A 13C NMR study of the carbon dioxide absorption and desorption equilibria by aqueous 2-aminoethanol and N-methyl-substituted 2-aminoethanol
Francesco
Barzagli
a,
Fabrizio
Mani
*a and
Maurizio
Peruzzini
b
aUniversity of Florence, Department of Chemistry, via della Lastruccia, 3, 50019, Sesto Fiorentino, Firenze, Italy. E-mail: fabrizio.mani@unifi.it
bICCOM CNR, via Madonna del Piano, 10, 50019, Sesto Fiorentino, Firenze, Italy
First published on 28th January 2009
Abstract
The 13C NMR experimental study presented investigates the absorption of CO2 by a series of primary, secondary and tertiary alkanolamines in aqueous solution. The absorption experiments were made at room temperature with four different amine concentrations in the range 0.167–0.667 M (1.01–5.88 wt%). As inferred by 13C NMR spectral analysis, the formation of carbamate increases with increasing amine concentration following the order secondary amine < primary amine. Moreover, it has been shown that carbamate reduces the CO2 absorption efficiency. A considerable physical absorption (10–20%) contributes to the loading capacity of the amines and partially compensates for the yield of chemical capture, which turned out to be poorer than was expected theoretically. Quite unexpectedly, carbamate was also produced by an endothermic reaction during the thermal CO2 desorption process which regenerated the amines (primary and secondary amines). In the case of the secondary amine 2-(methylamino)ethanol (MMEA), the amount of carbamate at the end of the desorption process is greater than the amount found at the end of the absorption step, thus reducing the desorption efficiency of the secondary amine in comparison to both primary and tertiary amines. Five cycles of absorption–desorption tests were carried out to verify the feasibility of regenerated amines for reuse. Our results indicate that absorption efficiency and loading capacity of the regenerated amine solutions remain essentially constant during the second to the fifth absorption–desorption experiments, but they both decrease slightly when compared to the initial amine.
Broader contextCarbon dioxide capture and sequestration (CCS) is of paramount importance to help control global climate changes due to anthropic CO2 emissions in the atmosphere. Alkanolamines are currently applied on industrial scale for CO2 uptake. In order to maximize the efficiency of the capture and to reduce the energy demand of the process, it is mandatory to rationalise the reactions occurring in both absorption and desorption processes. We apply 13C NMR spectroscopy to quantify the chemical species produced in the CO2/amine/H2O system. As a result of this study, we demonstrate that the formation of primary and secondary amine carbamates, in both absorption and desorption CCS steps, reduces their CO2 loading capacity and desorption efficiency in comparison to those of tertiary amines that can not give any carbamate. Such an effect is only partially balanced by a significant physical CO2 uptake. |
Introduction
The mitigation of global climate changes requires us to improve the efficiency of CO2 capture and sequestration (CCS),1 and to lower the amount of energy necessary for the process. Therefore, the ambitious goal of reducing the 1990 level2 of the European greenhouse gas emissions by 20% could become more reachable. Among the proposed technologies for chemical capture, a number of different aqueous alkanolamines have been studied for application on an industrial scale for CO2 absorption from natural gas extraction, gas refinery and exhaust gases produced by combustion of fossil fuels.3 Although CCS processes using alkanolamines are considered the most efficient and feasible technique, the energy demand for CO2 desorption and amine regeneration for reuse is still too high,4 thus lowering the net balance CO2(captured)–CO2(emitted).5 In order to maximize the efficiency of the alkanolamines CO2 capture and, consequently, lower the energy demand of the process, it is necessary to have a better understanding of the reactions occurring in both the absorption and the desorption processes through the identification of the chemical species produced in the CO2/amine/H2O system. 13C NMR spectroscopic analysis is the only direct experimental tool able to identify the species that are produced by the reaction of CO2 with different amines. Moreover, this experimental technique gives us the chance to evaluate the amount of the different carbon-containing compounds forming and equilibrating to each other during the reaction.Despite its practical importance and high potential, 13C NMR spectroscopy has scarcely been used to quantify the reactions occurring in different CO2/amine/H2O systems:6 quite surprisingly, it has never been applied to investigate the CO2 desorption processes, in spite of the crucial importance of the amine regeneration step in the CCS process. On this matter, we have previously reported in the 13C NMR speciation study of NH2CO2−, HCO3− and CO32− ions originating in the CO2/NH3/H2O system.7
In the present study we have applied our method7 to investigate the absorption and desorption equilibria taking place when CO2 is reacted with different aqueous alkanolamines such as 2-aminoethanol (monoethanolamine, MEA), 2-(methylamino)ethanol (N-methylethanolamine, MMEA) and 2-(dimethylamino)ethanol (N,N-dimethylaminoethanol, DMMEA), and also to provide a quantitative analysis of the distribution of the species in the different solutions. Noticeably, MEA, the less expensive of alkanolamines, has been the traditional absorbent for CO2 removal from flue gas stream as its reaction is fast even at low CO2 pressure. The main disadvantages that limit the generalized applicability of MEA as CCS agent, are the high heat of the reaction with CO2 and the relatively low absorption capacity caused by the formation of carbamate as the main reaction product when highly concentrated amine (20–30 wt%) solutions are used.8
The objective of our study is to compare the data resulting from the 13C NMR analysis to those obtained from the absorption–desorption experiments of CO2 with aqueous alkanolamines. This comparison will allow us (i) to quantify the main reactions occurring in the CO2/amine/H2O systems and (ii) to establish a correlation between the absorbent solutions and several parameters, such as CO2 absorption loading, capture efficiency and stripping capacity. The replacement of MEA with other sterically hindered alkanolamines has also been taken into consideration for the chemical absorption of CO2 from flue gas.9,10 In this context, the effect of both the methyl substitutes on the amine N-atom and of the amine concentration on the distribution of the chemical species forming in the absorption and desorption processes has also been taken into account.
