CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method
Takayuki
Abe
*,
Masaaki
Tanizawa
,
Kuniaki
Watanabe
and
Akira
Taguchi
Hydrogen Isotope Research Center, University of Toyama, 3190 Gofuku, Toyama, 930-8555, Japan. E-mail: tabe@ctg.u-toyama.ac.jp; Fax: +81-76-445-6931; Tel: +81-76-445-6933
First published on 14th January 2009
Abstract
Highly dispersed Ru nanoparticles loaded on a TiO2 support (Ru/TiO2(B)), which affects the hydrogenation of CO2 to CH4 (methanation), were prepared by employing a “dry” modification method using a barrel-sputtering instrument. The loaded Ru nanoparticles exhibited a narrow particle-size distribution with a mean diameter of ca. 2.5 nm. Methanation of CO2 on the Ru/TiO2(B) catalyst produced a 100% yield at ca. 160 °C, which is more than 200 °C below that required for Ru/TiO2 prepared by a conventional “wet” impregnation method. In addition, the methanation reaction over Ru/TiO2(B) proceeded at temperatures as low as room temperature with a reaction rate of 0.04 µmol min−1 g−1.
Broader contextCO2 is a recognized as a major cause of the so-called greenhouse effect. Catalytic approaches for CO2 fixation could play an important role, because CO2 can be artificially converted into reusable chemicals. Among the catalytic reactions of CO2, hydrogenation to form CH4 (methanation), which can recycle CO2 to a reusable fuel, is one candidate for such a fixation process.A barrel-sputtering method (cf. J. Phys. Chem. C, 2007, 112, 1479) allowed us to fabricate highly dispersed Ru nanoparticles on a TiO2 support by a “dry” impregnation method. The obtained Ru/TiO2 catalyst can efficiently carry out CO2 methanation with 100% yield at ca. 160 °C, which is more than 200 °C below that required for Ru/TiO2 prepared by a conventional “wet” impregnation method. Moreover, the Ru/TiO2 catalyst proceeds the methanation at temperatures as low as room temperature with a reaction rate of 0.04 µmol min−1 g−1. While there are some problems with the methanation reaction for practical use, such as hydrogen supply, the remarkable low temperature reaction could be of interest in CO2 recycling process where cheap and large amounts of by-product hydrogen can be utilized in a narrow as possible area (called ‘CO2 local recycling system’). |
Introduction
Carbon dioxide (CO2) is a recognized as a major factor responsible for the so-called greenhouse effect, the ever-increasing amounts of CO2 in the atmosphere are causing climate changes on a global scale.1 Therefore, a reduction in CO2 emission into the atmosphere is an urgent necessity. However, the levels of CO2 are increasing with increasing in the world's population and improvement in living standards. As a result, the control of CO2 emission by means of regulations such as those represented by the Kyoto Protocol, may no longer be feasible. Although various physical and chemical techniques have been proposed for the fixation of exhausted CO2, such as fixation in carbonates, geological or ocean storage, or afforestation,1 their immediate practical application has drawbacks in terms of economic factors, safety, efficiency, and reliability.Catalytic approaches for CO2 fixation can play an important role, because CO2 can be artificially converted into reusable chemicals.1 Among the catalytic reactions of CO2, hydrogenation of CO2, the so-called methanation reaction, is a suitable technique for the fixation of CO2; this technique can be used to convert exhausted CO2 into methane (CH4), which can be recycled for use as a fuel or a chemical.1e–g Temperatures in the range of 300–400 °C are generally applied to this reaction,2 and additional CO2 is generated for producing the energy required to maintain such temperatures. To overcome this drawback, a catalyst capable of affecting the methanation reaction at lower temperatures is required. Grätzel et al. reported the formation of CH4 over a Ru/TiO2 catalyst prepared by means of a wet process. They stated that the reaction started from room temperature at atmospheric pressure.3 However, Melsheimer et al. stated that the catalyst was characterized by a critical preparation technique.4 We also prepared samples under the prepared conditions described by Grätzel et al., and then we examined the physical and methanation properties of the samples. The results of our experiments are described later in this paper. Mechanical milling of a mixture of a transition metal and an oxide material under CO2 and H2 atmosphere at 80 kPa leads to the formation of CH4 at temperatures below 100 °C;5 however, the use of mechanical devices and the maintenance of a high-pressure environment leads to the generation of additional CO2.
Nanoparticles loaded on various supports exhibit high catalytic activity for several reactions.6 A number of “wet” techniques, including the sol-gel method, liquid precipitation, and colloidal micelles, have been developed for the production of uniformly sized nanoparticles of metals dispersed on powdery supports.6b,c However, in these techniques, heating is required for loading of the prepared nanoparticles onto the powdery supports, and the heating induces growth of the nanoparticles.
We have recently developed a “dry” technique for modifying the surfaces of powdery materials; this technique, which we have named “barrel-sputtering”, allows the fabrication of highly dispersed naked metal nanoparticles on a powdery support without any heating.7 In the present study, we attempted to load Ru nanoparticles onto a TiO2 support by the barrel-sputtering method, and studied the phyisical and catalytic properies of the prepaerd samples.
Experimental
Sample preparation
Titanium dioxide (TiO2 (anatase), ST-41, obtained from Ishihara Sangyo Kaisha, Ltd., Osaka, Japan) was used as a support after being dried overnight in an oven at 180 °C. Metallic Ru plates (purity; 99.9%, 50 × 100 mm2) were used as the sputtering target. Sputter deposition of the metal nanoparticles on the support was conducted by the barrel-sputtering equipment shown in Fig. 1.7a,b A hexagonal barrel that could be swung using a motor drive was placed in a vacuum chamber to hold a powdery support. The support loaded into the barrel was lifted up to a certain level along the wall of the barrel by swinging the barrel; the powdery support subsequently fell down along the barrel wall by its own weight and struck the wall. As a result, it was effectively stirred during the sputter deposition. The angle of the target stage could be freely varied to face the support, thereby allowing the sputter deposition efficiency to be enhanced. | ||
| Fig. 1 Schematic representation of the hexagonal barrel-sputtering system. | ||
In this study, 3.0 g of TiO2 powder was loaded into the barrel. The vacuum chamber was carefully evacuated up to 8.0 × 10−4 Pa, and then high-purity argon gas (purity: 99.995%) was gradually introduced into the chamber until the pressure increased up to 0.8 Pa. Radio-frequency (13.56 MHz) magnetron sputtering was performed for 25 min with an input power of 100 W, leading to a deposition of Ru on each particle surface in the powder. The amount of Ru loaded onto a support was adjusted to be approximately 0.8 wt% by controlling the sputtering duration. During sputtering, a swinging motion of ±75° at a speed of 4.2 rpm was provided to the hexagonal barrel. After sputtering, N2 gas (99.9998%) was gradually introduced to increase the pressure up to 1 atmosphere, whereupon the prepared samples could be extracted. The prepared TiO2 supported Ru, denoted as Ru/TiO2(B), was used as the catalyst for the methanation reaction without any pretreatment.
In order to allow the growth of Ru nanoparticles, samples were prepared by thermal treatment of Ru/TiO2(B) at 500, 700 or 800 °C under H2 flow (30 mL min−1) for 3 h: these samples are denoted as Ru/TiO2(B500), Ru/TiO2(B700), and Ru/TiO2(B800), respectively.
For comparison purposes, TiO2 supported Ru catalysts were prepared by a conventional wet method. The TiO2 supported Ru catalyst, denoted as Ru/TiO2(W), was prepared by the incipient wetness method. TiO2 (5.0 g) was added to a solution of RuCl3·3H2O (Acros, 0.1293 g) in 5 ml of water. The resultant slurry was dried at 80 °C for 16 h and then calcined in air at 350 °C for 8 h. The sample was reduced under flowing H2 (30.0 ml min−1) at 400 °C for 3 h in the reactor and subsequently used in the catalytic reaction without contact with the air.
In addition, the TiO2 supported Ru catalyst was prepared by following the Grätzel method.3a The obtained sample is denoted as Ru/TiO2(G). Prior to the catalysis, Ru/TiO2(G) was reduced under flowing H2 (3.0 mL/min−1) at 220 °C for 1 h in the reactor.
Characterization
Characterization of the prepared sample was carried out by means of X-ray diffraction (XRD; Philips, PW1825/00), inductive coupling plasma analysis (ICP; PerkinElmer, Optima 3300XL) and transmission electron microscopy (TEM; JEOL, JEM-2100). Furthermore, X-ray photoelectron spectroscopy (XPS) measurements were conducted (XPS; Shimadzu/Kratos, AXIS-HSX).Catalysis
The catalytic reaction was performed under atmospheric pressure in a fixed-bed flow reactor made of quartz and with an inner diameter of 6 mm. The reactor was heated in a furnace, and two thermocouples (one located in the catalyst in the fixed bed and the other located outside the quartz reactor) were used to control the temperature. Catalyst powder (1.0 g) was placed in the reactor; Ru/TiO2(G) was diluted by adding unloaded TiO2 in order to control the amount of Ru of 0.80 wt%. The reaction gas mixture consisting of CO2 and H2 in a molecular ratio of 1 : 4 was mixed with Ar and passed at a given feed rate (F/W). The reaction effluent was analyzed by using an online-gas chromatograph equipped with a thermal conductivity detector and an active carbon column. Subsequently, the feed gas was switched to pure Ar, and the temperature of the catalyst was increased at a rate of 3.0 °C min−1. This sequence was repeated starting from room temperature and then by increasing the temperature in steps of 10, 20 40 or 50 °C. In the range of temperatures examined, the products obtained were CH4 and H2O. The reaction effluent was separately analyzed by quadrupole mass spectroscopy (ANELVA, M-100QA): this proved that hydrocarbons other than CH4 were not produced.The catalytic activity of Ru/TiO2 was also evaluated through a batch test by using the same reactor by stopping the flow of reactant gas for a given time and by maintaining a certain temperature.
The methane yield and turnover number (TONCH4) are defined as
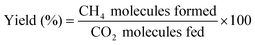
Here, NRu,atoms,cat. denotes the total number of Ru atoms loaded on the surface of 1.0 g of catalyst (see below).
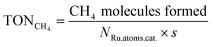
Results and discussion
Characterization
From the ICP measurments, the amount of Ru loaded on Ru/TiO2(B), Ru/TiO2(W) and Ru/TiO2(G) were determined to be 0.80, 0.75, and 3.60 wt%, respectively.The prepared Ru/TiO2catalysts were subjected to physical characterization. XRD measurements on Ru/TiO2(W) (shown in Fig. 2(C)) showed the presence of diffraction signals attributed to metallic Ru (JCPDS 06-0663) at 42.15 and 44.00° (CuKα), along with diffraction signals for TiO2 with the anatase structure (JCPDS 83-2243, Fig. 2(A)). Although definitive diffraction signals assignable to metallic Ru were observed in the XRD pattern of Ru/TiO2(G) (Fig. 2(D)), the signals were slightly broader than those of Ru/TiO2(W). Signals corresponding to Ru oxide were not observed. In the case of Ru/TiO2(B), on the other hand, only signals corresponding to TiO2 were obtained (Fig. 2(B)). However, XPS studies on Ru/TiO2(B) showed a signal with a binding energy of 279.5 eV; no signal attributed to Ru oxides4,8 was observed, indicating that metallic Ru was present on the TiO2.
 | ||
| Fig. 2 X-Ray diffraction patterns of (A) unsupported TiO2, (B) Ru/TiO2(B), (C) Ru/TiO2(W) and (D) Ru/TiO2(G). | ||
Fig. 3(A) , (B) and (C) show TEM images of Ru/TiO2(B), Ru/TiO2(W) and Ru/TiO2(G), respectively. In the case of Ru/TiO2(B), it can be observed that small dots were highly dispersed on the TiO2 supports. The XPS results suggest that these dots consist of metallic nanoparticles of Ru. The particle-size distribution, which is calculated from more than 100 nanoparticles on Ru/TiO2(B), is overlaid in Fig. 3(A). The sizes of the nanoparticles were distributed in the range 0.7–7.5 nm, and more than 80% of the nanoparticles were smaller than 3.0 nm. The mean particle size (D) was calculated to be about 2.5 nm (Table 1). The TEM images of Ru/TiO2 prepared by the wet method (Ru/TiO2(W)) are considerably different from those of Ru/TiO2(B). The sizes of metallic Ru particles on Ru/TiO2(W) are distributed over a wider range (1.4–29.0 nm, D = 9.5 nm). For Ru/TiO2(G), the particle size distribution and the mean particle size are found to be in the range 0.9–30.6 nm and 5.2 nm, respectively. The mean particle size of Ru/TiO2(G) is smaller than that of Ru/TiO2(W), however, it is apparent that Ru nanoparticles in Ru/TiO2(G) are more agglomerated as compared to those in Ru/TiO2(W). It can be observed that the barrel-sputtering method is advantageous, particularly for loading highly dispersed nanoparticles on a support. The absence of large particles of Ru for Ru/TiO2(B) explains the absence of Ru signals in the XRD measurements.7c
| Metal content [wt%] | Sample treatment | Mean particle size (D)/nm | Surface area of Ru (S.A.Ru.cat.)/m2 g-cat.−1 | Onset temperature of CH4 formation/°C | TONCH4 at 160 °C/s−1 | |
|---|---|---|---|---|---|---|
| a Value at F/W of 1.16 mL g−1 s−1. | ||||||
| Ru/TiO2(B) | 0.80 | as-prepared | 2.5 | 1.55 | 60 | 1.5 × 10−2 |
| 80a | 4.6 × 10−3a | |||||
| Ru/TiO2(B500) | 0.80 | 500 °C, 3h (H2) | 3.4 | 1.14 | 80 | 8.5 × 10−3 |
| Ru/TiO2(B700) | 0.80 | 700 °C, 3h (H2) | 5.0 | 0.77 | 100 | 4.8 × 10−3 |
| Ru/TiO2(B800) | 0.80 | 800 °C, 3h (H2) | 6.4 | 0.60 | 140 | 3.2 × 10−4 |
| Ru/TiO2(W) | 0.75 | 400 °C, 3h (H2) | 9.5 | 0.38 | 120 | 1.0 × 10−3 |
| RuTiO2(G) | 0.76 | 220 °C, 1 h (H2) diluted | 5.2 | 0.71 | 80 | 4.9 × 10−3 |
 | ||
| Fig. 3 TEM images of TiO2 supported Ru nanoparticles. (A) Ru/TiO2(B), (B) Ru/TiO2(W), (C) Ru/TiO2(G), (D) Ru/TiO2(B500), (E) Ru/TiO2(B700), (F) Ru/TiO2(B800) and (G) Ru/TiO2(B) after use in methanation reaction at 180 °C for one week. The particle size distribution obtained for more than 100 nanoparticles of Ru is overlaid. | ||
TEM images of the samples of Ru/TiO2(B500), Ru/TiO2(B700), and Ru/TiO2(B800) are shown in Fig. 3(D), (E) and (F), respectively. The sizes of the Ru nanoparticles increased with the treatment temperature, and the mean nanoparticle sizes of the samples were estimated to be 3.4, 5.0, and 6.4 nm, respectively (Table 1).
From the mean particle size of the Ru nanoparticles (D), the number of Ru atoms on the nanoparticle surface can be calculated. The weight (WRu,nanoparticle in g) and surface area (S.A.Ru,nanoparticle in m2) of an Ru nanoparticle are expressed as follows:
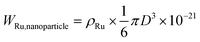
| S.A.Ru,nanoparticle = πD2 × 10−18 |
Here, the symbol ρRu denotes the density of Ru (12.41 g cm−3). The number of Ru nanoparticles in 1.0 g of the catalyst (NRu,nanoparticles in number) can be expressed in terms of the amount of Ru supported, Wcat., in wt%, as follows:

Thus, the total surface area of Ru nanoparticles in 1.0 g of catalyst (S.A.Ru,cat. in m2) is calculated as follows:
| S.A.Ru,cat. = NRu,nanoparticles × S.A.Ru,nanoparticle |
The surface density of Ru atoms in a closed-packing model, which corresponds to the number of Ru atoms in the (001) plane of the unit cell of a Ru hexagonal structure, is 0.1739 × 1020 (in number per m2, a0 = 0.27058 nm). Hence, the total number of Ru atoms on the surface of 1.0 g of catalyst (NRu,atom,cat. in number) can be expressed as follows:
| NRu,atom,cat. = S.A.cat. × 0.1739 × 1020 |
The values of NRu,atom,cat. in Ru/TiO2catalysts are listed in Table 1.
Methanation activity
Fig. 4(A) shows the relationship between the reaction temperature and the yield of CH4 over Ru/TiO2catalysts at a reaction-gas feed rate (F/W) of 0.24 mL g−1 s−1. With Ru/TiO2(W) pretreated at 400 °C for 3 h in H2, the formation of CH4 was observed at temperatures above 120 °C, where the yield of CH4 was 0.3% at 120 °C. The yield of CH4 increased with the reaction temperature and became quantitative at 380 °C. On the other hand, CH4 was formed over Ru/TiO2(B) at temperatures above 60 °C, where the yield was 0.3% at 60 °C even without any pretreatment: remarkably, the onset temperature for CH4 formation was reduced by about 60 °C as compared with the case of Ru/TiO2(W). The yield of CH4 increased with the reaction temperature, and it reached a 100% yield at 160 °C, which was about 220 °C less than the corresponding temperature for Ru/TiO2(W). Ru/TiO2(G) exhibited a lower onset temperature for methane formation (80 °C) and a lower complete reaction temperature (240 °C) as compared to those exhibited by Ru/TiO2(W). However, these values were still higher than those of Ru/TiO2(B). The turnover numbers (TONCH4) at 160 °C were calculated to be 1.5 × 10−2, 1.0 × 10−3 and 4.9 × 10−3 s−1 for Ru/TiO2(B), Ru/TiO2(W), and Ru/TiO2(G), respectively, as shown in Table 1. These data demonstrate that the catalytic activity of Ru/TiO2(B) was significantly higher than those of Ru/TiO2(W) and RuTiO2(G). It should be noted that in Fig 4(A), the yield of CH4 was found to be higher than 100%. However, the yield of methane gradually decreased and reached 100 ± 3% within 30 min, as shown in the time course of methanation by Ru/TiO2(B) at 180 °C shown in Fig. 4(B).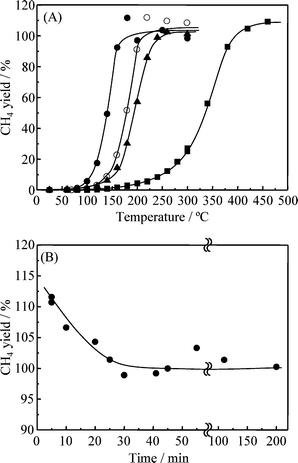 | ||
| Fig. 4 (A) Temperature dependence of the methane yield at methanation of CO2 over TiO2 supported Ru nanoparticles; (●) Ru/TiO2(B), (▲) Ru/TiO2(G), (■) Ru/TiO2(W) and (○) Ru/TiO2(B) diluted with unsupported TiO2. (B) Change of the methane yield as a function of reaction time in very early range at 180 °C. | ||
A study of the effect of F/W for Ru/TiO2(B) (Fig. 5) showed that an increase in F/W caused a slight increase in the onset temperature of the reaction and the temperature required for 100% yield. When the F/W was set at 1.16 mL g−1 s−1, the former and latter temperatures were 80 °C and 210 °C, respectively; these temperatures were still significantly lower than those for Ru/TiO2(W).
 | ||
| Fig. 5 The effect of F/W for Ru/TiO2(B) on methanation of CO2 as a function of reaction temperature; (○) 0.17, (●) 0.24, (△) 0.48 and (▲) 1.16 ml g−1 s−1, respectively. The reaction curve at F/W of 0.24 mL g−1 s−1 is the same with that in Fig. 4(A). | ||
A batch test was conducted to obtain a more accurate value of the onset temperature for CH4 formation (Fig. 6). In the case of Ru/TiO2(B) at 25 °C (room temperature), the formation of CH4 was clearly observed 5 min after the feed gas was introduced into the reactor. Since the amount of CH4 generated increased lineally with the reaction time, the rate of generation divided by the weight of the catalyst was calculated to be 0.04 µmol min−1 g−1. For Ru/TiO2(B) at 40 °C, the rate of generation was 0.11 µmol min−1 g−1, which was about three times that at room temperature. As for Ru/TiO2(G), on the other hand, the formation of CH4 was observed 60 min after the gas was introduced. The rate of CH4 generation was estimated to be 0.002 µmol min−1 g−1 at room temperature, and 0.01 µmol min−1 g−1 at 40 °C. In the case of Ru/TiO2(W) at room temperature or 40 °C, CH4 generation was not observed even 1 h after the feed gas was introduced: at 80 °C, 0.05 µmol min−1 g−1 of CH4 was observed.
 | ||
| Fig. 6 Change of the amount of methane as a function of the reaction time by a batch-type reaction; (●) Ru/TiO2(B) at 25 °C (room temperature), (○) Ru/TiO2(B) at 40 °C, (▲) Ru/TiO2(G) at 25 °C and (△) Ru/TiO2(G) at 40 °C, respectively. | ||
On the basis of the physical properties discussed above, the considerable difference in methanation activity between Ru/TiO2(B), Ru/TiO2(W) and Ru/TiO2(G) is considered from the viewpoint of the surface area of Ru nanoparticles (S.A.Ru.cat.). As listed in Table 1, the S.A.Ru,cat. values for Ru/TiO2(B), and Ru/TiO2(W) differ by a factor of 4.1. Therefore, we measured the dependency of the CH4 yield on the reaction temperature when the amount of Ru/TiO2(B) catalyst in the reactor was reduced to one quarter of the previous amount, as shown in Fig. 4(A). It should be noted that the total powder volume in the reactor was adjusted by adding unloaded TiO2. The formation of CH4 was observed at above 80 °C, and a 100% yield was attained at 210 °C. These temperatures were higher than those for Ru/TiO2(B), but the amount of CH4 formed at 160 °C remained about 20 times that formed on Ru/TiO2(W). Although the S.A.Ru,cat. values for Ru/TiO2(B) and Ru/TiO2(G) differ by a factor of about 2.2, the reaction curve of Ru/TiO2(B) catalyst reduced to one quarter is still higher than that of Ru/TiO2(G). These results indicated that the high catalytic activity of Ru/TiO2(B) cannot be attributed solely to the S.A.Ru,cat., and that another factor is involved. To clarify the effect of the size of nanoparticles, the catalytic activities of Ru/TiO2(B500), Ru/TiO2(B700), and Ru/TiO2(B800) were tested, as shown in Fig. 7(A). The same reaction curves of Ru/TiO2(B), Ru/TiO2(W), and Ru/TiO2(G) shown in Fig. 4(A) are included for comparison. The onset temperature for CH4 formation increased from 60 °C for Ru/TiO2(B) up to 140 °C for Ru/TiO2(B800). The temperature of 100% yield increased from 160 °C for Ru/TiO2(B) to 380 °C for Ru/TiO2(B800). In addition, the TONCH4 at 160 °C decreased from 1.5 × 10−2 s−1 for Ru/TiO2(B) to 3.2 × 10−4 for Ru/TiO2(B800) (Table 1). The catalytic activity of Ru/TiO2(B800) was almost the same as that of Ru/TiO2(W), and the activity of Ru/TiO2(B700) was similar to that of Ru/TiO2(G). These results strongly suggest that the methanation activity depends on the particle size of the loaded Ru nanoparticles (Table 1). Fig. 7(B) shows the dependencies of the onset temperature and TONCH4 on the mean particle diameters. The onset temperature decreased linearly with the particle diameter in the range of 2.5–6.0 nm, but remained almost constant at about 120 °C for particle diameters greater than about 6 nm. The particle-size dependency of the TONCH4 was a mirror image of that of the onset temperature. These results indicate that the remarkable reduction in the onset temperature and the higher activity of Ru/TiO2(B) can be attributed to the presence of Ru nanoparticles smaller than about 6.0 nm. In the case of Ru/TiO2(G), the particle size is distributed in a wide range of about 1–30 nm, showing a presence of nanoparticles smaller than about 6.0 nm (Fig. 3(C)). This unusual phenomenon in Ru/TiO2(G) is attributed to the predominant contribution of Ru nanoparticles smaller than 6 nm.
 | ||
| Fig. 7 (A) Change of the methane yield depends on the mean particle diameter as a function of reaction temperature; (○) Ru/TiO2(B500) with 3.4 nm, (△) Ru/TiO2(B700) with 5.0 nm and (□)Ru/TiO2(B800) with 6.4 nm. The reaction curves for (●) Ru/TiO2(B) with 2.5 nm, (▲) Ru/TiO2(G) with 5.2 nm and (■) Ru/TiO2(W) with 9.5 nm, the same as Fig. 4(A), are also included. (B) Relationships between (●) the onset temperature of methane formation or (○) TONCH4 at 160 °C and the mean particle diameter of Ru on TiO2. | ||
The activation energies of Ru/TiO2(W) and Ru/TiO2(G) for the reaction were calculated to be 44.5 and 59.8 kJ mol−1 from an Arrhenius plot of the reaction curve shown in Fig. 4(A). In the case of Ru/TiO2(B), however, two distinct activation energies were identified at temperatures in the range of 60–100 °C and 120–160 °C, and their values were estimated to be 76.1 and 59.2 kJ mol−1, respectively, suggesting that the reaction mechanism for Ru/TiO2(B) in the lower temperature range is different from that for Ru/TiO2(W) and Ru/TiO2(G). Furthermore, the frequency factor for Ru/TiO2(B) is about two orders of magnitude greater than that for Ru/TiO2(W). The most likely explanation for this is that the frequency factor is related to the dispersibility of Ru nanoparticles on TiO2. That is, because methanation is an exothermic reaction, the heat of the reaction generated on a certain nanoparticle is probably transmitted to neighboring particles and it accelerates subsequent reactions on these particles. However, a more detailed study aimed at the clarification of the mechanism is in progress.
It has been shown that Ru/TiO2(B) is a stable catalyst for the reactions at 80 and 140 °C for 24 h, and 180 °C for one week (Fig. 8). The catalytic activity remained constant at each temperature. TEM studies conducted on the catalyst used at 180 °C revealed no change in the size or size distribution of Ru nanoparticles as compared with the state of the as-prepared sample (Fig. 3(G) and (A)), indicating that Ru/TiO2(B) is sufficiently stable for long-term use in reactions.
 | ||
| Fig. 8 Time course of methane yield by hydrogenation of CO2 over Ru/TiO2(B) at (●) 180 °C, (▲) 140 °C and (■) 80 °C. | ||
Conclusions
The CO2 methanation reaction on Ru/TiO2(B) prepared by the barrel-sputtering method, by which highly dispersed Ru nanoparticles were deposited on the TiO2 support, produced a 100% yield of CH4 at 160 °C, which was significantly lower than that required in the case of Ru/TiO2(W) and Ru/TiO2(G). In addition, the methanation reaction over Ru/TiO2(B) proceeded at temperatures as low as room temperature with a reaction rate of 0.04 µmol min−1 g−1.We notice that there are two major problems of practical application of Ru/TiO2(B). The first problem is the fact that methane is one of the gases which promotes the greenhouse effect. However, the combination of the catalytic formation of methane from CO2 exhausted and the reuse of the formed methane can control the emission of CO2 into the atmosphere. The second problem is the manner in which hydrogen is obtained without CO2 being newly exhausted. The use of the by-product hydrogen obtained in ironworks and petrochemical plants appears to be a realistic solution to the problem. Thus, the CO2 recycling process by the Ru/TiO2(B) catalyst should be performed in an area as narrow as possible; the recycling system (called a ‘CO2 local recycling system’) should be situated at the place where cheap and large amounts of by-product hydrogen can be utilized. In addition, the use of Ru/TiO2(B) might be useful in confined spaces that can not obtain sufficient energy, for example, in spaceships and space stations.
References
- (a) T. Sakakura, J. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (b) R. Zevenhoven, S. Eloneva and S. Teir, Catal. Today, 2006, 115, 73 CrossRef CAS; (c) G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636 CrossRef CAS; (d) G. A. Olah, A. Goeppert and G. K. S. Parakashed, in Beyond Oil and Gas: The Methanol Economy, Wiley-VCH GmbH & Co. KGaA, Weinheim, 1st edn., 2006, ch. 12, pp. 209 Search PubMed; (e) K. Hashimoto, M. Yamasaki, S. Meguro, T. Sasaki, H. Katagiri, K. Izumiya, N. Kumagai, H. Habazaki, E. Akiyama and K. Asami, Corros. Sci., 2002, 44, 371 CrossRef CAS; (f) H. Arakawa et al., Chem. Rev., 2001, 101, 953 CrossRef CAS; (g) X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305 CrossRef.
- (a) P. J. Lunde and F. L. Kester, J. Catal., 1973, 30, 423 CrossRef CAS; (b) D. J. Dwyer and G. A. Somorjai, J. Catal., 1978, 52, 291 CrossRef CAS; (c) F. Solymosi, A. Erdöhelyi and M. Kocsis, J. Chem. Soc., Faraday Trans., 1981, 77, 1003 Search PubMed; (d) E. Zağli and J. L. Falconer, J. Catal., 1981, 69, 1 CrossRef CAS; (e) G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1982, 77, 460 CrossRef CAS; (f) A. Erdöhely, M. Pásztor and F. Solymosi, J. Catal., 1986, 98, 166 CrossRef CAS; (g) Z. Zhang, A. Kladi and X. E. Verykios, J. Catal., 1994, 148, 737 CrossRef CAS.
- (a) K. Ravindranathan Thampi, J. Kiwi and M. Grätzel, Nature, 1987, 327, 506 CrossRef CAS; (b) J. G. Highfieled, P. Ruterana, K. R. Thampi and M. Graetzel, Stud. Surf. Sci. Catal., 1989, 48, 469; (c) P. Ruterana, P.-A. Buffat, K. R. Thampi and M. Graetzel, Ultramicroscopy, 1990, 34, 66 CrossRef CAS.
- J. Melsheimer, W. Guo, D. Ziegler, M. Wesemann and R. Schlögl, Catal. Lett., 1991, 11, 157 CrossRef CAS.
- S. Mori, W.-C. Xu, T. Ishidzuki, N. Ogasawara, J. Imai and K. Kobayashi, Appl. Catal., A, 1996, 137, 255 CrossRef CAS.
- (a) M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175 CrossRef CAS; (b) J. D. Aiken and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 CrossRef; (c) M. Che and C. O. Bennett, Adv. Catal., 1998, 36, 55.
- (a) T. Abe, S. Akamaru and K. Watanabe, J. Alloys Compd., 2004, 377, 194 CrossRef CAS; (b) T. Abe, S. Akamaru, K. Watanabe and Y. Honda, J. Alloys Compd., 2005, 402, 227 CrossRef CAS; (c) M. Inoue, H. Shingen, T. Kitami, S. Akamaru, A. Taguchi, Y. Kawamoto, A. Tada, K. Ohtawa, K. Ohba, M. Matsuyama, K. Watanabe, I. Tsubone and T. Abe, J. Phys. Chem. C, 2007, 112, 1479.
- C. Elmasides, D. I. Kondarides, W. Grünert and X. E. Verykios, J. Phys. Chem. B, 1999, 103, 5227 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |