, K. Koyano and T. Tatsumi, Catal. Today, 1998, 44, 327–332 CrossRef CAS.
- S. G. Zhang, Y. Fujii, K. Yamashita, K. Koyano, T. Tatsumi and M. Anpo, Chem. Lett., 1997, 659–660 CrossRef.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- W. Lin and H. Frei, Stud. Surf. Sci. Catal., 2004, 153, 283–288 CAS.
- W. Y. Lin, H. X. Han and H. Frei, J. Phys. Chem. B, 2004, 108, 18269–18273 CrossRef CAS.
- W. Lin and H. Frei, C. R. Chim., 2006, 9, 207–213 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- X.-Q. Gong, A. Selloni, M. Batzill and U. Diebold, Nat. Mater., 2006, 5, 665 CrossRef CAS.
- X. Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS.
- X. Q. Gong, A. Selloni and A. Vittadini, J. Phys. Chem. B, 2006, 110, 2804–2811 CrossRef CAS.
- X. Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560–19562 CrossRef CAS.
- U. Diebold, N. Ruzycki, G. S. Herman and A. Selloni, Catal. Today, 2003, 85, 93–100 CrossRef CAS.
- N. Ruzycki, G. S. Herman, L. A. Boatner and U. Diebold, Surf. Sci., 2003, 529, L239–L244 CrossRef CAS.
- M. Lazzeri and A. Selloni, Phys. Rev. Lett., 2001, 87 Search PubMed.
- Y. Liang, S. P. Gan, S. A. Chambers and E. I. Altman, Phys. Rev. B, 2001, 63 Search PubMed.
- R. Hengerer, B. Bolliger, M. Erbudak and M. Gratzel, Surf. Sci., 2000, 460, 162–169 CrossRef CAS.
- W. Gopel, G. Rocker and R. Feierabend, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 3427–3438 CrossRef CAS.
- J. Nerlov, S. V. Christensen, S. Weichel, E. H. Pedersen and P. J. Moller, Surf. Sci., 1997, 371, 321–336 CrossRef CAS.
- A. Gutiérrez-Sosa, J. F. Walsh, R. Lindsay, P. L. Wincott and G. Thornton, Surf. Sci., 1999, 433–435, 538 CrossRef CAS.
- S. Krischok, O. Höfft and V. Kempter, Surf. Sci., 2002, 507–510, 69 CrossRef CAS.
- M. Brause and V. Kempter, Surf. Sci., 2001, 476, 78 CrossRef CAS.
- A. V. Emeline, G. V. Kataeva, A. V. Panasuk, V. K. Ryabchuk, N. V. Sheremetyeva and N. Serpone, J. Phys. Chem. B, 2005, 109, 5175–5185 CrossRef CAS.
- J. Nowotny, T. Bak, M. K. Nowotny and L. R. Sheppard, Int. J. Hydrogen Energy, 2007, 32, 2651–2659 CrossRef CAS.
- H. Yamashita, Y. Fujii, Y. Ichihashi, S. G. Zhang, K. Ikeue, D. R. Park, K. Koyano, T. Tatsumi and M. Anpo, Catal. Today, 1998, 45(1–4), 221 CrossRef CAS.
- K. Ikeue, H. Yamashita, M. Anpo and T. Takewaki, J. Phys. Chem. B, 2001, 105, 8350–8355 CrossRef CAS.
- K. Ikeue, S. Nozaki, M. Ogawa and M. Anpo, Cata. Lett., 2002, 80, 111 Search PubMed.
- H. Yamashita, H. Nishiguchi, N. Kamada, M. Anpo, Y. Teraoka, H. Hatano, S. Ehara, K. Kikui, L. Palmisano, A. Sclafani, M. Schiavello and M. A. Fox, Res. Chem. Intermed., 1994, 20, 815 CrossRef CAS.
- I. H. Tseng, W.-C. Chang and J. C. S. Wu, Appl. Catal., B, 2002, 37, 37–48 CrossRef CAS.
- J. C. S. Wu, H.-M. Lin and C.-L. Lai, Appl. Catal., A, 2005, 296, 194 CrossRef CAS.
- P. Pathak, M. J. Meziani, L. Castillo and Y.-P. Sun, Green Chem., 2005, 7, 667–670 RSC.
- O. Ozcan, F. Yukruk, E. Akkaya and D. Uner, Top. Catal., 2007, 44, 523 CrossRef CAS.
- C.-C. Lo, C.-H. Hung, C.-S. Yuan and J.-F. Wu, Sol. Energy Mater. Sol. Cells, 2007, 91, 1765–1774 CrossRef CAS.
- T.-V. Nguyen and J. C. S. Wu, Appl. Catal., A, 2008, 335, 112–120 CrossRef CAS.
- T.-V. Nguyen and J. C. S. Wu, Sol. Energy Mater. Sol. Cells, 2008, 92, 864–872 CrossRef CAS.
- T.-V. Nguyen, J. C. S. Wu and C.-H. Chiou, Catal. Commun., 2008, 9, 2073–2076 CrossRef CAS.
- G. Li, S. Ciston, Z. V. Saponjic, L. Chen, N. M. Dimitrijevic, T. Rajh and K. A. Gray, J. Catal., 2008, 253, 105–110 CrossRef CAS.
- H. Yamashita, N. Kamada, H. He, K. Tanaka, S. Ehara and M. Anpo, Chem. Lett., 1994, 855–858 CAS.
- S. Kaneco, Y. Shimizu, K. Ohta and T. Mizuno, J. Photochem. Photobiol., A, 1998, 115, 223 CrossRef CAS.
- S. Kaneco, H. Kurimoto, Y. Shimizu, K. Ohta and T. Mizuno, Energy, 1999, 24, 21 CrossRef CAS.
- T.-V. Nguyen and J. C. S. Wu, Sol. Energy Mater., 2008, 92, 864 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature (London, United Kingdom), 1979, 277, 637–638 Search PubMed.
- E. Fujita, Coord. Chem. Rev., 1999, 186, 373–384 CrossRef.
- M. Anpo and H. Yamashita, in Hetergeneous Photocatalysis, ed. M. Schiavello, John Wiley & Sons, Chichester, UK, 1997, p. 136 Search PubMed.
- H. Yamashita, K. Ikeue, T. Takewaki and M. Anpo, Top. Catal., 2002, 18, 95 CrossRef CAS.
- E. A. Wovchko and J. T. Yates Jr., J. Am. Chem. Soc., 1998, 120, 7544–7550 CrossRef CAS.
- V. M. Aroutiounian, V. M. Arakelyan and G. E. Shahnazaryan, Sol. Energy, 2005, 78, 581–592 CrossRef CAS.
- M. Paulose, G. K. Mor, O. K. Varghese, K. Shankar and C. A. Grimes, J. Photochem. Photobiol., A, 2006, 178, 8 CrossRef CAS.
- S. Ichikawa and R. Doi, Catal. Today, 1996, 27, 271 CrossRef CAS.
- I. H. Tseng and J. C. S. Wu, Catal. Today, 2004, 97, 113–119 CrossRef CAS.
- MCI, Mitsui Chemicals to Establish a Pilot Facility to Study a Methanol Synthesis Process from CO2, http://www.mitsuichem.com/release/2008/080825e.htm, accessed 01/09/2008 Search PubMed.
- F. S. Zeman and D. W. Keith, Philos. Trans. R. Soc. London, Ser. A, 2008, 366(1882), 3901–3918 CAS.
- F. Solymosi, J. Mol. Catal., 1991, 65, 337 CAS.
- S. G. Wang, D. B. Cao, Y. W. Li, J. G. Wang and H. J. Jiao, J. Phys. Chem. B, 2005, 109, 18956–18963 CrossRef CAS.
- D. H. Gibson, Coord. Chem. Rev., 1999, 185–186, 335–355 CrossRef CAS.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 227–273 CAS.
- J. Rasko and F. Solymosi, J. Phys. Chem., 1994, 98, 7147–7152 CrossRef CAS.
- K. O. Hartman and I. C. Hisatsune, J. Chem. Phys., 1966, 44, 1913–1918 CrossRef CAS.
- L. L. Perissinotti, M. A. Brusa and M. A. Grela, Langmuir, 2001, 17, 8422–8427 CrossRef CAS.
- R. S. Morrison, in The Chemical Physics of Surfaces, 1990 Search PubMed.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef CAS.
- A. A. Bonapasta and F. Filippone, Surf. Sci., 2005, 577, 59–68 CrossRef CAS.
- S. Krischok, O. Hofft and V. Kempter, Surf. Sci., 2002, 507–510, 69–73 CrossRef CAS.
- A. Markovits, A. Fahmi and C. Minot, Theochem, 1996, 371, 219–235 CrossRef CAS.
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 Search PubMed.
- G. Ramis, G. Busca and V. Lorenzelli, Mater. Chem. Phys., 1991, 29, 425–435 CrossRef CAS.
- J. Raskó, Catal. Lett., 1998, 56, 11–15 CrossRef CAS.
- M. Schmidt, in Carbon dioxide chemistry: Environmental issues, ed. J. P. Paul, Claire-Marie, RSC, Cambridge, 1994, pp. 22–30 Search PubMed.
- M. S. Chen and D. W. Goodman, Top. Catal., 2007, 44, 41–47 CrossRef CAS.
- R. G. S. Pala and F. Liu, J. Chem. Phys., 2006, 125, 144714 CrossRef.
- S. P. Raj Ganesh and L. Feng, J. Chem. Phys., 2006, 125, 144714 CrossRef.
- I. N. Remediakis, N. Lopez and J. K. Nørskov, Angew. Chem., Int. Ed., 2005, 44, 1824–1826 CrossRef CAS.
- S. Chrétien and H. Metiu, Catal. Lett., 2006, 107, 143 CrossRef CAS.
- D. P. Colombo, Jr. and R. M. Bowman, J. Phys. Chem., 1996, 100, 18445–18449 CrossRef.
- T.-f. Xie, D.-j. Wang, L.-j. Zhu, T.-j. Li and Y.-j. Xu, Mater. Chem. Phys., 2001, 70, 103 CrossRef CAS.
- R. D. McConnell, Renewable and Sustainable Energy Reviews, 2002, 6, 271 Search PubMed.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991 CrossRef CAS.
- L. Y. Chen, G. K. Chuah and S. Jaenicke, Catal. Lett., 1998, 50, 107–114 CrossRef CAS.
- Q. Yuan, A. Hagen and F. Roessner, Appl. Catal., A, 2006, 303, 81 CrossRef CAS.
- N. Ulagappan and H. Frei, J. Phys. Chem. A, 2000, 104, 7834–7839 CrossRef CAS.
- W. Y. Lin and H. Frei, J. Am. Chem. Soc., 2005, 127, 1610–1611 CrossRef CAS.
- K. Sogabe, A. Hasegawa, Y. Yamada and M. Miura, Bull. Chem. Soc. Jpn., 1972, 45(11), 3362–3366 CrossRef CAS.
- T. L. Thompson, O. Diwald and J. T. Yates Jr., J. Phys. Chem. B, 2003, 107, 11700–11704 CrossRef CAS.
- J. N. Wilson, S. D. Senanayake and H. Idriss, Surf. Sci., 2004, 562, L231–L237 CrossRef CAS.
- P. M. Kumar, S. Badrinarayanan and M. Sastry, Thin Solid Films, 2000, 358, 122 CrossRef CAS.
- J. C. S. Wu and H.-M. Lin, International Journal of Photoenergy, 2005, 7, 115–119 Search PubMed.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287–24293 CrossRef CAS.
- W. Y. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- N. Lewis and G. Crabtree, Report on the Basic Energy Sciences Workshop on Solar Energy Utilization, http://www.sc.doe.gov/bes/reports/files/SEU_rpt_print.pdf, accessed 11/30/2008 Search PubMed.
Footnote |
| † If we assume that the solar PV electricity displaces coal-fired power generation, a reduction of 50 Mt CO2 year−1 would imply a solar PV generation of 50 GWh(electric) year−1. Given the energy required for methanol synthesis (∼702 kJ mole−1 CO2), with a 10% efficiency for electricity-to-chemical energy conversion, we estimate the potential for CO2 conversion to methanol using solar PV electricity to be ∼1.1 Mt CO2 year−1. |
| This journal is © The Royal Society of Chemistry 2009 |