The results we report show that the capture of CO2 by aqueous amines is not as straightforward as previously thought, and also show that the generally accepted CO2 desorption mechanism is an oversimplification of the real process. As a final note, our study demonstrates that the use of dilute solutions of amines (1–1.5 wt%) in the regenerative absorption–desorption processes could reduce some of the disadvantages which hold back the diffusion of alkanolamines as CO2 absorbents, leaving their efficiency unaltered. A deeper understanding of the reaction mechanism, via an in-depth study of the speciation in solution, could make it easier for us to determine if, and which, other amines could be better as CO2 absorbents.
Results and discussion
Absorption of CO2 by aqueous MEA, MMEA and DMMEA
The absorption experiments summarized in Table 1 were carried out at 20 °C and atmospheric pressure with aqueous amines at concentrations 0.167 M (MEA, 1.01 wt%; MMEA, 1.24 wt%; DMMEA, 1.47 wt%), 0.334 M (MEA, 2.02 wt%; MMEA, 2.48 wt%; DMMEA, 2.94 wt%), 0.500 M (MEA, 3.03 wt%; MMEA, 3.72 wt%; DMMEA, 4.41 wt%), and 0.667 M (MEA, 4.04 wt%; MMEA, 4.96 wt%; DMMEA, 5.88 wt%). In every absorption experiment the amine solution was in a thermostatted home-built glass absorber – see Experimental Section – but not circulated, while the CO2/N2 gas mixture simulating the flue gas would continuously flow at the bottom of the absorber through a sintered-glass diffuser. The outlet gas was dried and purified before being GC-analysed. Samples of the solution (0.5 mL) were taken from the absorbent solution at increasing steps of CO2 absorption and checked by 13C NMR analysis. Changes of pH were also measured during the CO2 absorption processes, showing a progressive acidification of the working solution with values decreasing from 12.25–12.86 to 8.98–9.28 for CO2 loaded solutions. We aimed to compare the effects of the concentration of different amines on both CO2 loading capacity and absorption efficiency. The duration of each absorption experiment was therefore increased from one hour for the less concentrated to four hours for the more concentrated solutions, so that the overall CO2(flowed)/amine(starting) molar ratios remained basically unchanged between 1.15 and 1.30. With these experimental conditions, the CO2 loading capacity – expressed as molar ratio CO2(absorbed)/amine(starting) – ranges from 0.733 (0.167 M MEA) to 0.990 (0.167 M DMMEA), while the absorption efficiency, [CO2(absorbed)/CO2(flowed)]·100, ranges between 63.3% (0.667 M MEA) to 78.1% (0.167 M MMEA). Both the absorption efficiency (except for 0.167 M MMEA) and the loading capacity increase in the order MEA < MMEA < DMMEA and, for MEA and MMEA, with decreasing amine concentration. On the contrary, the absorption efficiency of DMMEA was not affected by the change in the concentration. These solutions presented a relatively low absorption efficiency (<80%) due to the CO2(flowed)/amine(starting) ratio (>1) at the end of every absorption experiment. As a matter of fact, the reduction of the CO2(flowed)/amine(starting) molar ratio to 0.6–0.7, causes an augmentation in the efficiency of CO2 absorption to more than 90%, at the expense of the loading capacity, which is reduced to 0.53–0.65.| AMH conc. (mol dm−3) | AMH/H2O (wt%) | CO2 (abs.) | CO2/AMH (mol/mol) | CO2 Physical absorption (%) | CO2(abs.) | CO2/AMH (mol/mol) | CO2(abs.) | CO2/AMH (mol/mol) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t (min)a | (%) | t (min)b | (%) | t (min)c | (%) | |||||||
| a Absorption time which makes an ideal compromise between loading capacity and absorption efficiency. b Absorption time necessary for having the best absorption efficiency. c Absorption time for having the best loading capacity. | ||||||||||||
| MEA | 0.167 | 1.01 | 60 | 72.1 | 0.818 | 6.1 | 30 | 93.8 | 0.532 | 120 | 41.8 | 0.910 |
| 0.334 | 2.02 | 120 | 66.8 | 0.798 | 9.0 | 60 | 90.4 | 0.540 | ||||
| 0.500 | 3.03 | 180 | 63.9 | 0.751 | 6.5 | 90 | 90.7 | 0.533 | ||||
| 0.667 | 4.04 | 240 | 63.3 | 0.733 | 7.8 | 120 | 92.4 | 0.535 | ||||
| MMEA | 0.167 | 1.24 | 60 | 78.1 | 0.918 | 9.6 | 30 | 95.7 | 0.562 | 90 | 53.5 | 0.993 |
| 0.334 | 2.48 | 120 | 72.8 | 0.900 | 14.6 | 60 | 94.6 | 0.585 | ||||
| 0.500 | 3.72 | 180 | 71.7 | 0.897 | 12.8 | 90 | 95.9 | 0.600 | ||||
| 0.667 | 4.96 | 240 | 69.5 | 0.867 | 9.2 | 120 | 95.6 | 0.596 | ||||
| DMMEA | 0.167 | 1.47 | 60 | 72.7 | 0.990 | 17.3 | 30 | 95.2 | 0.648 | 90 | 53.3 | 1.036 |
| 0.334 | 2.94 | 120 | 73.1 | 0.968 | 17.9 | 60 | 95.4 | 0.632 | ||||
| 0.500 | 4.41 | 180 | 73.7 | 0.941 | 15.4 | 90 | 96.3 | 0.615 | ||||
| 0.667 | 5.88 | 240 | 73.2 | 0.921 | 13.0 | 120 | 96.3 | 0.606 | ||||
Inspection of Table 1 shows that the maximum loading capacity occurred after 1.5–2.0 h of CO2 absorption by 0.167 M amine solutions [CO2(flowed)/amine(starting) in the range 1.86 and 2.17] with values of 0.910 for MEA, 0.993 for MMEA, and 1.036 for DMMEA. On the other hand, the CO2 absorption efficiency of the same solutions decreases to values in the range 42–54% (Table 1 and Fig. 1).
![Average CO2 loading [CO2(absorbed)/amine] (), and average absorption efficiency [CO2(absorbed)/CO2(flowed)] () at increasing steps (time in minutes) for 0.167 M solutions of the different alkanolamines. Estimated errors are less than 2%.](/image/article/2009/EE/b814670e/b814670e-f1.gif) | ||
Fig. 1 Average CO2 loading [CO2(absorbed)/amine] (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), and average absorption efficiency [CO2(absorbed)/CO2(flowed)] ( ), and average absorption efficiency [CO2(absorbed)/CO2(flowed)] (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) at increasing steps (time in minutes) for 0.167 M solutions of the different alkanolamines. Estimated errors are less than 2%. ) at increasing steps (time in minutes) for 0.167 M solutions of the different alkanolamines. Estimated errors are less than 2%. | ||
The apparently anomalous loading capacity of DMMEA, i.e. greater than the limiting value 1, is due to the physical absorption of CO2 which contributes to the overall CO2 capture (see later). The purpose of a CO2 capture process is clearly not to maximize the loading capacity at the expense of a low absorption efficiency, or vice-versa, but it is, instead, to obtain the best compromise between these two objectives. In order to rationalize the results obtained from the absorption steps with different amines tested at various concentrations, we have compared, taking into account the reactions occurring in the CO2/amine/H2O systems, the absorption experiments summarized in Table 1 with the 13C NMR analysis data of the absorbing solutions.
Although the reaction of CO2 with aqueous primary or secondary amines is characterized by several equilibria, a simplification of the system may be drawn from the analysis of the main reactions that describe the CO2/amine/H2O system as shown in eqn (1)–(7).
| AMH + CO2 + H2O ⇌ HCO3− + AMH2+ | (1) |
| HCO3− + AMH ⇌ AMCO2− + H2O | (2) |
| HCO3− + AMH ⇌ CO32− + AMH2+ | (3) |
| AMCO2− + CO2 + 2H2O ⇌ 2HCO3− + AMH2+ | (4) |
| CO2 + CO32− + H2O ⇌ 2HCO3− | (5) |
Reaction (1) and (2) and, respectively, (1) and (3) can be rewritten as
| 2AMH + CO2 ⇌ AMCO2− + AMH2+ | (6) |
| 2AMH + CO2 + H2O ⇌ CO32− + 2AMH2+ | (7) |
The solutions' composition mainly depends on the ratio between the free amine and the absorbed CO2. Both eqn (2) and (3) require an excess of amine as regard to the CO2 absorbed and, consequently, the concentrations of both AMCO2− and CO32− ions are expected to decrease when CO2 absorption increases. In contrast, a simultaneous increase in the HCO3− concentration may be anticipated, according to eqn (1), (4) and (5) and, at the end of each absorption experiment, the equilibrium (1) prevails and bicarbonate and protonated amine become the prevalent species in solution.
In order to evaluate the distribution of the species in the solution, we have recorded a series of 13C NMR spectra on several solutions at increasing CO2 loading. The 13C NMR spectra of the CO2 loaded solutions of MEA and MMEA are simple, displaying two (MEA) or three (MMEA) distinct couples of more intense resonances in the range 42.0–43.9 ppm, 58.4–62.0 ppm (MEA) and 33.2–35.8 ppm, 51.2–52.0 ppm, 57.1–60.8 ppm (MMEA) and two less intense resonances in the range 161–165 ppm. The signals between 42.0–42.5 ppm (MEA) and 51.2–52.0 ppm (MMEA) are attributed to the N–CH2 carbon atom of both protonated and free amine that are fast exchanging on the NMR time scale via proton scrambling. The signals in the range 58.4–61.1 ppm (MEA) and 57.1–59.7 ppm (MMEA) are assigned to CH2–OH carbon of the same species. The N–CH3 carbon signal of both free and protonated MMEA resonates between 33.2 and 34.6 ppm. Less intense signals at 43.9 ppm, 62.0 ppm (MEA) and 35.8 ppm, 51.2 ppm and 60.8 ppm (MMEA) are due to the same carbon atoms of the carbamate species. The chemical shifts of MEA are in substantial agree with those reported in literature.6 Remarkably, while the CH2–CH2 carbamate resonances are virtually unaffected by the CO2/amine ratio, the same carbon resonances of the freely exchanging AMH/AMH2+ pair move high-field by increasing the CO2 absorption. Finally, the low intensity resonances at 161 and 165 ppm are ascribed to the fast exchanging HCO3−/CO32− pair and, respectively, to the carbamate carbon, AMCO2−. Since the carbon atoms of the CH2–CH2 backbone are likely relaxing with comparable rates, we can safely assume that the relative amounts of carbamate with respect to the summed AMH/AMH2+ pair may be reasonably determined by NMR integration of the corresponding signals11 (estimated error less than 5%, see experimental). A similar approach was previously and successfully used for quantifying the species distribution in the CO2/NH3/H2O system7 and for quantitatively studying the carbamate speciation6 by 13C NMR spectroscopy.
The results of the 13C NMR analysis for both MEA and MMEA CO2-loaded solutions are reported in Table 2.
| AMH conc. (mol dm−3) | AMH/H2O (wt%) | AMCO2−a (%) | AMH b (%) | AMH2+c (%) | reaction (1)d (%) | reaction (6)e (%) | overall equil. f (%) | |
|---|---|---|---|---|---|---|---|---|
| a Carbamate percentage, mol. b Free amine percentage, mol. c Protonated amine percentage, mol. d Relative percentage of reaction (1), see text. e Relative percentage of reaction (6), see text. f Right hand shift of the overall reaction of CO2 absorption, see text. | ||||||||
| MEA | 0.167 | 1.01 | 10.7 | 12.5 | 76.8 | 86.1 | 13.9 | 82.5 |
| 0.334 | 2.02 | 17.8 | 10.7 | 71.5 | 75.1 | 24.9 | 81.6 | |
| 0.500 | 3.03 | 19.2 | 10.5 | 70.3 | 72.7 | 27.3 | 81.4 | |
| 0.667 | 4.04 | 22.3 | 10.1 | 67.6 | 67.0 | 33.0 | 81.0 | |
| MMEA | 0.167 | 1.24 | 0.0 | 17.0 | 83.0 | 100.0 | 0.0 | 83.0 |
| 0.334 | 2.48 | 6.2 | 16.9 | 76.9 | 92.0 | 8.0 | 80.1 | |
| 0.500 | 3.72 | 5.2 | 16.6 | 78.2 | 93.4 | 6.6 | 80.9 | |
| 0.667 | 4.96 | 7.4 | 14.4 | 78.2 | 90.6 | 9.4 | 82.6 |
At the end of any absorption experiment, the rise in the concentration of carbamate obtained, is strictly correlated to the increase in the amine concentration [reactions (2) and (6)]. Regardless of the amine concentration, a lower amount of carbamate forms in solution of MMEA, as to the amount resulting from MEA. Such finding may be deriving, in agreement with literature data,9 from the steric hindrance of the methyl group in MMEA, which reduces the carbamate stability. The speciation of the 0.667 M MEA and MMEA solutions as a function of the CO2 loading time (h) is plotted in Fig. 2.
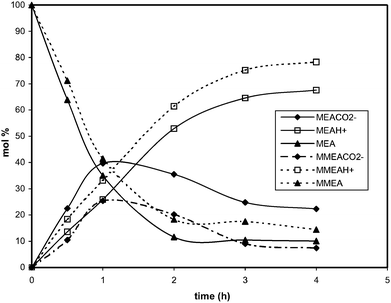 | ||
| Fig. 2 CO2 absorption by MEA and MMEA solutions (0.667 M): changes of the relative amounts (% on molar scale) of carbamate, free amine and protonated amine as a function of the absorption time (h). Estimated errors are less than 5%. | ||
These experimental speciation plots agree with those already shown for MEA and 2(ethylamino)ethanol6 and match with the modelling results.12 The observed drop off of carbamate after reaching a maximum concentration is due to reaction (4) prevailing over reaction (2) when the concentration of free amine decreases. These results prove that the smaller amount of amines free for CO2 capture, caused by the formation of carbamate, may significantly disfavour the loading capacity of the sorbent. That explains the lower absorption efficiency of MEA with respect to MMEA.
To clarify the mechanism of the overall CO2 capture process, we have also computed the relative contribution (%) of the two reactions (1) and (6) to the overall equilibrium that describes the CO2 capture process (Table 2). This calculation was based on the analysis of the relative amount of the species in solution obtained from 13C NMR data (see experimental for details) with reference to the amount of CO2 absorbed during the absorption experiments. To this purpose, the relative amount of rapidly equilibrating free and protonated amine in the absorbed solutions has to be known. Therefore, we did a quantitative 13C NMR study on D2O standard solutions of neat amine, protonated amine and their 1 : 1 molar mixture. Plotting of the chemical shifts of the 13C resonance due to either NCH2 or CH2OH groups of pure AMH, pure AMH2+ and 1 : 1 AMH/AMH2+ provides straight lines, suggesting that the resonance frequency of the carbon atoms of either CH2OH or NCH2 groups in the acid–base equilibrium AMH2+ ⇌ AMH + H+ is proportional to the relative concentration of each species. Such approach has been previously applied for the HCO3−/CO32− pair in the CO2/NH3/H2O system.7 For the 0.667 M MMEA solution, for example, we obtained 0.0148 mol of carbamate and 0.157 mol of protonated MMEA from the 13C NMR spectrum recorded at the end of the absorption process—the last being the same amount of chemically captured CO2, [eqn (1) and (6)]. The CO2 globally captured by a related absorption experiment is 0.173 mol. On the basis of these figures, we may figure out a 9.2% of physically captured CO2 at the end of the absorption process, while only 0.0296 moles of free amine remain in solution. Noticeably, previous studies have hypothesised that the physical capture of CO2 could occur for high values of CO2(absorbed)/amine molar ratio, although no experimental verification had been carried out.12 The percentage ratio between the amount of protonated MMEA arising from reaction (6) – the same amount of carbamate, 0.0148 mol – and the overall protonated MMEA (0.157 mol) is 9.4% and represents a reliable estimation of the impact of reaction (6). Finally, the yield of the entire process of chemically captured CO2 (82.6%) can be obtained from the theoretical value of CO2 loading (0.953), calculated on the relative percentages of reaction (1) and (6), and on the experimental value 0.787 (only considering the chemically captured CO2). The results obtained for all of the CO2 loaded MEA and MMEA solutions are summarized in Table 2.
Eqn (1) and (7) express the reactions of CO2 with the tertiary amine DMMEA in aqueous solution. The higher efficiency of DMMEA as CO2 absorbent than both MEA and MMEA could be traced back to the absence of carbamate in the CO2 loaded solutions of DMMEA. The experimental results obtained by 13C NMR analysis compared with those obtained by DMMEA absorption experiments give us an insight of the absorption mechanism of CO2 capture by this tertiary amine.
At low CO2 loading (<0.63), the amount of DMMEAH+ inferred by the 13C NMR spectra is higher than the CO2 captured in the absorption experiments, thus indicating that reaction (7) contributes to CO2 capture. This contribution decreases, as expected, at higher CO2 loading [reaction (5)] and becomes negligible at CO2 loading values over 0.63, when the amount of CO2 absorbed exceeds that of DMMEAH+. In this last case, the difference between CO2 and DMMEAH+ may safely be assumed as the contribution of physically absorbed CO2. The relevant results are summarized in Table 3.
| AMH conc. (mol dm−3) | AMH/H2O (wt%) | CO2(abs.) time (h) | CO2/AMH (mol/mol) | Physical absorption (%) | reaction (1)a (%) | reaction (7)b (%) | overall equil. c (%) |
|---|---|---|---|---|---|---|---|
| a Relative percentage of reaction (1), see text, and HCO3− (molar scale). b Relative percentage of reaction (7), see text, and CO32− (molar scale). c Right hand shift of the overall reaction of CO2 absorption. | |||||||
| 0.167 | 1.47 | 1 | 0.978 | 17.2 | 100.0 | 0.0 | 81.0 |
| 0.334 | 2.94 | 1 | 0.637 | 3.5 | 100.0 | 0.0 | 79.5 |
| 2 | 0.968 | 17.9 | 100.0 | 0.0 | |||
| 0.500 | 4.41 | 1 | 0.416 | 0.0 | 93.0 | 7.0 | 79.5 |
| 2 | 0.791 | 7.0 | 100.0 | 0.0 | |||
| 3 | 0.940 | 15.4 | 100.0 | 0.0 | |||
| 0.667 | 5.88 | 1 | 0.309 | 0.0 | 79.0 | 21.0 | 80.0 |
| 2 | 0.605 | 0.0 | 97.4 | 2.6 | |||
| 3 | 0.840 | 8.0 | 100.0 | 0.0 | |||
| 4 | 0.920 | 13.0 | 100.0 | 0.0 |
In the case of higher CO2 loading values (in the range 0.920–0.978) and with the residual free amine at only about 20% of the starting reagent, a physical process absorbs an appreciable amount of CO2 (in the range 13–18% of the overall amount). On the contrary, the chemical CO2 capture by reaction (7) is appreciable at the earlier stages of absorption. From these data we may conclude that DMMEA is a better absorbent than both MEA and MMEA since it does not form carbamate and gives rise to a greater CO2 physical capture.
Desorption of CO2 loaded MEA, MMEA and DMMEA solutions
The desorption of CO2 from the loaded solutions of alkanolamines, prepared as described above, was carried out at 115 °C and at room pressure until CO2 stopped being released from the solution. The desorption experiments lasted 50 to 65 min. The volume of overall desorbed CO2 was measured at 25 °C and at room pressure using the apparatus described in the experimental section. The desorption of chemically absorbed CO2 (stored as carbamate and/or bicarbonate) was inferred from the 13C NMR spectra by comparing the amounts of protonated amine still remaining in the desorbed solutions to those found in the solutions by the end of absorption experiments. In general, there is no direct correspondence between the desorption efficiency (in the range 73%–94.0% with respect to the chemically captured CO2), and chemical properties of the amines or their concentrations. To unfold the mechanism of the CO2 desorption, the composition of both MEA and MMEA solutions were monitored by 13C NMR spectroscopy at different intervals (15, 30 min and at the end of desorption) and then compared with the same solutions at the end of the absorption process. Surprisingly, the carbamate concentration plots (Fig. 3) of desorption are similar to those reported for the absorption process (Fig. 2): the carbamate concentration increases at an early stage of desorption (within 15 min), reaches a maximum and then decreases up to the end of desorption. | ||
| Fig. 3 CO2 desorption from MEA and MMEA loaded solutions: change of the relative amounts (% on molar scale) of carbamate with the desorption time (min), and solution concentration (wt%). Estimated errors are less than 5%. | ||
Differently from MEA solutions, in which the amount of carbamate is lower at the end of desorption than at the end of the absorption, in MMEA the formation of carbamate is higher at the end of each thermal desorption than at the end of the absorption step. These results are in contrast with the general belief that low temperature favours both the formation of carbamate, and, upon heating, the decomposition of carbamate in amine and carbon dioxide.13
Both desorption of CO2 and regeneration of the free amine are endothermic processes which are expected to occur via the reverse of reactions (1)–(5). On the other hand, the reverse of reaction (4) is just the endothermic reaction that produces carbamate in the desorption step.
| 2HCO3− + AMH2+ ⇌ AMCO2− + CO2 + 2H2O | (8) |
The stoichiometry of reaction (8) makes clear that only half of the captured CO2 stored as HCO3− is released, and no amine is regenerated. That lowers the overall efficiency of desorption. On this basis, the average desorption efficiency of CO2 loaded MMEA solutions (about 73%) – smaller than that of MEA solutions (about 85%) – may be due to the greater amount of carbamate in the desorbed MMEA solution compared to that found in the absorbed final solution (Fig. 3). The contrary occurs for the MEA solution. From a different point of view, since a part of the energy is used by MMEA desorption to produce a carbamate ion that still contains part of the absorbed CO2 and of the amine, further energy is required to decompose the carbamate for restoring free amine and for releasing CO2. It is interesting to note that all of the CO2 remaining in solution after the desorption of MEA is in a carbamate form, therefore confirming the high stability of the carbamate derivative of MEA. Considering the lack of carbamate in loaded DMMEA solution, the average desorption efficiency (about 84%) is not as high as it was expected on the basis of the aforementioned considerations and on the fact that DMMEAH+ is a stronger acid [the pKa of protonated amines decreases in the order MMEA (9.95) > MEA (9.65) > DMMEA (9.23)], so that the release of CO2 would be favoured according to the reverse of eqn (1). At present, this behaviour has not been fully explained. Our final consideration is meant to emphasise the importance of the loading capacity and of the absorption efficiency of the regenerated amine solutions concerning the consecutive absorption–desorption stages, rather than their values during a single stage of absorption.
Absorption–desorption tests with regenerate amine solutions
The primary goal of every regenerative process of CO2 capture is to maximize the regeneration of the sorbent solution in order to maintain high loading capacity and absorption efficiency during the following regeneration–absorption steps. Aiming at verifying the efficiency of regenerated amine solutions for reuses, we conducted five consecutive absorption–desorption cycles. For comparison purposes we used the less (0.167 M) and the more (0.667 M) concentrated solutions of MEA, MMEA and DMMEA for each test. In order to keep the absorption efficiency high (in the range 75–85% for the less concentrated solutions), the absorption experiments were interrupted when about 50 ± 5% of CO2 was absorbed by the residual amine solutions. The desorption experiments, carried out at 115 °C and at room pressure, were stopped when no more CO2 was released from the loaded solutions. In the five absorption–desorption cycles, the average removal efficiency of the more concentrated solution (0.667 M; 0.200 mol) is over 99.7% for MEA and MMEA and higher than 98% for DMMEA, because of the large excess of free amine that still remains in solution at the end of every absorption. On the contrary, the less concentrated solution (0.167 M, 0.050 mol), only contains a slight excess of amine in comparison with the overall amount of CO2 flowing during the absorption experiment (molar ratio CO2(flowed)/amine(starting) in the range 0.916–0.978). Also, the free amine at the end of the absorption is less than 15–20% of the total. In these absorption–desorption experiments, the CO2 removal efficiency of the initial “fresh” amine solution is always greater (in the range 80–92%) than that of the four following regenerated solutions (in the range 73–86%) (Fig. 4), that being caused by the presence of protonated amine (about 4–15%) and carbamate (about 4–6%, for MEA and MMEA) in the regenerated solutions, as it was confirmed by the 13C NMR spectroscopy. | ||
| Fig. 4 Absorption efficiency of 0.167 M solutions of MEA, MMEA, DMMEA during five consecutive absorption–desorption recycling tests. | ||
However, both loading capacity and removal efficiency of the regenerated amines remain fairly constant during the four additional absorption–desorption experiments and increase in the order MEA < MMEA < DMMEA (Table 4).
| Desorb. time (min) | Absorb. a (%) | CO2(abs.)/amine b (mol/mol) | AMCO2−c (%) | ||
|---|---|---|---|---|---|
| abs. | des. | ||||
| a Average CO2 absorption efficiency. b Average CO2 loading capacity. c Percentage on molar scale of carbamate in the absorbed and desorbed solutions at the end of the processes. | |||||
| MEA | 40 | 73.4 | 0.682 | 18.7 | 6.0 |
| MMEA | 40 | 79.8 | 0.791 | <5 | <5 |
| DMMEA | 40 | 85.4 | 0.853 | — | — |
Experimental
General information
All reagents were reagent grade. The alkanolamines, 2-aminoethanol (monoethanolamine, MEA), 2-(methylamino)ethanol (N-methylethanolamine, MMEA) and 2-(dimethylamino)ethanol (N,N-dimethylaminoethanol, DMMEA) (Sigma-Aldrich) were used as received. Pure CO2 and N2 (Rivoira) were used to simulate flue gas. pH measurements were carried out with a Crison GLP 22.02 model pH-meter calibrated with standard buffer solutions at pH 7.0 and 9.0. Flow rates of N2 and CO2 were measured with gas mass flow meters (Aalborg) equipped with a gas controller (Cole Parmer). The inlet and outlet CO2 concentrations in the flue gas mixture were measured with a Varian CP-4900 gas chromatograph calibrated with a 10% v/v CO2–N2 reference gas (Rivoira).The absorber device was a home-built glass cylinder with a diameter of 56 mm and a height of 300 mm. It was fitted with three polyethylene disks threaded on a 2-mm glass rod and equipped with a thermometer and a combined pH electrode.14 The absorber was charged with 0.300 dm3 of the absorbent solution. The absorption experiments were carried out at 20 °C with aqueous amines at concentrations 0.167 M (MEA, 1.01 wt%; MMEA, 1.24 wt%; DMMEA, 1.47 wt%), 0.334 M (MEA, 2.02 wt%; MMEA, 2.48 wt%; DMMEA, 2.94 wt%), 0.500 M (MEA, 3.03 wt%; MMEA, 4.18 wt%; DMMEA, 4.41 wt%), and 0.667 M (MEA, 4.04 wt%; MMEA, 4.96 wt%; DMMEA, 5.88 wt%). The temperature of the absorber was constantly kept under control with a thermostatted water bath (Julabo model F33-MC refrigerated bath) regulated at the required absorption temperature. The absorber was continuously fed through a sintered glass diffuser (16–40 µm pores) placed at the bottom of the absorbent solution. A gas mixture containing 12% (v/v) CO2 in N2, injected at a flow rate of 14 dm3 h−1, was used to imitate flue gas. The vent gas was released from the top of the absorber. Bubbling through water at the operating temperature before entering the absorbent reactor humidified the inlet gas mixture. The outlet gas was dried out by flowing, in turn, through a condenser cooled at −5 °C, a concentrated H2SO4 solution and a gas purification tower filled with P2O5, before being analysed by the gas chromatograph.
The release of pure CO2 during the desorption processes was measured using a gastight apparatus which comprises a 250 mL conical flask containing half of the different solutions obtained during the absorption steps. In this way a duplicate measurement of the CO2 release could be obtained for every solution. The conical flask was equipped with two condensers cooled at room temperature and connected to two 250 mL gas burettes equipped with a pressure-equalising device. Both burettes and pressure-equalising devices were filled with CO2 saturated water. Through three-way valves, one burette was filled with CO2 while the other was emptied, thus allowing a continuous collection of gas. The gas pressures inside the burette and the external pressure continuously balanced each other. The total volume measurements were about ±5 mL accurate. The release of CO2 took a maximum of 50–65 min at 115 °C. All these solutions were checked by both 13C NMR spectroscopy and pH measurement.
13C NMR spectroscopy
The 13C{1H} NMR spectra of the absorbed solutions were obtained with a Varian Gemini g300bb spectrometer operating at 75.46 MHz. Chemical shifts are relative to tetramethylsilane as an external standard at 0.00 ppm. CH3CN was used as internal reference (CH3, δ = 1.47 ppm). The standard pulse sequence with proton decoupling was used to acquire the 13C{1H} NMR spectra with the following acquisition parameters: pulse angle = 62.3°, at = 1.701 s, d1 = 0 s, nt = 30·103. Increasing the acquisition time (up to 150 s) and/or the relaxation delay (up to 60 s) does not produce substantial changes in the relative peak areas of the CH2–CH2 carbon atoms. The water solutions were added with about 10% (v/v) D2O (Aldrich) to allow enough signal for deuterium lock. NMR spectra were recorded in successive absorption and desorption steps and at the end of every absorption and desorption experiment. Normally, the integration of 13C NMR resonances does not grant a reliable quantification of species with carbon atoms in different environments, because the spin–lattice T1 relaxation time strongly depends on the number of protons attached to the carbon atom.11 In the species we have been dealing with, i.e. carbamate and the rapidly equilibrating (free amine)/(protonated amine), the 13C atoms of the CH2–CH2 backbone have the same number of directly-attached hydrogens, and it is conceivable that they exhibit similar T1, as shown by the similar peak integrals occurred in each CH2 resonance (estimated error <2%). On the other hand, the 13C atoms of HCO3−/CO32− pair and of CO2− functionality of the amines have no attached hydrogen and definitely show higher relaxation times than those of CH2 groups (which result in much lower peak intensities). For these reasons, the relative amounts of carbamate, free amine and protonated amine have been determined by NMR integration of the corresponding signals of the CH2–CH2 carbons, whose chemical shifts are strongly related to the chemical environment. The feasibility of this procedure was tested by carrying out several 13C NMR spectra on reference solutions prepared by dissolving different molar ratios of accurately weighted amounts free amine, fully protonated amine and amine carbamate in D2O. Using these standard solutions, we found a quantitative relationship (estimated error <5%) between the relative peak areas of CH2CH2 carbon atoms and the known concentrations of each species. The quantification method is therefore, on an empirical level, quite reliable, consequently reflecting similarities of the relaxation rate for similar carbons in both carbamate and the rapidly equilibrating (free amine)/(protonated amine). In order to measure the relative amounts of rapidly equilibrating free amine and protonated amine by NMR analysis, we have carried out a quantitative 13C NMR study on D2O standard solutions of free amine, protonated amine and their 1 : 1 molar mixture. The chemical shift of a single 13C-resonance caused by NCH2 or CH2OH groups is plotted as a straight line with limiting values assigned to free amine (CH2OH: MEA, 63.61 ppm; MMEA, 60.60 ppm; NCH2: DMMEA, 59.38 ppm) and to protonated amine (CH2OH: MEA, 57.61 ppm; MMEA, 56.42 ppm; NCH2: DMMEA, 55.14 ppm). As previously reported for the HCO3−/CO32− system,7 the straight lines featuring these plots point out that the frequency of the resonance for the carbon atom of the CH2CH2 unit is proportional to the relative concentration of the two species.Reaction contributions
The contribution of reactions (1) and (6) to the overall equilibrium of CO2 capture has been estimated as described below on the basis of the amounts of CO2 measured during the absorption experiments and of the amounts of protonated amine, free amine and carbamate calculated from 13C NMR analysis.If z is the total amount of protonated amine equilibrating in both reactions (1) and (6), we have, from the 13C NMR spectra:
| z = AMH2+(total) = CO2(absorbed) = x + y |
| reaction (6): x = AMCO2− = AMH2+ |
| reaction (1): y = HCO3 = AMH2+ |
Knowing the relative percentages of reactions (1) and (6) allows us to calculate the theoretical value for the CO2 captured by assuming a CO2(absorbed)/amine molar ratio 1 : 1 and 1 : 2 for reaction (1) and reaction (6), respectively. The molar ratio between z and this value represents the total equilibrium of the CO2 chemically captured by reaction (1) and (6).
Finally, the difference between the overall CO2 captured and z represents the physical absorption of CO2.
Conclusions
The separation of CO2 from flue gas through alkanolamines chemical absorption is a crucial technical process toward which a noticeable interest has grown, especially in view of CCS urgency. In the present study we have described how to apply a 13C NMR spectroscopy to investigate the species distribution during the CO2 capture by primary (MEA), secondary (MMEA) and tertiary (DMMEA) amine-containing aqueous solutions. The purpose was to improve our overall knowledge of the CO2 uptake mechanism, mainly trying to understand the scenario of CO2 reaction equilibria and to investigate the contribution of CO2 physical absorption. A careful comparison of the data obtained through a series of 13C NMR spectra with the results of related absorption experiments has allowed us to establish that: (i) the overall CO2 chemical capture ranges between 80 and 83%; (ii) the carbamate formation that accompanies the reaction with MEA and MMEA ranges between 0 and 27%, and (iii) the physical uptake of CO2, incrementing the overall CO2 uptake, may even reach an important value such as 18%.Our results confirm that the carbamate formation, which increases both with increasing concentration of primary and secondary amines and with decreasing steric hindrance on the amine N-atom, negatively affects the efficiency of CO2 absorption. Following these assertions, the tertiary amine DMMEA, which cannot be converted into carbamate, exhibits both the highest loading capacity and the highest absorption efficiency. The appreciable contribution from the CO2 physical absorption also contributes to the global loading value of 1.04 found for the 0.167 M DMMEA solution, in spite of the overall 81% reaction equilibrium of CO2 chemical capture. The physical absorption of CO2 increases with the CO2/amine molar ratio.
In addition, it is worth a mention the fact that our experimental conditions well match the primary objective of an efficient process for CO2 capture, which is not to maximise the loading capacity at the expense of low absorption efficiency, but instead to approach the best compromise between these two properties. In detail, the best results have been achieved for the less concentrated solutions of the amine, exhibiting quite an interesting CO2(absorbed)/amine molar ratio in the range 0.82–0.99 and an overall absorption efficiency in the range 72–78%.
Both for MEA and MMEA-loaded solutions, the results of the desorption processes show an increase in the carbamate formation during the early stages of the thermal desorption process. This fact contrasts with the previous belief of carbamate being formed by an exothermic reaction during the absorption process and decomposing in the desorption step. On the contrary, we give clear evidence that the amount of carbamate is higher at the end of the desorption step of MMEA loaded solution, than at the end of the absorption process. As a consequence of the formation of carbamate, less CO2 is released and less free amine is regenerated, therefore reducing the desorption efficiency. The endothermic formation of carbamate in the desorption step, from a thermodynamic point of view, is a negative element, since it increases the process' energy demand—a major point within the entire process of CO2 capture. It is also indicated that the formation of carbamate in the desorption step is more detrimental than its formation during the absorption step.
The results of five consecutive absorption–desorption cycles confirm the feasibility of amine regeneration for reuse, as the regenerated solutions maintain high loading capacity and absorption efficiency, even if slightly lowered than the starting “fresh” amine solutions (<6–9%). Among the three amines, the regenerated solution of 0.167 M DMMEA is the most efficient CO2 absorbent in terms of both overall loading capacity and absorption efficiency. The 0.167 M solution of MEA, although less efficient for CO2 uptake, has a slightly greater desorption efficiency, presumably deriving from the neat decrease of carbamate at the end of desorption step of the loaded solution of MEA.
Finally, a fair dilution of these amines (1.01–1.47 wt%) reduces their corrosive properties but not their high reactivity towards carbon dioxide.
On the basis of the results we have achieved, we are currently pursuing the application of alkanolamines to CO2 removal from flue gas and we are also investigating amines and techniques for CO2 capture which may reveal themselves more efficient.
Acknowledgements
Financial support from MIUR (Rome, Italy) and Ente Cassa di Risparmio di Firenze (Florence, Italy), through Florence Hydrolab Project, is gratefully acknowledged.References
- Intergovernmental Panel on Climate Change Special Report on Carbon Dioxide Capture and Storage, Ed. B. Metz, O. Davidson, H. C. de Coninck, M. Loos, L. A. Meyer, 2005, Cambridge University Press, Cambridge, UK and New York, NY, USA, pp. 442 . See: http://www.ipcc.ch/ipccreports/srccs.htm Search PubMed.
- Chemistry and Energy; The role of the chemical sciences in the European energy policy. See: http://www.euchems.org/binaries/030882EnergyReport_tcm23-118847.pdf.
- P. M. Blauwhoff, G. F. Versteeg and W. P. M. van Swaaij, Chem. Eng. Sci., 1984, 39, 207–225 CrossRef CAS; G. Astarita, D. W. Savage and A. Bisio, Gas Treating with Chemical Solvents, John Wiley & Sons, New York, 1984 Search PubMed; D. A. Glascock, J. E. Critchfield and G. T. Rochelle, Chem. Eng. Sci., 1991, 46, 2829–2845 Search PubMed; O. Erga, O. Juliussen and H. Lidal, Energy Convers. Manage., 1995, 36, 387–392 CrossRef; C. Mathonat, V. Mayer, A. E. Mather and J. P. Grolier, Ind. Eng. Chem. Res., 1998, 37, 4136–4141 CrossRef CAS; M. K. Abu-Arabi, A. Tamini and A. M. Al-Jarrah, J. Chem. Eng. Data, 2001, 46, 1125–1129 CrossRef CAS; J.-Y. Park, S. J. Yoon, H. Lee, J.-H. Yoon, J.-G. Shim, J. K. Lee, B.-Y. Min, H.-M. Eum and M. C. Kang, Fluid Phase Equilib., 2002, 202, 359–366 CrossRef CAS.
- A. B. Rao and E. S. Rubin, Ind. Eng. Chem. Res., 2006, 45, 2421–2429 CrossRef CAS; B. A. Oyenekan and G. T. Rochelle, Ind. Eng. Chem. Res., 2006, 45, 2457–2466 CrossRef CAS; M. S. Jassin and G. T. Rochelle, Ind. Eng. Chem. Res., 2006, 45, 2465–2472 CrossRef.
- CO2(emitted) represents the overall amount of CO2 released in the atmosphere by burning fossil fuels to produce all forms of energy (electrical, thermal and mechanical) necessary to sustain the entire CO2-removal cycle, from the production of the reagents to the final transport of CO2 and its disposal.
- T. Suda, T. Iwaki and T. Mimura, Chem. Lett., 1996, 25, 777–778 CrossRef; S. J. Yoon and H. Lee, Chem. Lett., 2003, 32, 344–345 CrossRef CAS; J. Y. Park, S. J. Yoon and H. Lee, Environ. Sci. Technol., 2003, 37, 1670–1675 CrossRef CAS.
- F. Mani, M. Peruzzini and P. Stoppioni, Green Chem., 2006, 8, 995–1000 RSC.
- J. T. Yeh and H. W. Pennline, Energy Fuels, 2001, 15, 274–278 CrossRef CAS; R. Idem, M. Wilson, P. Tontiwachwuthikul, A. Chakma, A. Veawab, A. Aroonwilas and D. Gelowitz, Ind. Eng. Chem. Res., 2006, 45, 2414–2420 CrossRef CAS; E. F. da Silva and H. F. Svendsen, Ind. Eng. Chem. Res., 2006, 45, 2497–2504 CrossRef CAS.
- G. Sartori and D. W. Savage, Ind. Eng. Chem. Fundam., 1983, 22, 239–249 CrossRef CAS.
- A. K. Saha, S. S. Bandyopadhyay, P. Saju and A. K. Biswas, Ind. Eng. Chem. Res., 1993, 32, 3051–3055 CrossRef CAS; B. P. Mandal, A. K. Biswas and S. S. Bandyopadhyay, Chem. Eng. Sci., 2003, 58, 4137–4144 CrossRef CAS.
- E. Breitmaier and W. Voelter, Carbon-13 NMR Spectroscopy, 3rd edn, VCH, Weinheim, Germany, 1990 Search PubMed.
- D. M. Austgen, G. T. Rochelle, X. Peng and C. Chen, Ind. Eng. Chem. Res., 1989, 28, 1060–1073 CrossRef; Y. Liu, L. Zhang and S. Watanasiri, Ind. Eng. Chem. Res., 1999, 38, 2080–2090 CrossRef CAS; A. Aboudheir, P. Tontiwachwuthikul, A. Chakma and R. Idem, Chem. Eng. Sci., 2003, 58, 5195–5210 CrossRef CAS; E. Blanchon le Bouhelec, P. Mougin, A. Barreau and R. Solimando, Energy Fuels, 2007, 21, 2044–2055 CrossRef CAS.
- R. J. Hook, Ind. Eng. Chem. Res., 1997, 36, 1779–1790 CrossRef CAS; D. Bonenfant, M. Mimeault and R. Hausler, Ind. Eng. Chem. Res., 2003, 42, 3179–3184 CrossRef.
- F. Mani, M. Peruzzini and P. Stoppioni, Energy Fuels, 2008, 21, 1714–1719 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |