Palladium in fuel cell catalysis
Ermete
Antolini
Scuola di Scienza dei Materiali, Via 25 aprile 22, 16016, Cogoleto, Genova, Italy
First published on 7th May 2009
Abstract
Carbon supported platinum is commonly used as anode and cathode electrocatalyst in low-temperature fuel cells fuelled with hydrogen or low molecular weight alcohols. The cost of Pt and the limited world supply are significant barriers to the widespread use of these types of fuel cells. Moreover, platinum used as anode material is readily poisoned by carbon monoxide, present in the reformate gas used as H2 carrier in the case of polymer electrolyte fuel cells, and a byproduct of alcohol oxidation in the case of direct alcohol fuel cells. In addition, Pt alone does not present satisfactory activity for the oxygen reduction reaction when used as cathode material. For all these reasons, binary and ternary platinum-based catalysts and non-platinum-based catalysts have been tested as electrode materials for low temperature fuel cells. Palladium and platinum have very similar properties because they belong to the same group in the periodic table. The activity for the oxygen reduction reaction (ORR) of Pd is only slightly lower than that of Pt, and by addition of a suitable metal, such as Co or Fe, the ORR activity of Pd can overcome that of Pt. Conversely, the activity for the hydrogen oxidation reaction (HOR) of Pd is considerably lower than that of Pt, but by adding of a very small amount (5 at%) of Pt, the HOR activity of Pd attains that of pure Pt. This paper presents an overview of Pd and Pd-containing catalysts, tested both as anode and cathode materials for low-temperature fuel cells.
 Ermete Antolini Ermete Antolini | Ermete Antolini is Research Professor at Scuola Scienza Materiali, Genova, Italy. He received his PhD in Chemistry from the University of Genova, Italy. His research interests focus on the development of materials for heterogeneous catalysis with emphasis on supported catalysts for low-temperature fuel cells. |
Broader contextFuel cells are widely considered to be a sustainable energy conversion system and are a key technology for the development of a hydrogen economy. Low-temperature fuel cells have been undergoing rapid development for mobile applications and in particular for the transport sector. Platinum is commonly used as anode and cathode catalyst in low-temperature fuel cells. The cost of platinum, however, and the limited world supply are significant barriers to the widespread use of these types of fuel cells. To reduce the cost of the fuel cells, one of the important challenges is the development of platinum-free catalysts or catalysts with a lower content of Pt. For all these reasons, binary and ternary platinum-based catalysts and non-platinum-based catalysts have been tested as electrode materials for low temperature fuel cells. This work shows that palladium, which has costs lower than those of platinum, and is at least fifty times more abundant on Earth than Pt, can be substituted for Pt both as anode and cathode material without worsening fuel cell performance. |
1. Introduction
Low-temperature fuel cells, generally conceived around an acid electrolyte (phosphoric acid fuel cells, PAFC), an alkaline electrolyte (alkaline fuel cells, AFC) or a proton electrolyte membrane (polymer electrolyte membrane fuel cells, PEMFC), seem able to be used for a large range of power applications. Compared with other fuel cell systems, they have the advantage of high power densities at relatively low operating temperatures (80–200 °C); moreover they are also small and lightweight. A fuel cell consists of an anode, to which hydrogen fuel is commonly supplied, and a cathode to which oxygen (or air) is supplied, separated by the electrolyte.1,2 Alcohols, mainly methanol and ethanol, are widely proposed as possible hydrogen alternative fuels for mobile applications such as electric vehicles.3,4 The use of methanol as fuel has several advantages in comparison to hydrogen: it is a cheap liquid fuel, easily handled, transported, and stored, and with a high theoretical energy density5,6 However, the question of the toxicity of methanol remains crucial. Methanol has for a long time been considered as a toxic product, in addition to possible environmental problems in relation to its large miscibility in water. Ethanol offers an attractive alternative as a fuel in low-temperature fuel cells because it can be produced in large quantities from agricultural products and it is the major renewable biofuel from the fermentation of biomass. By comparing the performance of fuel cells employing a H3PO4-doped polybenzimidazole membrane and Pt–Ru as anode catalyst operating on various methanol-alternative fuels, Wang et al.7 found that ethanol is a promising alternative fuel with an electrochemical activity comparable to that of methanol. Other low molecular weight alcohols, such as propanol and ethylene glycol, have been tested as fuel, particularly for alkaline fuel cells.Platinum is commonly used as an electrocatalyst for both hydrogen oxidation and oxygen reduction in phosphoric acid fuel cells and proton exchange membrane fuel cells. Because catalysis is a surface effect, the catalyst needs to have the highest possible surface area. So, the active phase is dispersed on a conductive support such as high surface area carbon powders. But because of the elevated price and limited resources, Pt cannot be used for large-scale applications and alternative materials are needed. In addition, platinum used as anode material, at room or moderate temperatures is readily poisoned by carbon monoxide, present in the reformate gas used as H2 carrier, and a byproduct of alcohol oxidation in direct alcohol fuel cells. Moreover, Pt alone does not present satisfactory activity for the oxygen reduction reaction (ORR) when used as cathode material. For all these reasons, binary and ternary Pt-based catalysts and non-platinum-based catalysts have been tested as electrode materials for low temperature fuel cells.8–20
Pt and Pd have very similar properties (same group of the periodic table, same fcc crystal structure, similar atomic size). The cost of palladium, however, is lower than that of platinum, so it could be a good substitute for Pt as the catalyst in fuel cells. Pd is interesting as it is at least fifty times more abundant on the earth than Pt. For these reasons, Pd has been tested in fuel cells as a Pt cocatalyst (as anode and cathode material in acid media) and as a Pt-free catalyst (as anode material in alkaline media and in direct formic acid fuel cells, and as cathode material).
This paper presents an overview of Pd and Pd-containing catalysts, tested both as anode and cathode materials for low-temperature fuel cells.
2. Structural characteristics of Pd and Pt–Pd alloys
Alloy formation between two metals depends on the heat of mixing. If mixing is exothermic, the metals tend to form alloys. Depending on the extent of their exothermic mixing behavior, one can talk about strong or weak alloying. Park and Lee21 estimated a heat of mixing of Pd and Pt of −0.03 eV at−1, supporting a slightly exothermic behavior upon mixing. So, it can be supposed that Pd–Pt systems may form weak alloys at all compositions. Bulk Pt–Pd alloys are continuous solid solutions, i.e. structures in which the atoms are randomly mixed for all compositions.22 The pure elements and the bulk alloy phases exhibit fcc packing of atoms and cubic symmetry (L12).23 The values of the lattice parameter of pure Pt and Pd metals are 0.3923 nm 0.3890 nm, respectively. The lattice parameters of carbon supported Pt–Pd catalysts, commonly used in fuel cells, are higher than the value for pure Pd but smaller than that for pure Pt, indicative of alloy formation.24–27 The degree of alloying of carbon supported Pt–Pd catalysts depends on the preparation method. Fig.1 shows the dependence of the lattice parameter of carbon supported Pt–Pd alloy catalysts, prepared by the polyol process in ethylene glycol solution, on Pt content in the catalyst, from data of Li et al.24,25 As can be seen in Fig. 1, the lattice parameter of these supported Pt–Pd alloy catalysts increases almost linearly with Pt content in the catalyst, in agreement with the results obtained for bulk alloys.28,29 The surface composition of alloys, and especially the composition of the topmost surface layer, is generally different from the bulk one due to segregation processes. By low-energy ion scattering (LEIS) measurements, Rousset et al.30 observed that the surfaces of Pd–Pt particles with Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt atomic ratio 65:35 and 17:83 and sizes in the range 1.8–4.2 nm are strongly enriched in Pd, and the surface Pd concentration increases as the size increases. Analogously, Massen et al.31 observed segregation in Pt–Pd clusters, with most having Pt-rich cores and Pd-rich surfaces. This was explained in terms of the lower surface energy of Pd and the higher cohesive energy of Pt. Van den Oetelaar et al.32 investigated the metal surface composition of supported bimetallic catalysts and alloys. Indeed, the metal–support interaction may influence surface segregation by preferential anchoring of one of the constituents to the support at the cluster–support interface.33 They found that for carbon supported Pd–Pt catalysts, with low metal dispersions of about 0.3 and 0.8, Pd surface segregation does take place to approximately the same extent as in the Pd–Pt bulk alloys. They believed that the interaction of Pd and Pt with the carbon support does not differ significantly to affect surface segregation. In agreement with these results, Garcia et al.,27 either by cyclic voltammetry or CO stripping experiments, suggested the presence of a Pd layer on the surface of carbon supported Pd–Pt particles. This layer may present distinct hydrogen and CO adsorption properties, as compared to bulk Pd.
:Pt atomic ratio 65:35 and 17:83 and sizes in the range 1.8–4.2 nm are strongly enriched in Pd, and the surface Pd concentration increases as the size increases. Analogously, Massen et al.31 observed segregation in Pt–Pd clusters, with most having Pt-rich cores and Pd-rich surfaces. This was explained in terms of the lower surface energy of Pd and the higher cohesive energy of Pt. Van den Oetelaar et al.32 investigated the metal surface composition of supported bimetallic catalysts and alloys. Indeed, the metal–support interaction may influence surface segregation by preferential anchoring of one of the constituents to the support at the cluster–support interface.33 They found that for carbon supported Pd–Pt catalysts, with low metal dispersions of about 0.3 and 0.8, Pd surface segregation does take place to approximately the same extent as in the Pd–Pt bulk alloys. They believed that the interaction of Pd and Pt with the carbon support does not differ significantly to affect surface segregation. In agreement with these results, Garcia et al.,27 either by cyclic voltammetry or CO stripping experiments, suggested the presence of a Pd layer on the surface of carbon supported Pd–Pt particles. This layer may present distinct hydrogen and CO adsorption properties, as compared to bulk Pd.
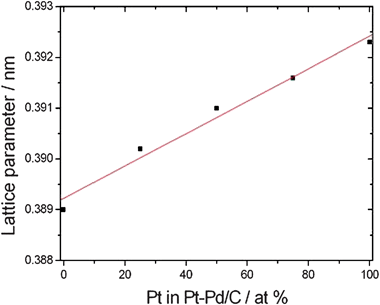 | ||
| Fig. 1 Dependence of the lattice parameter of carbon supported Pt–Pd alloy catalysts, prepared by the polyol process in ethylene glycol solution, on Pt content in the catalyst.24,25 | ||
Generally the particle size of carbon supported metals increases from Pt → Pt–Pd → Pd.24,25,27,34–38Fig. 2 shows the dependence of the metal particle size of carbon supported Pt–Pd catalysts on Pd content.24,27,36,38 Independently of the preparation method, metal particle size increases with increasing Pd content in the catalyst. The increase in metal particle size is more significant for Pd content >50 at%. The different dispersion of metal nanoparticles on the carbon support is mainly due to the different nucleus-growth mechanism of metal nanoparticles in Pt/C and Pd/C catalysts. The larger particle size of Pd and Pt–Pd, than of Pt, decreases the active surface area of the catalyst, so can negatively influence the catalytic activity.
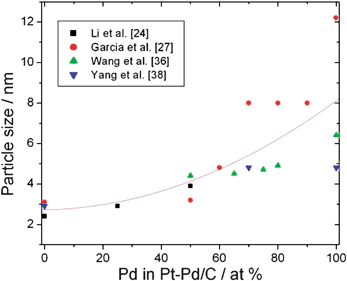 | ||
| Fig. 2 Dependence of metal particle size of carbon supported Pt–Pd catalysts on Pd content in the catalyst. | ||
3. Activity of Pt–Pd, Pd–Pt, Pd and Pd-based catalysts as anode materials in acid media
3.1 Catalytic activity for the hydrogen oxidation reaction (HOR)
Platinum presents appreciably high activity for the hydrogen oxidation reaction (HOR), but, because of its high cost, the substitution of pure Pt with other catalysts, such as binary and ternary Pt-based or Pd-based alloys, is necessary to allow wide commercialisation of the fuel cells. Binary palladium–platinum catalysts can be separated into two groups, Pd-rich catalysts, named Pd–Pt, with Pd/Pt atomic ratio >1, and Pt-rich catalysts, named Pt–Pd, with Pd/Pt atomic ratio <1. Stonehart39 reported promising results regarding the use of Pt–Pd catalysts as anode materials in PAFCs, with an optimum Pd content for HOR of 50 at%. Palladium alone has poor activity for HOR but the addition of very low amounts of Pt (5–10 at%) to Pd attains HOR activity, in PEMFCs fuelled with pure hydrogen, similar to that of pure Pt.27,40,41 Cho et al.40 compared the performance of PEMFCs with Pd, Pd–Pt (19:1) and Pt as anode material. The cell with Pd/C as anode material presented a poor performance, but by adding a very small amount of Pt (5 at%) to Pd, the cell with Pd–Pt/C attained the performance of the cell with Pt/C as anode catalyst. Papageorgopoulos et al.41 investigated the potential use of carbon supported PtPdy (where y = 1–6) electrocatalysts in PEMFCs. The voltage–current characteristics, with pure hydrogen as the fuel, of these Pd–Pt catalysts are shown in Fig. 3. Similar j/V characteristics to those of Pt were attained, with small deviations occurring, especially at higher current densities as the Pd/Pt fraction increases. In contrast, carbon supported Pd performed poorly compared to Pt. Thus, even a small amount of Pt in the catalyst was considered necessary, but it was also enough to enhance comparable PEMFC performance to that of a platinum anode. According to Garcia et al.,27 these results have to be ascribed to a change of HOR kinetics on the Pd skin layer present on the surface of Pd–Pt particles, which when compared to bulk Pd, is most probably related to a change of the Pd–H bond strength. Regarding Pt-rich Pt–Pd catalysts, Gamez et al.42 reported the exchange current at 150 mV vs. RHE for HOR (iex) of Pt–Pd with various Pt:Pd atomic ratios. As shown in Table 1, the value of iex per unit gram of Pt increases with increasing Pd content in the catalyst. The presence of Pd enhances HOR of Pt, which may be due to the effect of alloying on Pt electronic structure.
| Catalyst | i/A mPt−1 | i o/1010 A cmPt−2 |
|---|---|---|
| Pt/C | 411 | 0.2 |
| PtPd/C (9:1) | 412 | 4.3 |
| PtPd/C (82:18) | 542 | — |
| PtPd (76:24) | — | 1.6 |
| PtPd/C (64:36) | 569 | 0.8 |
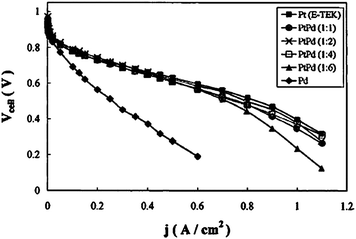 | ||
| Fig. 3 Current density–cell voltage curves of cells operated on pure H2 with anode catalysts consisting of carbon supported Pt (E-TEK), PtPd, PtPd2, PtPd4, PtPd6 and Pd at a pressure of 1.5 bar. Tcell = Thum = 80 °C. Reprinted from ref. 41, ©2002, with permission from The Electrochemical Society. | ||
3.2 Catalytic activity for the HOR in the presence of CO
For practical reasons, hydrogen for the fuel cell anode is obtained by processing fossil fuels, and the product gas in these processes contains carbon monoxide. Platinum is susceptible to CO poisoning, particularly at low temperature. Indeed, since the rate controlling step for hydrogen oxidation is dual-site dissociation of the hydrogen molecule on the catalyst surface, being that the activation energy for this reaction is low, 4–5 kcal mol−1,39 the reaction rate for HOR is not expected to increase significantly with temperature. The fact that hydrogen and carbon monoxide mixtures show significant enhancement in apparent hydrogen reaction rate with increasing temperature means that the adsorption isotherm for carbon monoxide on the platinum electrocatalyst surface is sensitive to temperature.39 Therefore, relatively small increases in temperature produce large increases in the apparent reaction rate for hydrogen oxidation in the presence of carbon monoxide. So, the performance of PEMFCs with Pt as anode material, operating at lower temperatures (<100 °C) than PAFCs (∼180 °C), is more seriously affected by trace amounts of CO, because CO strongly adsorbs on the Pt anode catalyst surface, causing a drastic decrease of the available active Pt surface sites for H2 electrooxidation. Although Pd possesses very low CO tolerance, inferior to that of pure Pt, tests in PEMFCs fuelled with H2/CO revealed that the presence of Pd increases the CO tolerance of Pt and Pt–Ru catalysts. He et al.43 tested carbon supported Pt, Pt–Ru (Pt:Ru = 5:2 and 1:1) and Pt–Ru–Pd (2:2:1) catalysts in phosphoric acid fuel cell conditions with H2 containing 1 mole percent CO. As shown in Fig. 4, the Pt–Ru–Pd electrode had the best tolerance for CO, followed by Pt–Ru and Pt electrodes. The HOR activity of Pt–Ru in the presence of CO increased following Pd addition. The decrease of the Pt–Ru lattice parameter by addition of Pd was indicative of Pd incorporation in the Pt–Ru crystal structure. The effect of Pd is different than is the effect of Ru, indeed, the addition of Ru to Pt supports CO oxidation (a bifunctional effect from the presence of RuOx) while Pd addition to Pt decreases CO coverage on catalyst particles (an electronic effect by Pd alloying). The potential use of carbon supported Pd-rich Pd–Pt electrocatalysts, with Pd/Pt 1–6, as CO-tolerant anodes for proton exchange membrane fuel cell applications was investigated by Papageorgopoulos et al.41 Cyclic voltammetry experiments at 80 °C, the operating temperature of the PEMFC, revealed that upon CO saturation, a lower fraction of surface sites were poisoned in the case of Pd–Pt compared with Pt, resulting in higher amounts of adsorbed hydrogen. Tests in PEMFCs showed that the cells with anodes consisting of Pd–Pt catalysts enhanced CO tolerance compared to the cell with Pt as anode material, with Pd4Pt providing the best results. As shown in Fig. 5, HOR activity of Pd4Pt/C at different CO concentrations was compared to that of a carbon supported well-alloyed Pt–Ru catalyst (E-TEK) and a non-alloyed Pt–Ru catalyst, prepared at the Energy Research Center of the Netherlands, with suggested better oxidation properties.44 The Pd4Pt catalyst exhibited higher current densities than both Pt–Ru systems. The tolerance of the Pd4Pt catalyst to carbon monoxide poisoning improved with higher amounts of CO in the fuel stream. An improvement in current close to 15% was seen for CO concentrations higher than 200 ppm. This indicated that palladium-rich Pd–Pt electrocatalysts are promising candidates for replacing PtRu for PEMFC applications. Garcia et al.27 investigated the performance of H2/O2 proton exchange membrane fuel cells fed with CO-contaminated hydrogen for anodes with Pd-rich Pd–Pt/C and Pd–Pt–Ru/C electrocatalysts. The results showed that the performance of the systems drops considerably when CO-containing H2 is introduced to the anode, particularly for Pd/C and Pt/C. On the other hand, the results for the Pd–Pt/C and Pd–Pt–Ru/C electrodes showed that the performance with H2 + 100 ppm CO was much better than for the pure noble metal catalysts. Based on the above facts they concluded that the main characteristic of the Pd–Pt/C catalysts was to present a reduced degree of coverage of CO on the Pd surface layer, enough for the anode to provide currents of the order of 1 A cm−2without too much potential losses. According to the authors, this effect was related to an electronic effect of Pt on Pd and vice versa, because this was done in a range of potentials where no CO2 formation was detected in the anode outlet. For Pd–Pt–Ru/C the CO tolerance also presented a contribution to the bifunctional mechanism as shown by the presence of CO2 in the anode outlet.
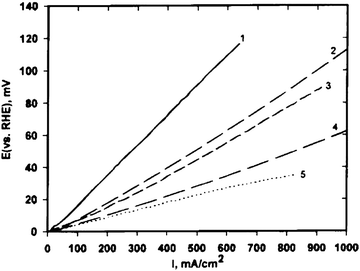 | ||
| Fig. 4 Comparison of CO tolerance of different Pt-based electrodes (with IR correction). Pt loading for all electrodes: 0.5 mgPt cm−2. (1) Pt electrode, H2/(1% CO), 177 °C cell temperature, 62 °C saturator temperature. (2) Pt/Ru (1:1) electrode, H2/(1% CO), 180 °C cell temperature, 77 °C saturator temperature. (3) Pt/Ru (5:2) electrode, H2/(1% CO), 177 °C cell temperature, 62 °C saturator temperature. (4) Pt/Ru/Pd (2:2:1) electrode, H2/(1% CO), 177 °C cell temperature, 62 °C saturator temperature. (5) Pt electrode, H2, 177 °C cell temperature, 62 °C saturator temperature. Reprinted from ref. 43, ©1997, with permission from The Electrochemical Society. | ||
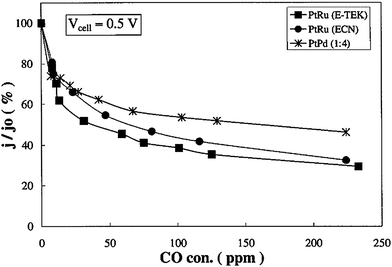 | ||
| Fig. 5 Current density ratios j/jo, where jo is the current density obtained in pure H2, vs. CO concentration for cells operated with anode catalysts consisting of carbon supported PtRu (E-TEK), PtPd4, and prepared PtRu at a pressure of 1.5 bar and a cell voltage of 0.5 V. Tcell = Thum = 80 °C. Reprinted from ref. 41, ©2002, with permission from The Electrochemical Society. | ||
Recently, Ueda et al.45,46 proposed the use of Pt-free Pd/SnO2/C and Pd/TiO2/C catalysts as anode material for PEMFCs. According to the authors, modification of the Pd/C anode with SnO2 or TiO2 nanoparticles enhances the cell performance in pure H2 and in H2/CO. The enhancement in the cell performance of Pd/SnO2/C and Pd/TiO2/C was mainly due to decreases in Pd particle size. The added oxides in the Pd/C catalyst may act as an anchor for the adjacent Pd particles to hinder the agglomeration of Pd particles, which leads to an increase in the active surface area of the catalyst.
3.3 Catalytic activity for alcohol oxidation
Among low molecular weight alcohols, methanol and ethanol are the most promising fuel for direct alcohol fuel cells in acid medium. Although a large number of other single metal electrodes were investigated, of these, platinum appears to be the best electrocatalyst for methanol and ethanol oxidation reactions (MOR and EOR, respectively) in acid medium.47 However, the electrooxidation of platinum is complicated by poisoning of intermediates and therefore the catalytic activity diminishes with time. Bagotzki et al.48 proposed a reaction pathway for methanol oxidation in acid medium at Pt electrodes, based on the successive dehydrogenation of methanol molecules with formation of strongly bound intermediates occupying 1–3 Pt sites (x) as shown in the following scheme:| CH3OH → CH2OH (x) → CHOH (xx) → COH (xxx) → CO (x) | (1) |
It was suggested that adsorbed CO can be oxidized to CO2 and adsorbed species like CH2OH and CHOH would be intermediates leading also to the formation of formaldehyde and formic acid, respectively. However, MOR activity of platinum is low and not suitable for use in direct methanol fuel cells (DMFCs). On the other hand, Pd is completely inactive for electrooxidation of methanol in acid solutions.49 Few works have reported the activity for alcohol oxidation in acid media of Pt–Pd catalysts. Kardigan et al.50 investigated MOR activity of Pt–Pd alloys in acid, neutral and alkaline solutions. Alloy electrodes were prepared by electrolytic co-deposition of Pt and Pd precursors. In alkaline solution the exchange current density for MOR displayed a pronounced maximum for a surface composition of about 15 at% in palladium. This synergistic effect is relatively important, since the exchange current densities are up to 10 times greater than for platinum. Conversely, in acid and neutral media there is no such maximum and the exchange current densities decrease monotonically from pure platinum to pure palladium. Nevertheless, this corresponds to a synergistic effect, since the activity of alloys is higher than the weighted activity of the two metals. According to the authors, the enhancement of the overall reaction rate in acid medium has to be ascribed to decreasing electrode poisoning. As previously stated, the platinum electrode is blocked by the formation of a strongly bound species, such as (COH)ads, which requires three neighbouring Pt sites to be adsorbed. As far as this requirement is fulfilled, the electrode surface is slowly poisoned, and the electrocatalytic activity decreases according to the following mechanism for the overall oxidation process:51
| Pt + CH3OH → Pt–(COH)ads + 3H+ + 3e− | (2a) |
| Pt–(COH)ads + H2O → Pt + CO2 + 3H+ + 3e− | (2b) |
The alloying of platinum with palladium, which is inactive in acid medium, results in the diluting of platinum sites, which prevents the presence of the three adjacent sites necessary for the formation of the strongly bound intermediates. For alloy electrodes of surface composition >33 at% Pd, it is statistically difficult to find three adjacent Pt sites. Only two neighbouring Pt sites are now available for CH3OH adsorption, which favours the adsorbed species (CO)ads. This leads to the other methanol oxidation path, according to the mechanism:52
| Pt + CH3OH → Pt–(CO)ads + 4H+ + 4e− | (3a) |
| Pt–(CO)ads + H2O → Pt + CO2 + 2H+ + 2e− | (3b) |
The rate of reaction (3a) is lower than that of reaction (2a), but the rate of reaction (3b) is higher than that of reaction (2b). Consequently, the overall reaction rate is increased. For greater content of palladium, the electroactivity of the alloy electrode decreases because of the inactivity of the palladium sites. Thus, the electrocatalytic activity must be enhanced for surface composition comprising between 0 and 33 at% Pd. More recently, Xu and Lin53 and Selvaray et al.54 studied the methanol oxidation in acid media on Pt and Pt–Pd nanoparticles electrodeposited on a glass carbon electrode and incorporated on polypyrrole, respectively. In both cases, the Pt–Pd catalysts showed significantly higher catalytic activity for the methanol oxidation reaction with respect to Pt only.
Wang et al.55 prepared highly ordered Pd/Pt core–shell nanowire arrays (Pd/Pt NWAs) by coating a Pt thin film on the surface of Pd NWA core using a magnetron sputtering method. Pt loading of the sputtered Pt film was 0.03 mg cm−2. For comparison, a Pt film electrode on Ti foil prepared by magnetron sputtering and a conventional 33 wt% PtRu/C (Pt:Ru = 2:3, E-TEK) with the same Pt loadings were also tested under the same conditions. The Pd/Pt NWA electrode showed a higher electrochemical active surface area and higher electrocatalytic activity for methanol electrooxidation in acid medium than the other catalysts. This demonstrated the significant promotion effect of the ordered Pd NWA core to the electrooxidation of methanol because of the oxophilic nature of Pd when compared to Pt, and the high surface-to-volume ratio and excellent current collector ability of the Pd NWA support.
Regarding ethanol oxidation, Zhou et al.56 tested the activities for EOR of carbon supported Pt and bimetallic Pt–M catalysts (M = Sn, Ru, Pd and W, Pt:M = 1:1) in a H2SO4/CH3OH solution. The ethanol oxidation activity of Pt–Pd (1:1) was higher than that of Pt, but lower than that of all the other Pt-based binary catalysts. Tests in direct ethanol fuel cells (DEFCs) showed that the performance of the single cell with Pt/C as anode catalyst is similar to that of a single cell with Pt1Pd1/C. The single cell test demonstrated that, from a practical point of view, Pt/C and Pt–Pd/C do not seem to be good catalysts for ethanol oxidation. Lopes et al.26 investigated the activity for EOR of carbon supported Pt–Pd (3:1) and Pt catalysts by linear sweep voltammetry measurements in a H2SO4/CH3CH2OH solution. For fuel cell applications, the working potentials of the anode were located between 0.3 and 0.5 V vs. RHE to obtain a cell voltage from 0.6 to 0.4 V assuming a potential of about 0.9–1.0 V vs. RHE for the cathode, with the highest current density. As can be seen in Fig. 6, at room temperature, 60 °C, and 90 °C for potentials vs. RHE lower than 0.5 V the current density (jEOR) for EOR for Pt–Pd/C is very low and almost the same as that of Pt/C.
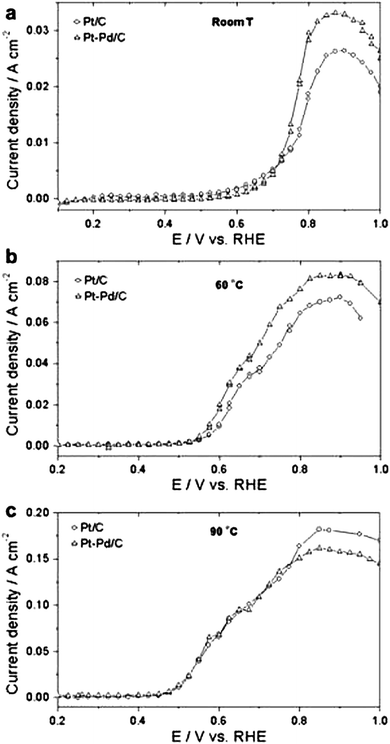 | ||
| Fig. 6 Slow scan voltammograms for ethanol oxidation on Pt–Pd/C (3:1) and Pt/C electrocatalysts in 1.0 M ethanol solution at (a) room temperature, (b) 60 °C and (c) 90 °C. Sweep rate 1 mV s−1. Reproduced from ref. 26, ©2008, with permission from Elsevier. | ||
3.4 Catalytic activity for formic acid oxidation
Formic acid has been investigated as an alternative fuel to hydrogen and methanol in PEMFC. Formic acid is a strong liquid electrolyte, hence, is expected to facilitate both electronic and proton transport within the anode compartment of the fuel cell. The theoretical open circuit potential (OCP) or emf of a formic acid–oxygen fuel cell, as calculated from the Gibbs free energy, is 1.45 V.57 The feasibility of direct formic acid fuel cells (DFAFCs) based on proton exchange membranes has been demonstrated by Masel et al.57–59 Wang et al.60 have also reported that the rate of formic acid crossover can be reduced by a factor of 5 compared to that of methanol and thus higher performance can be obtained when formic acid is used to replace methanol under the same conditions. Indeed, the penetration efficiency of formic acid through the Nafion membrane is much lower than that of methanol due to the repulsion between HCOO− and SO3− ions in the Nafion membrane.61 Moreover, the optimal operating concentration of formic acid can be as high as 20 M,62,63 while the best concentration of methanol in a DMFC is only about 2 M. To increase the catalytic activity of formic acid electrooxidation, efforts have been devoted to the development of catalysts. Yu and Pickup64 review recent advances in DFAFCs, focusing mainly on anodic catalysts for electrooxidation of formic acid. On platinum, formic acid oxidation occurs via a dual pathway mechanism.65 The most desirable reaction pathway for direct formic acid fuel cells is via a dehydrogenation reaction, which does not form CO as a reaction intermediate. The formic acid oxidation pathway (4) forms CO2 directly as:| HCOOH → CO2 + 2H+ + 2e− | (4) |
The product CO2 is formed by circumventing the adsorbed CO intermediate poisoning step, thereby enhancing the overall turnover rate. The second reaction pathway via dehydration is somewhat similar to that of methanol oxidation, forming adsorbed carbon monoxide (CO) as a reaction intermediate. The formic acid reaction pathway is shown in steps (5–8):
| HCOOH + Pt0 → Pt⋯CO + H2O | (5) |
| Pt0 + H2O → Pt⋯OH + H+ + e− | (6) |
| Pt⋯CO + Pt⋯OH → 2Pt0 + CO2 + H+ + e− | (7) |
| Overall: HCOOH → CO2 + 2H+ + 2e− | (8) |
Nanostructured Pt catalysts for electrooxidation of formic acid are poisoned by the adsorbed CO intermediate of the reaction.63,66,67 This suggests that the decomposition of HCOOH on platinum nanoparticles is likely to proceed via a dual path mechanism. It has been demonstrated that the modification by palladium, as a Pt–Pd alloy,68 Pd “decorated” polycrystalline Pt surfaces,69 and Pt single crystals coated by palladium electrolytically,70 produce increased reactivity. Capon and Parson68 using chronoamperometry measurements at 0.5 V on Pt, Pd and a Pd–Pt alloy (bulk 87.5% Pd, surface 55% Pd), found that the initial current on the mixed Pt + Pd electrode was greater than that on either of the two constituent metals, attesting that the Pt + Pd dual site electrode enhances the rate of HCOOH oxidation through a weakly bound intermediate pathway. Lu et al.69 deposited palladium on platinum to produce palladized polycrystalline platinum surfaces. Unambiguously, the Pt/Pd surface showed higher activity for formic acid oxidation than the clean Pt surface, especially at potentials below 0.5 V. On the Pt/Pd surface, the current density maximum was located near 0.27 V; 0.3 V lower than on a clean Pt surface. Baldauf and Kolb70 studied the oxidation of formic acid on thin, epitaxially-grown Pd overlayers on Au and Pt single crystal electrodes as a function of overlayer thickness ranging from submonolayer coverage up to the equivalent of 10 monolayers. The results were compared with those for massive Pd single crystal surfaces. The electrocatalytic properties of the Pd overlayers were found to depend markedly on their thickness and crystallographic orientation. Pd(100) showed the highest catalytic activity of all three low-index faces investigated, and Pd films on Pt(hkl), even when two or three layers thick, generally exhibited much higher activity than Pd films on Au(hkl) or Pd(hkl). In all cases high resistivity of the Pd surfaces against poisoning by CO was observed.
More recently, with the increasing interest in DFAFCs, many studies have addressed formic acid oxidation in Pt–Pd catalysts.17,71–74 The performance of Pt, Pt–Pd and Pt–Ru as catalysts for direct formic acid fuel cells was investigated by Rice et al.17 Analysis of data indicated that the addition of palladium enhanced the rate of formic acid electrooxidation via a direct reaction mechanism, while ruthenium additions suppressed the direct pathway and enhanced electrooxidation via a reactive CO intermediate. All the catalysts showed stable operation during tests of several hours. Waszczuk et al.71 used spontaneous deposition to decorate platinum nanoparticles with controlled amounts of palladium and palladium/ruthenium. Among the catalysts investigated the Pt–Pd catalyst showed the best performance. Notwithstanding, the Pt–Pd catalyst required the highest potential to remove CO; it was the most active for HCOOH oxidation. It can be inferred, therefore, that within the dual path mechanism of formic acid oxidation, the direct CO2 formation channel on Pt–Pd is much less affected by CO chemisorption than that on Pt and on Pt–Pd–Ru catalysts. A kinetic study of electrooxidation of formic acid on spontaneously-deposited Pt–Pd nanoparticles carried out by Zhao et al.72 confirmed the results of Waszczuk et al.71 They found that the Pt–Pd catalysts are a factor of thirty more active than Pt under the conditions examined. The catalysts also continued to function and showed reasonable activity as CO built up on the catalyst surface. Thus, the catalysts were CO tolerant. In the same way, Larsen and Masel73 performed a kinetic study of CO tolerance during electrooxidation of formic acid on spontaneously deposited Pt–Pd and Pt–Ru nanoparticles. They found that, while the oxidation of formic acid on Pt and Pt–Ru is poisoned by CO, the electrooxidation of formic acid on Pt–Pd is not greatly inhibited by adsorbed CO. Therefore, it was suggested that formic acid electrooxidation occurs on a reaction site that is not inhibited by CO. Li and Hsing74 prepared carbon supported PtxPd1−x catalysts by a co-deposition procedure or sequential-deposition; depositing Pd (or Pt) onto Pt/C (or Pd/C). Physicochemical and electrochemical characterization showed that co-deposited PtxPd1−x/C had higher catalytic activity than sequential-deposition catalysts, due to a probable synergistic effect between Pt and Pd. Furthermore, it was revealed that at a lower potential, the formic acid oxidation current increases with increased Pd surface concentration on PtxPd1−x/C catalysts.
Pt/Ru and Pt/Pd alloys were able to diminish the CO poisoning effect to some extent, but CO poisoning still significantly limited their catalytic activities for formic acid oxidation. Capon and Parson49 observed that little or no strongly adsorbed intermediates of the type found on Pt are present on Pd during the oxidation of HCOOH. So it can be inferred that formic acid is oxidized directly to CO2. While no activity for methanol oxidation was observed on Pd metal, a fast direct oxidation was operative for formic acid oxidation. The negatively charged C atom in CH3OH was much more difficult to adsorb onto Pd sites than HCOOH because Pd is a poor electron acceptor due to its saturated valence electron structure of 4d10. It has to be remarked that the charge density of H in the O–H bond is more positively charged than other atoms, which make it favored for adsorption on Pd, thus facilitating the direct oxidation of HCOOH on Pd. However, it is very difficult to compare catalytic activities of Pt and Pd towards HCOOH oxidation because of the difference in competing reaction pathways. As reported by Arenz et al.75 the interaction of the formic acid molecule with Pt and Pd atoms is completely different. While Pd has in the entire potential region a propensity to break only the O–H bonds of the HCOOH molecule, Pt has a propensity to break both the C–O and/or C–H bond (at low potential) as well as the O–H bond (at higher potential). Consequently, HCOOH oxidation on Pd atoms proceeds exclusively through the dehydrogenation reaction step, while on Pt at low potentials the dehydration reaction pathway is predominant. Jayashree et al.76 reported on the preparation, structural characterization, and electrochemical activity of electrodeposited dendritic Pt, spontaneously-deposited Pd on dendritic Pt, co-electrodeposited Pt/Pd (1:1), and electrodeposited Pd catalyst structures towards formic acid oxidation. Pd-containing catalysts were found to be significantly more active than pure Pt catalyst structures. Chronoamperometry at low anode potentials, which correspond to high cell potential in a fuel cell, Pt structures modified by spontaneously-deposited Pd were found to be the most active electrocatalysts, while pure Pd structures showed higher activity at higher anode potentials, i.e. at lower cell potentials. So, pure Pd catalyst structures may be a good choice for fuel cells continuously operating under high load. On this basis, Masel et al. tested unsupported Pd77 and carbon supported Pd/C78 as anode catalysts in DFAFCs. They found that palladium catalysts produced significant performance enhancement. DFAFCs operated with dry air and zero back-pressure could generate power densities of 255 ∼ 230 mW cm−2 at relatively high voltages of 0.40–0.50 V at a concentration range of formic acid from 3.0 to 15.0 M at room temperature (20 °C). This is not very different from a hydrogen–air polymer exchange membrane fuel cell with power density of 320 mW cm−2 obtained under comparable conditions, and much higher than a direct methanol fuel cell with power density of 50 mW cm−2. Liu et al.35 compared the performance of DFAFCs with Pt/C and Pd/C as anode catalysts. They observed that the cell with Pd/C performs better than that with Pt/C. The higher activity of Pd can be explained by the fact that on a Pd catalyst formic acid follows a different oxidation reaction pathway from the reaction pathway on a Pt catalyst. Pd promotes dehydrogenation without the formation of a surface poisoning COads intermediate. Some workers79–81 report that the addition of other metals, such as Au, Ir and Co, to Pd further improves performance of Pd-based catalysts.
Palladium catalysts are quite active for formic acid electrooxidation, but the activity of pure palladium nanoparticles decays slowly with time. Larsen et al.82 investigated the properties of palladium on various supports to see if the activity could be stabilized. They found that submonolayer Pd films deposited on V, Mo, W, and Au foils showed activity that is stable. Moreover, as can be seen from Fig. 7, the activity of Pd–V for formic acid oxidation outperforms that of conventional Pt–Ru catalyst at 0.3 V vs. RHE by three orders of magnitude. More recently, Ge et al.83 prepared a Pd nanocatalyst using HVO42− as the stabilizer agent. HVO42−, a tridentate oxoanion with a C3 symmetry structure and O–O distance closely matching Pd–Pd distance, was predicted to be an excellent stabilizer for Pd nanoparticles. Dispersed Pd nanoparticles supported on carbon black were obtained with average size of 4.8 nm and narrow size distribution. The HVO42− adsorbed on the Pd particles after preparation could be simply removed by washing with excessive water. The catalyst showed better performance for formic acid oxidation compared to the Pd/C catalyst prepared by common methods. The stability was also dramatically improved. Both improvement in catalytic activity and stability could be attributed to the small size and the uniform distribution of the Pd nanocatalyst with HVO42− as stabilizer.
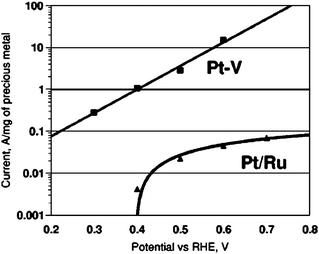 | ||
| Fig. 7 Tafel plot of Pd–V (θPd = 0.6) and HighSpec 6000 Pt–Ru catalyst toward formic acid electrooxidation. The measurements were done by immersing the catalysts in a solution containing 5 M HCOOH and 0.1 M H2SO4 at open-cell potential, stepping up to the indicated potential, and measuring the current as a function of time for 2 h, then recording the steady state current. Reprinted from ref. 82, ©2006, with permission from The Electrochemical Society. | ||
4. Activity of Pd and Pd–M catalysts for alcohol oxidation in alkaline media
A major contribution to the relatively low direct alcohol fuel cell (DAFC) performance is from kinetic constraints in the alcohol oxidation reaction in acid media. Improved alcohol oxidation kinetics can be facilitated using basic media. As reported by Tripkovic et al.,84 the kinetics for methanol oxidation are much higher in alkaline than in acid solution; at 60 °C and at 0.5 V, these were a factor of 30 for Pt and a factor of 20 for Pt2Ru3. The pH effect was attributed to the pH competitive adsorption of oxygenated species with anions from supporting electrolytes. Accordingly, the approach was to develop an alkali analogue of the DAFC. Several DAFCs which use an anion exchange membrane (AEM) as a polymer electrolyte membrane have already been reported. Direct fuel cells which use methanol,85–89 ethanol,85,89 ethylene glycol,88–90 and hydrazine91 are typical examples. These previous studies showed much better kinetics for fuel oxidation in alkaline medium than in acidic medium. Direct 2-propanol fuel cells have attracted more and more attention as 2-propanol is the smallest secondary alcohol, less toxic than methanol and its electrochemical oxidation is of great interest due to its particular molecular structure.92Generally, the activity of Pd for alcohol oxidation is very low in acid but relatively high in alkaline media. Pd presents poor activity for methanol and ethylene glycol (EG) oxidation, lower than that of Pt.93,94 As reported by Xu et al.,94 the current density for methanol oxidation at −0.3 V vs. Hg/HgO for Pt and Pd supported on carbon microspheres (CMS) was 11.4 and 2.5 mA cm−2, respectively, while the current density for EG oxidation was 22.8 and 3.6 mA cm−2, respectively. The activity order of alcohol oxidation on Pd/C and Pd/CMS was ethanol > EG > methanol. Recently, the same research group90 compared the electrocatalytic activity of Pd for methanol and propanol oxidation in alkaline media. They found that the current densities of 1-propanol and 2-propanol oxidation on Pd electrode are much higher at corresponding potentials than that of methanol oxidation, and the onset potentials of 1-propanol and 2-propanol oxidation on Pd electrode are more negative compared to that of methanol oxidation. The activity of Pd for propanol oxidation was higher than that of Pt.95,96 Ye et al.96 investigated Pd and Au as electrocatalysts for 2-propanol oxidation and compared their activity with that of the conventional Pt catalyst in alkaline medium. The results showed that the activity for the electrooxidation of 2-propanol of Pd is higher than that of Pt and Au. Xu et al.97 observed that addition of Au can significantly increase the catalytic activity and stability of palladium for 2-propanol oxidation. Pd–Au/C in the Pd:Au atomic ratio of 4:1 has higher electrocatalytic activity and better stability for electrooxidation of 2-propanol than Pd/C. Pd possesses remarkably higher activity for ethanol oxidation in alkaline media than Pt, and is a promising material for use in direct ethanol alkaline fuel cells.93,94 The effect of addition of various oxides to Pd/C catalysts on alcohol oxidation was investigated by Xu et al.93,98–100 Addition of oxides like CeO2, NiO, Co3O4 and Mn3O4 significantly promoted catalytic activity, and stability of the Pd/C electrocatalysts for alcohol electrooxidation. The effect of CeO2 and NiO content in Pt/C and Pd/C catalysts on ethanol electrooxidation in 1.0 M KOH / 1.0 M ethanol solution is shown in Fig. 8. In Fig. 8, the positive effect of oxide presence on EOR activity is more significant for Pd than for Pt, and for NiO than CeO2.
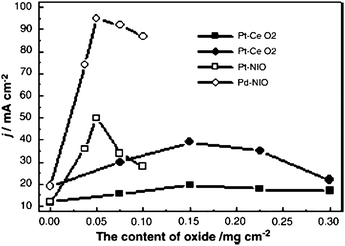 | ||
| Fig. 8 Effect of the content of oxide in Pt/C and Pd/C catalysts for ethanol electrooxidation in 1.0 M KOH solution containing 1.0 M ethanol with a sweep rate of 50 mV s−1, Pt or Pd loading: 0.3 mg cm−2. Reproduced from ref. 99, ©2007, with permission from Elsevier. | ||
Ru can promote the catalytic activity of Pt toward methanol and ethanol oxidation by providing oxygen-containing species at more negative potentials. Thus, Chen et al.101 investigated whether such a promotion effect was also effective for Pd catalysts in alkaline medium. They found that the catalytic activity of Pd–Ru was considerably higher than that of Pd toward the oxidation of methanol, ethanol, and ethylene glycol. The incorporation of Ru gave rise to a negative shift of 0.15 V in the onset potential. The activity sequence of Pd–Ru toward the alcohol oxidation was ethanol > ethylene glycol > methanol, and Pd–Ru with 1:1 atomic ratio exhibited the highest activity. They also compared the activities of Pd-based catalysts and the Pt–Ru catalyst in alkaline media toward alcohol oxidation. For the oxidation of methanol and ethylene glycol, the activity of Pd–Ru is higher than that of Pd but still lower than that of Pt–Ru. However, for ethanol oxidation, Pd–Ru exhibits exceptionally high activity. At potentials ranging from 0.3 to 0.4 V, the mass specific activity of Pd–Ru is almost 4 times that of Pt–Ru. After 0.45 V, the anodic current on Pt–Ru starts to decline; while on Pd–Ru, the current continues to increase and reaches a mass specific activity of 1.5 A mg−1 at 0.5 V.
Wang et al.102 prepared highly ordered Pd nanowire arrays by a template-electrodeposition method using an anodic aluminum oxide template. The Pd nanowire arrays presented high electrochemical active surface and showed excellent catalytic properties for ethanol electrooxidation in alkaline media. The activity of Pd nanowire arrays for ethanol oxidation was not only higher that of Pd film, but also higher than that of commercial E-TEK PtRu(2:1 by weight)/C. The micrometer sized pores and channels in nanowire arrays act as structural units. They make liquid fuel diffuse into, and products diffuse out of, the catalyst layers much easier, therefore, the utilization efficiency of catalysts gets higher. Repetitive potential cycling in 1 M KOH / 1 M CH3CH2OH indicated that Pd nanowire arrays possess good stability.
5. Activity of Pt–Pd, Pd–Pt, Pd and Pd-based catalysts for oxygen reduction
5.1 Principles
Platinum has the highest catalytic activity for oxygen reduction of any of the pure metals, and when supported on a conductive carbon serves as a state-of-the-art electrocatalyst in low temperature fuel cell cathodes.103 However, due to kinetic limitations of ORR, the cathodic overpotential losses amount to 0.3–0.4 V under typical PEFC operating conditions.103 The development of more active oxygen reduction electrocatalysts than Pt has been the subject of extensive research for a number of decades. The search for ORR catalysts more active, less expensive, and with greater stability than Pt has resulted in the development of Pt alloys. The alloys of transition metals, such as V, Cr, Co, Ti and Ni, with platinum have been found to exhibit significantly higher electrocatalytic activities towards ORR than platinum alone in low temperature fuel cells.12,13,104–111 The activity enhancement observed when using supported Pt alloy electrocatalysts has been ascribed to geometric factors (decreased Pt–Pt bond distance),12 dissolution of the more oxidisable alloying component,105 change in surface structure106 or electronic factors (increased Pt d-band electron vacancy of the Pt skin layer originating from the bulk alloys).13The effect of Pd modification of Pt surfaces on Pt ORR activity was studied by Schmidt et al.112 They found that the kinetics of ORR is significantly enhanced by modification of Pt(hkl) surfaces with Pd. Even on the highly active Pt(111) surface the kinetics can be improved by a factor of approximately two to four due to Pd modification. Recently, Zhang et al.113,114 reported that monolayer Pt deposited on Pd(111) could enhance ORR activity greatly with very low Pt loading. In the same way Nilekar et al.115 synthesized and successfully tested for the ORR monolayers of Pt deposited on different late transition metals (Au, Pd, Ir, Rh, or Ru); the Pd-supported Pt monolayer had the highest ORR activity. The amount of Pt used was further decreased by replacing part of the Pt monolayer with a third late transition metal (Au, Pd, Ir, Rh, Ru, Re, or Os). Several of these mixed Pt monolayers deposited on Pd single crystal or on carbon supported Pd nanoparticles exhibited up to 20-fold increases in ORR activity on a Pt-mass basis when compared to conventional Pt electrocatalysts. Density functional theory (DFT) calculations showed that the superior activity originated from the interaction between the Pt monolayer and the Pd substrate and from reduced OH coverage on Pt sites, the result of enhanced destabilization of Pt–OH induced by the oxygenated third metal.
Among various metals, Pd possesses ORR activity close to that of Pt.14 So, electroactive platinum-free Pd and Pd-alloy catalysts have been proposed as cathode materials for ORR in acid medium.116,117
Regarding direct methanol fuel cells, one of the major problems is of methanol crossover through the polymer electrolyte. This feature has been extensively studied.118–122 Methanol adsorbs on Pt sites in the cathode for the direct reaction between methanol and oxygen. The mixed potential, which results from ORR and methanol oxidation occurring simultaneously, reduces the cell voltage, generates additional water and increases the required oxygen stoichiometric ratio. On the other hand, in direct ethanol fuel cells, Song et al.123 found that the ethanol that permeated to the cathode exhibited a less serious effect on cell performance compared to methanol because of, both, its lower permeability through Nafion® membrane and its slower electrochemical oxidation kinetics over Pt/C cathode. The influence of ethanol crossover on DEFC performance, however, is not negligible.124,125 The problem of alcohol crossover could be solved either by using electrolytes with lower alcohol permeability or by developing new cathode electrocatalysts with higher alcohol tolerance than Pt. The current direction is to test the activity for ORR in the presence of alcohol on some Pt-alloys, which present a higher activity for ORR than platinum in low temperature fuel cells operated with hydrogen.126 Alternatively, platinum-free catalysts have shown improved selectivity for ORR and tolerance to the presence of alcohol molecules in acid medium. Among them, Pd and Pd–M catalysts present very important selectivity for ORR in acid medium in the presence of alcohol, thus maintaining high activity as cathode catalysts for the electroreduction of oxygen.
In the following paragraphs an overview of ORR activity of Pt–Pd, Pd–Pt, Pd and Pd–M catalysts in acid medium, both in the absence and in the presence of alcohol is presented.
5.2 Activity of platinum–palladium catalysts for the ORR
Firstly, ORR activity of Pt-rich Pt–Pd catalysts in acid medium was investigated by Gamez et al.42 They found that all Pt–Pd catalysts investigated presented higher ORR activity than Pt. A small amount of Pd (9 at%) gave rise to the highest increase in ORR activity. Above 9 at% Pd, the exchange current decreased with increasing Pd content in the catalyst, see Table 1. In agreement with the results of Gamez et al.,42 more recent work showed that the addition of Pd to Pt increased the ORR activity of platinum,24,26,38,54,127,128 and that the dependence of ORR activity on Pd content goes through a maximum. Guerin et al.,127 by high-throughput synthesis and screening of thin film materials observed an enhancement in activity for a range of PtPd alloy compositions over either of the pure elements. They found a maximum in activity in the composition range of 50–90% Pt that was potential-dependent. At the higher potential (lower overpotential for ORR) where the currents are more sensitive to kinetic control, the maximum in activity was more clearly defined in a composition range between 70 and 90% Pt. Li et al.24 prepared Pt–Pd/C (Pt:Pd = 3:1 and 1:1) and Pt/C catalysts by a modified polyol process method. In agreement with the results of Guerin et al.,127 they found that the catalytic activity for ORR of Pt–Pd/C at the Pt:Pd atomic ratio 3:1 was improved compared with that of Pt/C or Pt–Pd/C (1:1). The enhancement of Pt ORR activity in the presence of Pd has been proved by theoretical calculations. DFT calculations showed that the Pt atom bears a negative charge while Pd is positively charged in PtmPdn clusters and that the negatively charged Pt atoms in PtmPdn facilitate the dissociation of O2 on the Pt surface, thus enhancing ORR activity. Ye and Crooks128 prepared Pt–Pd bimetallic nanoparticles containing an average of 180 atoms, and which were composed of seven different Pt:Pd ratios within sixth-generation, hydroxyl-terminated, poly(amidoamine) dendrimers. Cyclic voltammetry and rotating disk voltammetry measurements showed that the Pt:Pd ratio of the nanoparticles determined their efficiency for ORR. The maximum activity for the ORR occurs at a Pt:Pd ratio of 5:1, which corresponds to a relative mass activity enhancement of 2.4 compared to otherwise identical monometallic Pt nanoparticles. As the percentage of Pd increases, the catalytic performance monotonically decreases. Xu and Lin54 found that an electrodeposited Pt–Pd (9:1) catalyst presented significantly higher stability and catalytic activity for ORR than the correspondingly electrodeposited Pt. Lopes et al.26 observed by ORR tests in H2SO4 solution at room temperature that the activity for ORR of a Pt–Pd/C (3:1) catalyst is slightly higher than that of a commercial Pt/C electrocatalyst. Finally, Yang et al.38 investigated the activity for ORR in acidic solutions of carbon supported Pd-rich pseudo-core–shell Pd–Pt (Pd@Pt/C) nanoparticles, with Pt coverage close to a monolayer, prepared from a simple galvanic replacement reaction between Pd/C particles and PtCl42− at 100 °C. Fig. 9a shows ORR polarization curves for Pd/C, Pt/C, and various Pd@Pt/C catalysts. For monometallic Pd/C and Pt/C catalysts, the half-wave potentials are 0.652 and 0.700 V, respectively. The half-wave potential for Pd70@Pt30/C of 0.717 V is 65 mV more positive than that of Pd/C, and 17 mV more positive than that of Pt/C. The Pt shell in the pseudo-core–shell Pd70@Pt30/C catalyst was therefore more active than bulk Pt in ORR. The inset in Fig. 9a shows the relationship between the half-wave potential and the Pt ratio in the pseudo-core–shell Pd@Pt/C particles. The general increase in the half-wave potential relative to Pd is easily understood as Pt is a more active ORR metal. More interesting was the observation of enhanced ORR activity of Pt, which began at a Pt/(Pd + Pt) ratio of 0.20 and reached a maximum value at ∼0.30, corresponding to the formation of a monolayer coverage of Pt on Pd. The enhancement effect is lower after this value, when Pt begins to form multilayers on the surface or diffuses into the Pd core. The current densities at 0.8 V were used to compare the performance of the various catalysts (Fig. 9b). Regardless of whether total metal (Pd + Pt) or Pt was used as the basis of normalization, the Pd70@Pt30/C catalyst always showed the highest activities (1.22 and 2.8 times relative to Pt/C, respectively). When currents were normalized by electrochemically active surface areas, the Pd70@Pt30/C still came out on top (2.45 compared to 2.16 A m−2 for Pt/C). This corroborateds the presence of more active ORR sites on Pd70@Pt30/C, and that the Pt monolayer on Pd is different from normal Pt. The authors explained the enhanced ORR activity of pseudo-core–shell Pd70@Pt30/C relative to Pt/C in terms of the weaker OHads binding on the Pt sites, and the combination of favorable electronic and strain effects.
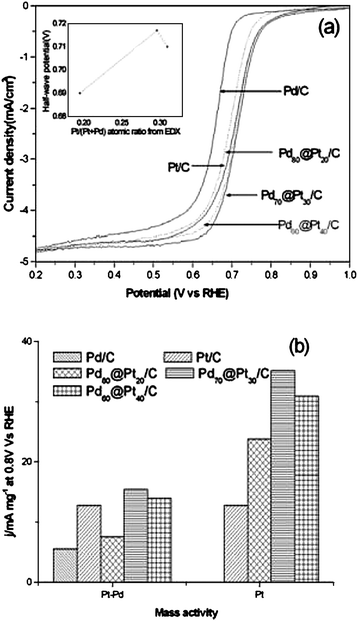 | ||
| Fig. 9 Linear voltammograms of Pd/C, Pt/C, and various Pd@Pt/C catalysts in oxygen-saturated 0.1 M HClO4 showing the negative-going scans. (a) Sweep rate: 20 mV s−1, room temperature, 1600 rpm. The inset shows the relation between the half-wave potential and the Pt/(Pt + Pd) ratio; specific mass current density at 0.8 V for various catalysts. (b) The left panel shows normalization by the total metals, the right panel shows normalization by Pt only. Reprinted from ref. 38, ©2008, with permission from The Electrochemical Society. | ||
5.3 Activity of platinum–palladium catalysts for the ORR in the presence of alcohol
Few works deal with alcohol tolerant oxygen reduction platinum–palladium catalysts. Generally, Pd-rich Pd–Pt catalysts were tested as ORR methanol tolerant catalysts, being Pd inactive for methanol oxidation. Li et al.25 tested a Pd-rich carbon supported Pd–Pt (3:1) alloy catalyst as cathode material in DMFC. They found that the palladium-rich Pd–Pt/C catalyst enhances DMFCs cathode performance, due to its selective ORR activity in the presence of methanol. In the same way, Yang et al.38 investigated the ORR activity of Pd/C, Pt/C, and Pd70@Pt30/C in the presence of methanol in a 0.1 M HClO4/0.1 M CH3OH solution at room temperature in the potential range of 1.0–0.2 V. The activities of Pd70@Pt30/C were higher than that of Pt/C in the entire potential region, attesting to its being a more methanol tolerant ORR catalyst than Pt. Pd70@Pt30/C also outperformed Pd/C in the high potential region (>0.7 V). Wang et al.36 investigated ORR activity of various Pd-rich Pd–Pt catalysts in the presence and absence of methanol. They found that a Pd3Pt1/C alloy catalyst showed a comparative ORR activity with the Pt/C in 0.1 M HClO4 electrolyte. Instead, all the Pd–Pt/C catalysts exhibited much higher ORR activity than the Pt/C in the presence of methanol. The enhanced methanol tolerance of Pd-rich Pd–Pt catalysts, than that of Pt alone, can be ascribed to the presence of MOR inactive Pd and to the formation of a PdPt alloy. Regarding the core–shell structure, the generally weaker binding of adsorbed species on the Pd70@Pt30/C surface, related to electronic and strain effects, facilitated the oxidative removal of CO-like intermediates.
Lopes et al. tested platinum-rich Pt–Pd alloy catalysts with Pt/Pd atomic ratio 9129 and 326 as cathode material in direct ethanol fuel cells. In both cases, the performance of the cells with Pt–Pd as cathode material was better than that of the cell with Pt. In the presence of ethanol a higher increase in overpotential of ORR was found on pure Pt than on Pt–Pd, indicating the higher ethanol tolerance of the binary catalyst. Tests in DEFC at 60 °C showed that the performance of the cell with Pt–Pd/C is about the same as that of the cell with Pt/C as cathode material. When Pt–Pd/C was used as cathode material at 90 °C an enhancement of the cell performance was observed with respect to the cell with Pt/C, shown in Fig. 10. Considering that at 60 °C the ethanol crossover is negligible, the improvement of DEFC performance at 90 °C was ascribed to the higher ethanol tolerance of Pt–Pd/C. In this case, the enhanced ethanol tolerance of Pt-rich Pt–Pd catalysts can be ascribed mainly to the so-called “ensemble effect”. The ensemble effect occurs where the dilution of the active component with catalytically inert metals by alloying changes the distribution of active sites opening different reaction pathways.130 The dissociative chemisorption of alcohol requires the existence of several adjacent Pt ensembles131,132 and the presence of atoms of the second metal around Pt active sites can block alcohol adsorption on Pt sites due to the dilution effect. Consequently, alcohol oxidation on the binary-component electrocatalyst was suppressed. On the other hand, oxygen adsorption, which can usually be regarded as dissociative chemisorption, requires only two adjacent sites and is not affected by the presence of the second metal. The reduced ethanol adsorption can be also related to the decreased Pt–Pt bond distance and to electronic effects by alloying.
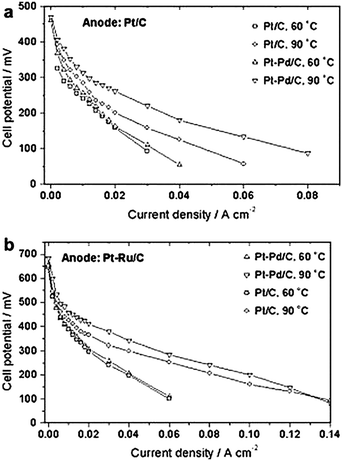 | ||
| Fig. 10 Polarization curves in a single DEFC with Pt–Pd/C and Pt/C electrocatalysts as cathode materials for oxygen reduction at 60/1 atm and 90 °C/3 atm O2 pressure using a 1 M ethanol solution. Cathode Pt loading 1 mg cm−2. (a) Pt/C E-TEK as anode material; (b) Pt–Ru/C (1:1) E-TEK as anode material. Anode Pt loading 1 mg cm−2. Reproduced from ref. 26, ©2008, with permission from Elsevier. | ||
5.4 Activity of Pd, Pd–M (M = Co, Fe, Cu, Ni, Au, …) and Pd–Co–M catalysts for the ORR
As reported by Nørskov et al.133 using density functional theory calculations, Pt and Pd stand out as the metals with the smallest overpotentials for ORR. The activity of pure Pd is slightly lower than that of pure Pt. DFT calculations have shown that Pt alloys with, for example, Ni, Co, Fe, and Cr (where Pt will segregate to the surface) have smaller oxygen binding energies than pure Pt.134,135 In the same way a thermodynamic method using DFT was reported by Fernandez et al.136 to correlate ORR activity of Pd–M alloys with their d-band centers. By rapid scanning electrochemical microscopy (SECM) techniques they found that non-platinum electrocatalyst systems, such as Pd–Ti and Pd–Co–Au attain and overcome ORR activity of Pt. The results of both classical rotating disk electrode (RDE) experiments and the SECM screening technique, used to evaluate the electrocatalytic activity of carbon supported binary catalysts, showed excellent agreement. Pd–Co/C electrodes (10–30% Co) exhibited remarkable activity for ORR catalysis, close to that of carbon supported Pt.137 On this basis Shao et al.138 synthesized Pd2Co/C alloy electrocatalyst nanoparticles and tested ORR activity. The activity of the non-Pt Pd2Co/C was comparable to that of commercial Pt-containing catalysts. The kinetics of ORR on this electrocatalyst predominantly involves a four-electron step reduction with the first electron transfer being the rate-determining step. The downshift of the d-band center of the Pd “skin”, which constitutes the alloy surface due to the strong surface segregation of Pd at elevated temperatures, determined its high ORR activity. In a similar work, the same research group139 correlated the electrocatalytic activity of intrinsic Pd and Pt surfaces and Pd and Pt overlayers on several substrates with their electronic properties, and established the volcano-type dependence of O2 reduction activity on the binding energy of oxygen and the d-band center of the top metal layer. Intrinsic Pd and Pt surfaces bind oxygen too firmly to allow efficient removal of the adsorbed reaction intermediates. Therefore, they do not have the highest activity and are not at the peak of the volcano plot. A Pd overlayer on a Pd3Fe(111) alloy, was predicted to lie at the peak on the volcano plot, and thus, it appears to be the most active catalyst among those investigated. The reason for its good activity is that this overlayer affords a moderate bond with oxygen that balances well the two competing influences, i.e., the first electron transfer or O–O bond-breaking, and the removal of O and OH.Many works report the effect of Co addition on ORR activity of Pd catalysts.116,137,138,140–149 Pd–Co with various Pd:Co atomic ratios were tested for ORR, and in many cases they presented catalytic activity comparable with that of Pt. Different values of the optimum Co content for ORR activity were reported.141–144 The difference in the optimum Pd/Co composition can depend on the different degree of alloying. Indeed, Liu and Manthiram,145 compared ORR activity of Pd–Co catalysts in the Pd:Co atomic ratio of 70:30 with different particle size and degree of alloying, with a controlled crystallite size of about 8 nm, the activity was found to decrease with increasing degree of alloying from 18 to 30 at% Co. Conversely, the highest ORR activity for Pd–Co catalysts (30–40 at% Co) was obtained by Zhang et al.146 for the highest Co content in the alloy, i.e. with the lowest Pd–Pd interatomic distance. Another positive effect of Co presence is the decrease in metal particle size with increasing Co content in the Pd–Co/C catalyst, which occurs independently of the preparation method resulting in an increase of catalyst surface area,143,144 see Fig. 11. Regarding the stability of Pd–Co catalysts, Liu and Manthiram147 observed that annealing at 350 and 500 °C enhanced the stability (durability) of carbon-supported nanostructured Pd4Co alloys significantly, due to an increasing degree of alloying between Pd and Co as well as an increasing particle size. Compared to the single element (Pd and Pt) electrocatalysts, the increase in alloying also partly counteracts the decrease in catalytic activity caused by the increase in particle size during annealing at 350 and 500 °C. The sample annealed at 350 °C exhibited a combination of high catalytic activity for ORR and good stability.
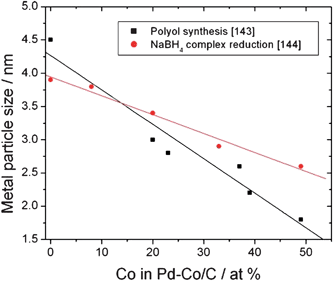 | ||
| Fig. 11 PdCo particle size vs. Co content in the catalysts from data reported in ref. 143 and 144. | ||
The activity for oxygen reduction of ternary Pd–Co-based catalysts has also been investigated. Raghuveer et al.136,150 reported the effect of Au addition to a Pd–Co catalyst. The resulting Pd–Co–Au presented ORR activity comparable or better, depending on the preparation method, than that of a commercial Pt/C catalyst. In a similar work, Raghuveer et al.151 found that a Pd–Co–Mo catalyst with a Pd:Co:Mo atomic ratio of 70:20:10 exhibited higher catalytic activity more like the Pd–Co–Au catalyst than the commercial Pt catalyst, but with excellent chemical stability unlike the Pd–Co–Au catalyst. In the same way, Mathiyarasu and Phani152 investigated ORR activity of ternary Pd–Co–M (M = Pt, Au, Ag) catalysts. Fig. 12 shows ORR polarization curves for carbon supported Pd–Co, Pd–Co–Pt, Pd–Co–Au, Pd–Co–Ag, and Pt catalysts in an oxygen saturated solution of 0.5 M sulfuric acid obtained using a rotating ring disk electrode at 1600 rpm. The Pd–Co–Pt curve shows an approx. 100 mV shift to more positive potentials compared to other Pd alloy systems. When compared to commercial Pt/C, there was not much difference in the half-wave potential value. The activity of Pd–Co–Pt/C is higher than that of the other alloy compositions and equal to the Pt/C electrocatalyst.
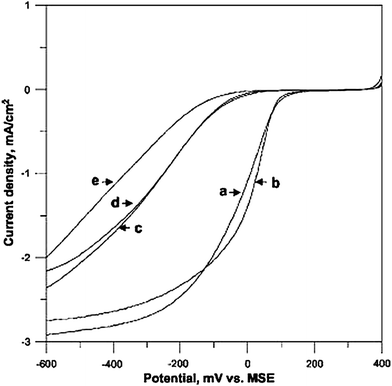 | ||
| Fig. 12 Comparison of polarization curves for ORR on carbon supported (a) Pt/C alloy, (b) Pd–Co–Pt/C, (c) Pd–Co–Ag/C, (d) Pd–Co/C, and (e) Pd–Co–Au/C catalyst in 0.5 M sulfuric acid solution, scan rate 5 mV s−1; rotation rate 1600 rpm; room temperature. Reprinted from ref. 152, ©2007, with permission from The Electrochemical Society. | ||
The ORR activity of Pd–Fe catalysts has also been extensively investigated.153–156 In all cases the highest ORR activity was presented by the catalyst with a Pd/Fe atomic ratio of 3. The ORR activity of these catalysts was higher or comparable to that of Pt. According to Shao et al.153 and Song et al.,154 the increase in ORR activity was ascribed to alloy formation. However, according to Tarasevich et al.,155 the occurrence of iron can influence catalyst activity in two ways (i) through the stabilization of palladium nanoparticles, despite the high temperature of pyrolysis during the synthesis of the catalyst, and (ii) through hindering oxide–palladium formation. Finally, Wang et al.156 investigated the effect of Ir addition on ORR activity of binary Pd–Fe catalysts. Compared to pure Pd/C and Pd3Fe/C, results showed that ORR activity of Pd–Fe–Ir/C was the highest.
Other binary Pd-based catalysts, as Pd–Cu,157 Pd–Ni,158,159 Pd–Cr,158 Pd–P,160 Pd–Au,161 Pd–Se,162 Pd–S,162 Pd–Mo163 and Pd–W164 were tested for ORR in acid medium. Apart from the Pd–S catalyst, ORR activity of these binary catalysts was higher than that of pure Pd and in some case comparable to that of Pt. Fig. 13 shows the current density for ORR at 0.85 V vs. RHE for various Pd–M catalysts (M = Co, Ni and Cr) as a function of the alloy composition. The highest electrocatalytic activity was observed for alloy composition ca. 60 at% Pd in all Pd alloys. The high catalytic activity of these binary Pd-based catalysts is attributed to the formation of Pd–M alloys and to the modification of the palladium surface.
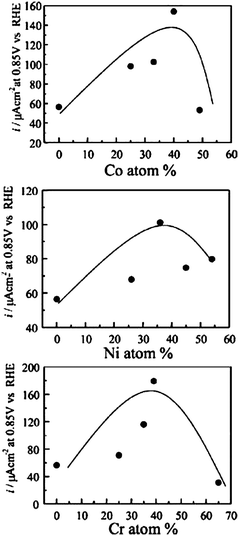 | ||
| Fig. 13 Current density for ORR at 0.85 V vs. RHE for different Pd–M catalysts (M = Co, Ni and Cr) as a function of alloy composition. Reprinted from ref. 158, ©2006, with permission from The Electrochemical Society. | ||
5.5 Activity of Pd and Pd–M catalysts for the ORR in the presence of alcohol
Pd and Pd–M catalysts present higher methanol tolerance than Pt. Indeed, unlike platinum, palladium is inactive for methanol electrooxidation in acid solution.44 Li et al.144,165 compared ORR activity of Pd/C and Pd4Co1/C with that of Pt/C, with and without methanol in the electrolyte. They found that although Pt/C presents superior ORR performance than Pd/C and Pd4Co1/C in pure 0.5 M HClO4 electrolyte; in the methanol containing solution, ORR activity of Pt/C was much lower, while than of Pd/C and Pd4Co1/C was stable.Excellent selectivity for the ORR in the presence of methanol was shown by Pd2Co/C138 and Pd3Fe1/C148 nanoparticles. Lee et al.158 investigated palladium-based alloys, such as Pd–Co, Ni, and Cr, as novel methanol-tolerant oxygen reduction electrocatalysts for direct methanol fuel cells. In the absence of methanol, the order of ORR activity was Pd–Cr > Pd–Co > Pd–Ni > Pd. In the presence of methanol, the order of ORR activity was Pd–Ni > Pd–Co > Pd–Cr > Pd. The Pd–Ni alloy exhibited better ORR activity than Pd–Co and Pd–Cr in the presence of methanol. The ORR activity for Pd and Pd alloys decreased in the presence of methanol compared to the absence of methanol. The current ratio iH2SO4 + CH3OH to iH2SO4, which indicates the effect of methanol for ORR catalytic activity, was ordered: Pd–Ni > Pd–Co > Pd–Cr > Pd. The effect of methanol on Pd and Pd–Cr alloy for decreasing ORR was greater than that on the Pd–Ni and Pd–Co alloys. In general, the rate of ORR depends strongly on the degree of interaction of oxygen molecules with the adsorption sites on the electrocatalyst surface. During the ORR process on an electrocatalyst surface, the oxygen molecules compete strongly with methanol molecules for the adsorption sites in the electrolyte containing methanol. Thus, the decrease in ORR activity observed on Pd and Pd alloys could be caused by the adsorption of methanol molecules on the active sites.
Savadogo and Varela166 compared the ORR activity of Pt, Pd and Pd–Co, in the Pd:Co atomic ratio 85:15 in the presence of ethanol and found that the best performance in an H2SO4 + C2H5OH solution, in terms of selectivity towards ORR, was given by the Pd–Co catalyst. The presence of alcohol had an effect on the Tafel slopes, ORR overpotentials and current densities of the Pt catalyst. Contrarily, ethanol had no negative effect on the Tafel slopes, ORR overpotentials and current densities of the Pd and Pd–Co based catalysts. Although the Pd–Co alloy composition may not be optimum for significantly higher ORR activity, than Pd or Pt, the results presented clearly show that tolerance to ethanol by these catalysts is significant even at concentrations of alcohol as high as 4 M. The selective behaviour of Pd and Pd–Co for the ORR can in part be attributed to a slow rate of adsorption of the alcohol and/or reaction intermediates on the catalytic surface. According to the authors, the high activity of Pd and Pd–Co catalysts for the ORR, as well as their high degree of tolerance to ethanol, make them suitable candidates for direct ethanol fuel cell applications.
6. Conclusions and outlook
6.1 Pt-containing palladium catalysts
A comparison of the catalytic activity of binary palladium–platinum catalysts with that of Pt and other Pt-based binary catalysts is shown in Table 2. Alone Pd presents poor activity for hydrogen oxidation but by adding a low amount of Pt to the Pd, the HOR activity of the resulting Pd-rich Pd–Pt attains that of Pt. Conversely to pure Pd and Pt metals, Pd-rich Pd–Pt catalysts possess high CO tolerance in the H2/CO stream. Also, Pd–Pt binary catalysts are more resistant to sulfur poisoning by sulfur-containing impurities in the fuel than either of the pure metals.167–169 Yasuda and Yoshimura167 observed that the coexistence of Pt and Pd in USY zeolite strongly improved sulfur tolerance dependent on the Pd/Pt ratio, and reached a maximum value at a Pd:Pt molar ratio of 4:1. The high sulfur tolerance of the Pd–Pt system was attributed to structural and electronic effects rather than to the degree of metal dispersion. The strong resistance to poisoning by the sulfur compound of the Pd–Pt catalyst with respect to its monometallic counterparts in the hydrogenation reactions was interpreted by Navarro et al.168 in terms of the electron-deficient platinum species (isolated Pt clusters on the Pd surface) in the resulting PtPd particles.
| Oxidation | Catalyst composition Pt/Pd atomic ratio | Activity |
|---|---|---|
| Hydrogen oxidation | Pt/Pd > 0.05 | PtPd ≈ Pt |
| Hydrogen oxidation, CO presence | Pt/Pd ≤ 1 | PtPd > Pt, PtRu |
| Methanol oxidation, acid media | Pt/Pd = 9,1 | PtRu,PtSn > PtPd ≥ Pt |
| Ethanol oxidation, acid media | Pt/Pd = 3,1 | PtRu,PtSn > PtPd ≥ Pt |
| Formic acid oxidation | n.d. | PtPd > Pt, PtRu |
| Oxygen reduction | Pt/Pd ≥ 3 | PtPd > Pt |
| Oxygen reduction, methanol presence | Pt/Pd = 0.33, 0.43 | PtPd > Pt |
| Oxygen reduction, ethanol presence | Pt/Pd = 3 | PtPd > Pt |
Slightly higher activity for oxygen reduction was observed on Pt-rich Pt–Pd catalysts with Pt/Pd atomic ratio >3 than on pure Pt. Also the activity for oxygen reduction in the presence of alcohol on Pt–Pd is higher than that on Pt. All these results can be explained by PtPd alloy formation. Electronic and geometric effects related to alloy formation reduce CO and alcohols adsorption, resulting in an increase of CO and alcohol tolerance in these catalysts. Also in the case of oxygen reduction, the activity enhancement observed when using Pt–Pd alloy electrocatalysts was ascribed to geometric or electronic factors by alloying.
Pt–Pd has similar activity for methanol oxidation, but higher activity for formic acid oxidation than Pt. In this case, the enhancement in formic acid oxidation by Pd addition to Pt does not correlate with CO tolerance. Within the dual path mechanism of formic acid oxidation, the addition of palladium to Pt enhances the rate of formic acid electrooxidation via the direct CO2 formation channel, without the formation of CO intermediate.
6.2 Platinum-free Pd and Pd–M catalysts
Comparison of the catalytic activity of palladium and palladium alloy catalysts with that of Pt is shown in Table 3. Pd alone presents poor activity for hydrogen oxidation, lower than that of Pt both in pure H2 and in H2/CO. Conversely, the activity of Pd for formic acid oxidation and ethanol and propanol oxidation in alkaline media is higher than that of Pt. The ORR activity of Pd is slightly lower than that of Pt, but by addition of suitable metals, such as Co and Fe, its catalytic activity can overcome than of Pt. In the presence of alcohols, however, all Pd-based catalysts outperformed Pt, so their use as cathode alcohol tolerant materials in DAFCs is recommended.| Reaction | Catalyst composition | Activity |
|---|---|---|
| Hydrogen oxidation | Pd | Pd < Pt |
| Hydrogen oxidation, CO presence | Pd | Pd < Pt |
| Formic acid oxidation | Pd, PdIr, PdAu | PdIr, PdAu > Pd ≥ Pt |
| Methanol oxidation, alkaline media | Pd | Pd < Pt |
| Ethanol oxidation, alkaline media | Pd | Pd > Pt |
| Propanol oxidation, alkaline media | Pd, PdAu | PdAu > Pd > Pt |
| Ethylene glycol, alkaline media | Pd | Pd < Pt |
| Oxygen reduction | Pd, PdM (M = Co, Ni, Se, P, Cu, Fe, Au, Mo, W, Cr), PdCoM (M = Au,Mo) | PdCoM > PdM ≥ Pt > Pd |
| Oxygen reduction, methanol presence | Pd, PdM (M = Co,Fe,Au…) | PtM,Pd > Pt |
| Oxygen reduction, ethanol presence | Pd, Pd85Co15 | Pd85Co15 > Pd > Pt |
In summary, Pd-rich Pd–Pt catalysts with very low Pt content can substitute Pt and Pt–Ru as anode catalyst in PEMFC with H2 and H2/CO as the fuel. This finding opens up a new avenue to develop potentially less expensive electrocatalysts. Comparison of PEMFC performances demonstrate the principal possibility of partial replacement of Pt by Pd on the hydrogen electrode of PEMFC. Binary Pd–M and ternary Pd–Co–M catalysts, in particular Pd–Co–Au and Pd–Co–Mo, are promising materials when used as cathode material in fuel cells, particularly those fuelled with alcohol. Indeed, these catalysts possess a similar/higher ORR than Pt, but a considerably higher alcohol tolerance than Pt. In this case Pd could fully substitute Pt as cathode material in fuel cells. So, on the basis of the PEMFC power densities reported in ref. 40 for Pt and Pd–Pt (as anode materials), and calculated from the steady-state polarization curves in ref. 151 for Pt and Pt–Co–Mo (as cathode materials), in the reasonable hypothesis that (a) anode and cathode performances are independent and (b) the relative performance of anode and cathode are independent of fuel cell operating conditions, a PEMFC fuel cell with a Pd-rich Pd–Pt (5 at% Pt) catalyst as anode material and a Pt-free Pd–Co–Mo (75:20:05) catalyst as cathode material, i.e. a “quasi platinum-free fuel cell”, should attain a higher performance than a fuel cell with Pt as anode and cathode material, see Fig. 14.
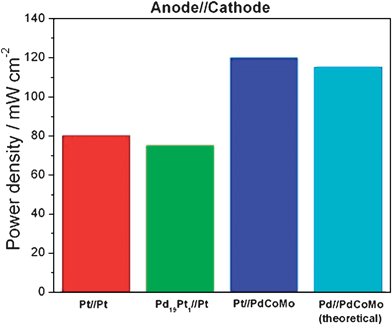 | ||
| Fig. 14 Experimental power density of PEMFCs with (1) Pt/C as anode and cathode materials,40,151 (2) Pd19Pt1/C as anode and Pt/C as cathode40 and (3) Pt/C as anode and Pd–Co–Mo/C as cathode,151 and (4) theoretical power density of a PEMFC with Pd19Pt1/C as anode and Pd–Co–Mo/C as cathode materials. | ||
Generally, Pd-based catalysts present larger particle size than Pt-based catalysts. As a consequence they have a lower active surface area than Pt-based catalysts, which decreases their catalytic activity. So, a target in Pd-based catalyst research is the development of synthesis methods which lead to high surface area Pd. Incidentally, the addition of a second metal, such as Co and Fe, to Pd decreased metal particle size.143,144,153 Future work should look also into nanostructured materials, such as Pd nanowire arrays,55 in view of their high stability and high surface area.
Pure Pd is a suitable material for use in direct formic acid fuel cells and in alkaline fuel cells fuelled with ethanol and propanol, but our understanding of this type of fuel cell has to be improved. Studies are currently in progress to further explore direct formic acid fuel cells and direct alcohol alkaline fuel cell systems.
References
- S. Litster and G. McLean, J. Power Sources, 2004, 130, 61 CrossRef CAS.
- E. Antolini, J. Appl. Electrochem., 2004, 34, 563 CrossRef CAS.
- C. Lamy, E. M. Belgsir and J.-M. Léger, J. Appl. Electrochem., 2001, 31, 799 CrossRef CAS.
- E. Peled, T. Duvdevani, A. Aharon and A. Melman, Electrochem. Solid-State Lett., 2001, 4, A38 CrossRef CAS.
- A. Hamnett, Catal. Today, 1997, 38, 445 CrossRef CAS.
- E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani, E. S. Smotkin and T. E. Mallouk, Science, 1998, 280, 1735 CrossRef CAS.
- J. Wang, S. Wasmus and R. F. Savinell, J. Electrochem. Soc., 1995, 142, 4218 CAS.
- M. Gotz and H. Wendt, Electrochim. Acta, 1998, 43, 3637 CrossRef CAS.
- W. Zhou, Z. Zhou, S. Song, W. Li, G. Sun, P. Tsiakaras and Q. Xin, Appl. Catal., B, 2003, 46, 273 CrossRef CAS.
- E. Antolini, Mater. Chem. Phys., 2003, 78, 563 CrossRef CAS.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. McBreen, J. Electrochem. Soc., 1995, 142, 1409 CAS.
- V. Jalan and E. J. J. Taylor, J. Electrochem. Soc., 1983, 130, 2299 CAS.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750 CrossRef CAS.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef CAS.
- P. Strasser, Q. Fan, M. Devenney, W. H. Weinberg, P. Liu and J. K. Nørskov, J. Phys. Chem. B, 2003, 107, 11013 CrossRef CAS.
- J. S. Cooper and P. J. McGinn, J. Power Sources, 2006, 163, 330 CrossRef CAS.
- C. Rice, S. Ha, R. I. Masel and A. Wieckowski, J. Power Sources, 2003, 115, 229 CrossRef CAS.
- E. Antolini, J. Power Sources, 2007, 170, 1 CrossRef CAS.
- C. Lamy, S. Rousseau, E. M. Belgsir, C. Coutanceau and J.-M. Léger, Electrochim. Acta, 2004, 49, 3901 CrossRef CAS.
- S. Wasmus and A. Kuver, J. Electroanal. Chem., 1999, 461, 14 CrossRef CAS.
- B. Park and H. Lee, J. Mater. Res., 1999, 14, 281 CrossRef CAS.
- F. R. de Boer, R. Boom, W. C. M. Mattens, A. R. Miedeama and A. K. Niessen, Cohesion in Metals: Transition Metal Alloys, Elsevier, Amsterdam, 1988 Search PubMed.
- W. B. Pearson, The Crystal Chemistry and Physics of Metals and Alloys, Wiley, New York, 1972 Search PubMed.
- H. Li, G. Sun, N. Li, S. Sun, D. Su and Q. Xin, J. Phys. Chem. C, 2007, 111, 5605 CrossRef CAS.
- H. Li, Q. Xin, W. Li, Z. Zhou, L. Jiang, S. Yang and G. Sun, Chem. Commun., 2004, 2776 RSC.
- T. Lopes, E. Antolini and E. R. Gonzalez, Int. J. Hydrogen Energy, 2008, 33, 5563 CrossRef CAS.
- A. C. Garcia, V. A. Paganin and E. A. Ticianelli, Electrochim. Acta, 2008, 53, 4309 CrossRef CAS.
- J. B. Darby and K. M. Miles, Metal Trans., 1972, 3, 653 Search PubMed.
- F. J. Kuijers, B. M. Tieman and V. Ponec, Surf. Sci., 1978, 75, 657 CrossRef CAS.
- J. L. Rousset, A. J. Renouprez and A. M. Cadrot, Phys. Rev. B, 1998, 58, 2150 CrossRef CAS.
- C. Massen, T. V. Mortimer-Jones and R. L. Johnston, J. Chem. Soc., Dalton Trans., 2002, 4375 RSC.
- L. C. A. van den Oetelaar, O. W. Nooij, S. Oerlemans, A. W. D. van der Gon, H. H. Brongersma, L. Lefferts, A. G. Roosenbrand and J. A. R. van Veen, J. Phys. Chem. B, 1998, 102, 3445 CrossRef CAS.
- O. L. J. Gijzeman, Appl. Surf. Sci., 1993, 64, 9 CrossRef CAS.
- S. A. Grigoriev, E. K. Lyutikova, S. Martemianov and V. N. Fateev, Int. J. Hydrogen Energy, 2007, 32, 4438 CrossRef CAS.
- Z. Liu, L. Hong, M. P. Tham, T. H. Lim and H. Jiang, J. Power Sources, 2006, 161, 831 CrossRef CAS.
- W. Wang, Q. Huang, J. Liu, Z. Zou, Z. Li and H. Yang, Electrochem. Commun., 2008, 10, 1396 CrossRef CAS.
- J. B. Joo, Y. J. Kim, W. Kim, N. D. Kim, P. Kim, Y. Kim and J. Yi, Korean J. Chem. Eng., 2008, 25, 770 Search PubMed.
- J. Yang, J. Y. Lee, Q. Zhang, W. Zhou and Z. L. Yang, J. Electrochem. Soc., 2008, 155, B776 CrossRef CAS.
- P. Stonehart, Int. J. Hydrogen Energy, 1984, 9, 921 CrossRef CAS.
- Y.-H. Cho, B. Choi, Y.-H. Cho, H.-S. Park and Y.-E. Sung, Electrochem. Commun., 2007, 9, 378 CrossRef CAS.
- D. C. Papageorgopoulos, M. Keijzer, J. B. J. Veldhuis and F. A. de Bruijn, J. Electrochem. Soc., 2002, 149, A1400 CrossRef CAS.
- A. Gamez, D. Richard and P. Gallezot, in Resume des Journees d'Etude Piles a Combustible, Paris, France, 1999, p. 401 Search PubMed.
- C. He, H. R. Kunz and J. M. Fenton, J. Electrochem. Soc., 1997, 144, 970 CrossRef.
- J. W. Long, R. M. Stroud, K. E. Swider-Lyons and D. R. Rolison, J. Phys. Chem. B, 2000, 104, 9772 CrossRef CAS.
- T. Takeguchi, Y. Anzai, R. Kikuchi, K. Eguchi and W. Ueda, J. Electrochem. Soc., 2007, 154, B1132 CrossRef CAS.
- E. N. Muhamad, T. Takeguchi, G. Wang, Y. Anzai and W. Ueda, J. Electrochem. Soc., 2009, 156, B32 CrossRef CAS.
- A. K. Vijh, J. Catal., 1975, 37, 410 CrossRef CAS.
- V. S. Bagotzky, Yu. B. Vassiliev and O. A. Khazova, J. Electroanal. Chem., 1977, 81, 229 CrossRef.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 44, 239 CrossRef CAS.
- F. Kardigan, B. Beden, J. M. Leger and C. Lamy, J. Electroanal. Chem., 1981, 125, 89 CrossRef.
- V. S. Bagotzky and Yu. B. Vassiliev, Electrochim. Acta, 1967, 12, 1323 CrossRef.
- T. Biegler, J. Phys. Chem., 1968, 7, 1571 CrossRef.
- Y. Xu and X. Lin, J. Power Sources, 2007, 170, 13 CrossRef CAS.
- V. Selvaraj, M. Alagar and I. Hamerton, J. Power Sources, 2006, 160, 940 CrossRef CAS.
- H. Wang, C. Xu, F. Cheng, M. Zhang, S. Wang and S. P. Jiang, Electrochem. Commun., 2008, 10, 1575 CrossRef CAS.
- W. Zhou, Z. Zhou, S. Song, W. Li, G. Sun, P. Tsiakaras and Q. Xin, Appl. Catal., B, 2003, 46, 273 CrossRef CAS.
- C. Rice, S. Ha, R. I. Masel, P. Waszczuk, A. Wieckowski and T. Barnard, J. Power Sources, 2002, 111, 83 CrossRef CAS.
- S. Ha, B. Adams and R. I. Masel, J. Power Sources, 2004, 128, 119 CrossRef CAS.
- Y. Zhu, S. Ha and R. I. Masel, J. Power Sources, 2004, 130, 8 CrossRef CAS.
- X. Wang, J. M. Hu and I. M. Hsing, J. Electroanal. Chem., 2004, 562, 73 CrossRef CAS.
- A. V. Tripkovic, K. Dj. Popovic, R. M. Stevanovic, R. Socha and A. Kowal, Electrochem. Commun., 2006, 8, 1492 CrossRef CAS.
- Y. W. Rhee, S. Ha, C. Rice and R. I. Masel, J. Power Sources, 2003, 117, 35 CrossRef CAS.
- J. Jiang and A. Kucernak, J. Electroanal. Chem., 2002, 520, 64 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 45, 205 CrossRef CAS.
- S. Park, Y. Xie and M. J. Weaver, Langmuir, 2002, 18, 5792 CrossRef CAS.
- J. D. Lovic, A. V. Tripkovic, S. Lj. Gojkovic, K. Dj. Popovic, D. V. Tripkovic, P. Olszewski and A. Kowal, J. Electroanal. Chem., 2005, 581, 294 CrossRef CAS.
- D. Capon and R. Parsons, J. Electroanal. Chem., 1975, 65, 285 CrossRef.
- G.-Q. Lu, A. Crown and A. Wieckowski, J. Phys. Chem. B, 1999, 103, 9700 CrossRef CAS.
- M. Baldauf and D. M. Kolb, J. Phys. Chem., 1996, 100, 11375 CrossRef CAS.
- P. Waszczuk, T. M. Barnard, C. Rice, R. I. Masel and A. Wieckowski, Electrochem. Commun., 2002, 4, 599 CrossRef CAS.
- M. Zhao, C. Rice, R. I. Masel, P. Waszczuk and A. Wieckowski, J. Electrochem. Soc., 2004, 151, A131 CrossRef CAS.
- R. Larsen and R. I. Masel, Electrochem. Solid-State Lett., 2004, 7, A148 CrossRef CAS.
- X. Li and I.-M. Hsing, Electrochim. Acta, 2006, 51, 3477 CrossRef CAS.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Phys. Chem. Chem. Phys., 2003, 5, 4242 RSC.
- R. S. Jayashree, J. S. Spendelow, J. Yeom, C. Rastogi, M. A. Shannon and P. J. A. Kenis, Electrochim. Acta, 2005, 50, 4674 CrossRef CAS.
- Y. Zhu, Z. Khan and R. I. Masel, J. Power Sources, 2005, 139, 15 CrossRef.
- S. Ha, R. Larsen and R. I. Masel, J. Power Sources, 2005, 144, 28 CrossRef CAS.
- R. Larsen, S. Ha, J. Zakzeski and R. I. Masel, J. Power Sources, 2006, 157, 78 CrossRef CAS.
- X. Wang, Y. Tang, Y. Gao and T. Lu, J. Power Sources, 2008, 175, 784 CrossRef CAS.
- R. Wang, S. Liao and S. Ji, J. Power Sources, 2008, 180, 205 CrossRef CAS.
- R. Larsen, J. Zakzeski and R. I. Masel, Electrochem. Solid-State Lett., 2005, 8, A291 CrossRef CAS.
- J. Ge, Y. Zhang, C. Liu, T. Lu, J. Liao and W. Xing, J. Phys. Chem. C, 2008, 112, 17214 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovic, B. N. Grgur, B. Blizanac, P. N. Ross and N. M. Markovic, Electrochim. Acta, 2002, 47, 3707 CrossRef CAS.
- A. Verma and S. Basu, J. Power Sources, 2005, 145, 282 CrossRef CAS.
- E. H. Yu and K. Scott, J. Power Sources, 2004, 137, 248 CrossRef.
- K. Scott, E. Yu, G. Vlachogiannopoulos, M. Shivare and N. Duteanu, J. Power Sources, 2008, 175, 452 CrossRef CAS.
- C. Coutanceau, L. Demarconnay, C. Lamy and J.-M. Léger, J. Power Sources, 2006, 156, 14 CrossRef CAS.
- J. R. Varcoe, R. C. T. Slade, E. Lam How Yee, S. D. Poynton and D. J. Driscoll, J. Power Sources, 2007, 173, 194 CrossRef CAS.
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Power Sources, 2005, 150, 27 CrossRef CAS.
- K. Asazawa, K. Yamada, H. Tanaka, A. Oka, M. Taniguchi and T. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 8024 CrossRef CAS.
- P. T. A. Sumodjo, E. J. da Silva and T. Rabockai, J. Electroanal. Chem., 1989, 271, 305 CrossRef CAS.
- C. Xu, L. Cheng, P. K. Shen and Y. Liu, Electrochem. Commun., 2007, 9, 997 CrossRef CAS.
- C. Xu, Y. Liu and D. Yuan, Int. J. Electrochem. Sci., 2007, 2, 674 Search PubMed.
- J. Liu, J. Ye, C. Xu, S. P. Jiang and Y. Tong, J. Power Sources, 2008, 177, 67 CrossRef CAS.
- J. Ye, J. Liu, C. Xu, S. P. Jiang and Y. Tong, Electrochem. Commun., 2007, 9, 2760 CrossRef CAS.
- C. Xu, Z. Tian, Z. Chen and S. P. Jiang, Electrochem. Commun., 2008, 10, 246 CrossRef CAS.
- P. K. Shen and C. Xu, Electrochem. Commun., 2006, 8, 184 CrossRef CAS.
- C. Xu, P. K. Shen and Y. Liu, J. Power Sources, 2007, 164, 527 CrossRef CAS.
- C. Xu, Z. Tian, P. K. Shen and S. P. Jiang, Electrochim. Acta, 2008, 53, 2610 CrossRef CAS.
- Y. Chen, L. Zhuang and J. Lu, Chinese J. Catal., 2007, 28, 870 Search PubMed.
- H. Wang, C. Xu, F. Cheng and S. Jiang, Electrochem. Commun., 2007, 9, 1212 CrossRef CAS.
- S. Gottesfeld and T. A. Zawodzinski, Polymer Electrolyte Fuel Cells in Advances in Electrochemical Science and Engineering, (ed.) R. C. Alkire, H. Gerischer, D. M. Kolb, C. W. Tobias, Wiley-VCH, Weinheim, 1st edn , 1997, Vol. 5, p. 195 Search PubMed.
- B. C. Beard and P. N. Ross, J. Electrochem. Soc., 1986, 133, 1839 CrossRef CAS.
- M. T. Paffett, J. G. Berry and S. Gottesfeld, J. Electrochem. Soc., 1988, 135, 1431 CrossRef CAS.
- B. C. Beard and P. N. Ross, J. Electrochem. Soc., 1990, 137, 3368 CAS.
- S. Mukerjee and S. Srinivasan, J. Electroanal. Chem., 1993, 357, 201 CrossRef CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659 CAS.
- M. Min, J. Cho, K. Cho and H. Kim, Electrochim. Acta, 2000, 45, 4211 CrossRef CAS.
- U. A. Paulus, A. Wokaun, G. G. Scherer, T. J. Schmidt, V. Stamenkovic, N. M. Markovic and P. N. Ross, J. Phys. Chem. B, 2002, 106, 4181 CrossRef CAS.
- E. Antolini, R. R. Passos and E. A. Ticianelli, Electrochim. Acta, 2002, 48, 263 CrossRef CAS.
- T. J. Schmidt, V. Stamenkovic, M. Arenz, N. M. Markovic and P. N. Ross Jr., Electrochim. Acta, 2002, 47, 3765 CrossRef CAS.
- J. Zhang, Y. Mo, M. B. Vukmirovic, R. Klie, K. Sasaki and R. R. Adzic, J. Phys. Chem. B, 2004, 108, 10955 CrossRef CAS.
- J. Zhang, Y. Mo, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2131.
- A. U. Nilekar, Y. Xu, J. Zhang, M. B. Vukmirovic, K. Sasaki, R. R. Adzic and M. Mavrikakis, Top. Catal., 2007, 46, 276 CrossRef CAS.
- O. Savadogo, K. Lee, K. Oishi, S. Mitsushima, N. Kamiya and K.-I. Ota, Electrochem. Commun., 2004, 6, 105 CrossRef CAS.
- O. Savadogo, K. Lee, S. Mitsushima, N. Kamiya and K.-I. Ota, J. New Mat. Electrochem. Syst., 2004, 7, 77 Search PubMed.
- B. Gurau and E. S. Smotkin, J. Power Sources, 2002, 112, 3339.
- P. M. Urban, A. Funke, J. T. Muller, M. Himmen and A. Docter, Appl. Catal., A, 2001, 221, 459 CrossRef CAS.
- A. Heinzel and V. M. Barragan, J. Power Sources, 1999, 84, 70 CrossRef CAS.
- K. Ramya and K. S. Dhathathreyan, J. Electroanal. Chem., 2003, 542, 109 CrossRef CAS.
- J. Cruickshank and K. Scott, J. Power Sources, 1998, 70, 40 CrossRef CAS.
- S. Song, W. Zhou, Z. Liang, R. Cai, G. Sun, Q. Xin, V. Stergiopoulos and P. Tsiakaras, Appl. Catal., B, 2005, 55, 65 CrossRef CAS.
- G. Andreadis and P. Tsiakaras, Chem. Eng. Sci., 2006, 61, 7497 CrossRef CAS.
- S. Song, G. Wang, W. Zhou, X. Zhao, G. Sun, Q. Xin, S. Kontou and P. Tsiakaras, J. Power Sources, 2005, 140, 103 CrossRef CAS.
- E. Antolini, T. Lopes and E. R. Gonzalez, J. Alloys Compd., 2008, 461, 253 CrossRef CAS.
- S. Guerin, B. E. Hayden, C. E. Lee, C. Mormiche and A. E. Russell, J. Phys. Chem. B, 2006, 110, 14355 CrossRef CAS.
- H. Ye and R. M. Crooks, J. Am. Chem. Soc., 2007, 129, 3627 CrossRef CAS.
- T. Lopes, E. Antolini and E. R. Gonzalez. 211th ECS Meeting. 2007 May 6–10, Chicago, Illinois Search PubMed.
- N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 2002, 45, 121 CrossRef CAS.
- C. Lamy, A. Lima, V. Le Rhun, C. Coutanceau and J. M. Leger, J. Power Sources, 2002, 105, 283 CrossRef CAS.
- H. A. Gasteiger, N. M. Markovic, P. N. Ross and E. J. Cairns, Electrochim. Acta, 1994, 39, 1825 CrossRef CAS.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B, 1999, 59, 15900.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120, 10240 CrossRef CAS.
- J. L. Fernandez, V. Raghuveer, A. Manthiram and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 13100 CrossRef CAS.
- J. L. Fernandez, D. A. Walsh and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 357 CrossRef CAS.
- M. H. Shao, T. Huang, P. Liu, J. Zhang, K. Sasaki, M. B. Vukmirovic and R. R. Adzic, Langmuir, 2006, 22, 10409 CrossRef CAS.
- M. Shao, P. Liu, J. Zhang and R. R. Adzic, J. Phys. Chem. B, 2007, 111, 6772 CrossRef CAS.
- J. L. Fernández, J. M. White, Y. Sun, W. Tang, G. Henkelman and A. J. Bard, Langmuir, 2006, 22, 10426 CrossRef CAS.
- W. E. Mustain, K. Kepler and J. Prakash, Electrochim. Acta, 2007, 52, 2102 CrossRef CAS.
- W. E. Mustain and J. Prakash, J. Power Sources, 2007, 170, 28 CrossRef CAS.
- W. Wang, D. Zheng, C. Du, Z. Zou, X. Zhang, B. Xia, H. Yang and D. L. Akins, J. Power Sources, 2007, 167, 243 CrossRef CAS.
- X. Li, Q. Huang, Z. Zou, B. Xia and H. Yang, Electrochim. Acta, 2008, 53, 6662 CrossRef CAS.
- H. Liu and A. Manthiram, Electrochem. Commun., 2008, 10, 740 CrossRef CAS.
- L. Zhang, K. Lee and J. Zhang, Electrochim. Acta, 2007, 52, 7964 CrossRef CAS.
- H. Liu and A. Manthiram, Energy Environ. Sci., 2009, 2, 124–132 RSC.
- L. Zhang, K. Lee and J. Zhang, Electrochim. Acta, 2007, 52, 3088 CrossRef CAS.
- V. Di Noto, E. Negro, S. Lavina, S. Gross and G. Pace, Electrochim. Acta, 2007, 53, 1604 CrossRef CAS.
- V. Raghuveer, P. J. Ferreira and A. Manthiram, Electrochem. Commun., 2006, 8, 807 CrossRef CAS.
- V. Raghuveer, A. Manthiram and A. J. Bard, J. Phys. Chem. B, 2005, 109, 22909 CrossRef CAS.
- J. Mathiyarasu and K. L. N. Phani, J. Electrochem. Soc., 2007, 154, B1100 CrossRef CAS.
- M.-H. Shao, K. Sasaki and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 3526 CrossRef CAS.
- S. Song, Y. Wang, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2008, 78, 381 CrossRef CAS.
- M. R. Tarasevich, G. V. Zhutaeva, V. A. Bogdanovskaya, M. V. Radina, M. R. Ehrenburg and A. E. Chalykh, Electrochim. Acta, 2007, 52, 5108 CrossRef CAS.
- R. Wang, S. Liao, Z. Fu and S. Ji, Electrochem. Commun., 2008, 10, 523 CrossRef CAS.
- X. Wang, N. Kariuki, J. T. Vaughey, J. Goodpaster, R. Kumar and D. J. Myers, J. Electrochem. Soc., 2008, 155, B602 CrossRef CAS.
- K. Lee, O. Savadogo, A. Ishihara, S. Mitsushima, N. Kamiya and K. Ota, J. Electrochem. Soc., 2006, 153, A20 CrossRef CAS.
- G. Ramos-Sánchez, H. Yee-Madeira and O. Solorza-Feria, Int. J. Hydrogen Energy, 2008, 33, 3596 CrossRef CAS.
- L. Cheng, Z. Zhang, W. Niu, G. Xu and L. Zhu, J. Power Sources, 2008, 182, 91 CrossRef CAS.
- M. Nie, P. K. Shen and Z. Wei, J. Power Sources, 2007, 167, 69 CrossRef CAS.
- A. A. Serov, S.-Y. Cho, S. Han, M. Min, G. Chai, K. H. Nam and C. Kwak, Electrochem. Commun., 2007, 9, 2041 CrossRef CAS.
- A. Sarkar, A. Vadivel Murugan and A. Manthiram, J. Phys. Chem. C, 2008, 112, 12037–12043 CrossRef CAS.
- A. Sarkar, A. Vadivel Murugan and A. Manthiram, J. Mater. Chem., 2009, 19(1), 159–165 RSC.
- H. Li, G. Sun, Q. Jiang, M. Zhu, S. Sun and Q. Xin, J. Power Sources, 2007, 172, 641 CrossRef CAS.
- O. Savadogo and F. J. R. Varela, J. New Mater. Electrochem. Syst.., 2008, 11, 69 Search PubMed.
- H. Yosuda and Y. Yoshimura, Catal. Lett., 1997, 46, 43 CrossRef CAS.
- R. M. Navarro, B. Pawelec, J. M. Trejo, R. Mariscal and J. L. G. Fierro, J. Catal., 2000, 189, 184 CrossRef CAS.
- A. Stanislaus and B. H. Cooper, Catal. Rev. Sci. Eng., 1994, 36, 75 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |