Water splitting into H2 and O2 over niobate and titanate photocatalysts with (111) plane-type layered perovskite structure
Yugo
Miseki
,
Hideki
Kato†
and
Akihiko
Kudo
*
Department of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjyuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.kagu.tus.ac.jp; Fax: +81-35261-4631; Tel: +81-35228-8267
First published on 13th January 2009
Abstract
Photophysical and photocatalytic properties of A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12 with layered perovskite structures, in which a plane in parallel with (111) of a simple perovskite structure was exposed at interlayer, were investigated. These oxides were obtained by a polymerizable complex method at 973–1473 K though only A5Nb4O15 (A = Sr and Ba) were prepared by a solid state reaction even at 1673 K. The shapes of these complex metal oxides were plate-like derived from the perovskite layered structure. These band gaps were estimated to be 3.7–4.1 eV from the onsets of diffuse reflection spectra. These oxides showed photoluminescence at 77 K. These oxides loaded with NiO cocatalysts showed activities for water splitting under UV irradiation. NiOx/BaLa4Ti4O15 and NiOx/Ba5Nb4O15 showed the highest activities among the titanates and niobates tested in the present study. NiOx/BaLa4Ti4O15 and NiOx/Ba5Nb4O15 gave 15% and 17% of quantum yields at 270 nm, respectively. Photocatalytic activities of ALa4Ti4O15 (A = Ca, Sr, and Ba) strongly depended on the alkaline earth metal ion. Pt, Au, Ni, and PbO2 were selectively photodeposited on basal or edge plane of the BaLa4Ti4O15 plate-like powder while these were randomly loaded on CaLa4Ti4O15. It was suggested that this difference in the surface property was the one of the important factors affecting photocatalytic ability for ALa4Ti4O15 (A = Ca, Sr, and Ba).
Broader contextThere is no doubt that solar hydrogen production from water is highly desirable. Water splitting using a particulate photocatalyst is one of the candidates for the solar hydrogen production. The photocatalyst system is not at the stage of practical use. Therefore, basic research is still important in this research field. The construction of a photocatalyst library and clarification of factors dominating photocatalytic activities are important in order to establish a guiding principle of photocatalyst design. In the present study, A5Nb4O15 (A = Sr, Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, Ba), and La4Ti3O12 with a (111) plane-type layered perovskite structure in which alkaline earth metal and lanthanum cations are ordered or not at interlayer and A-site were found to be new niobate and titanate photocatalyst materials for water splitting. 15% of the quantum yield was obtained. The ordering caused anisotropic characters of photocatalytic properties that were revealed by DRS, SEM, photodeposition of metals, and photoluminescence. The relationship between photocatalytic activities, and crystal and energy structures are systematically discussed in detail. These results and discussion will be valuable in this research field of not only water splitting but also photocatalytic degradation of hazardous organic compounds. |
1. Introduction
Photocatalytic water splitting using a heterogeneous semiconductor powder has been studied as an ideal H2 production method which does not give any burdens to the environment. Many oxide photocatalysts consisting of d0 and d10 metal ions show photocatalytic activities for water splitting under UV irradiation.1–35 Each material possesses unique character even if they seem similar to each other. So, finding a new material for the attractive and tough reaction, and studying its science is meaningful. Recently, metal oxynitride and nitride photocatalysts,36,37 and Z-scheme-type photocatalysts accompanied with two-photon process38–42 have been reported for water splitting under visible light irradiation. The final target of this research is efficient solar hydrogen production from water. The present efficiency is not satisfying for practical use. Photocatalytic water splitting is an attractive and challenging reaction. Therefore, basic research is still important in this research field. The construction of a photocatalyst library and clarification of factors affecting photocatalytic abilities are important in order to establish a guiding principle for design of a photocatalyst.4NiO/NaTaO3 photocatalyst shows high activity for water splitting under UV irradiation.5 Moreover, the activity of NiO/NaTaO3 is remarkably improved by La doping (1–2%).6 La-doping gives nano-sized NaTaO3 particles and characteristic surface step structure. Active sites for H2 evolution are separated from those for O2 evolution by the surface nano-step structure. So, the surface morphology is important. On the other hand, the metal oxides with layered structures, such as K4Nb6O17,7 K2La2Ti3O10,8 A2A′2O7 (A = Ca and Sr, A′ = Nb and Ta),12,14,30 RbNdTa2O7,15 La2Ti2O7,19 H2La2/3Ta2O7,23 K2Sr1.5Ta3O10,32 H1.8Sr0.81Bi0.19Ta2O7,33 and LiCa2Ta3O1034 show high activities for water splitting under UV irradiation. The interlayer can function as reaction sites and separate oxidation reaction sites from reduction reaction sites in hydrated layered compounds. The anisotropy derived from the layered structure may also affect the photocatalytic ability. Therefore, it is important to examine photocatalytic properties of various compounds with surface morphology and characteristic crystal structure like a layered structure in order to obtain further information on photocatalytic water splitting.
There are many kinds of compounds with layered perovskite structure consisting of various elements, and perovskite framework and interlayer structures. Layered perovskite compounds are attractive materials as photocatalysts. Layered perovskite structure can be classified into (100) and (110) plane structures due to the plane directions of interlayer. The (110) plane structure can be classified further into (110) and (111) plane-type structures due to the plane defect structure of perovskite crystal structure. In the present paper, layered perovskite structures are classified into (100), (110), and (111) plane-type structures due to the plane defect structure (Fig. 1). Here, (100), (110), and (111) plane-types mean that the interlayer is in parallel with (100), (110), and (111) planes of a bulk type perovskite structure, respectively. These structures can also be distinguished from each other by the exposed part of MO6 octahedra in interlayer, corner, edge, and face of the octahedra. It has been reported that K2La2Ti3O10,8 Sr4Ti3O10,16 A2A′Ta2O7 (A = H and K, A′ = La2/3 and Sr),18,23 La2Ti3O9,19 Sr3Ti2O7,29 and LiCa2Ta3O1034 with the (100) plane-type layered perovskite structure and La4CaTi5O17,12 A2A′2O7 (A = Ca and Sr, A′ = Nb and Ta),12,14,30 La2Ti2O7,19 and H1.81Sr0.81Bi0.19Ta2O733 with the (110) plane-type layered perovskite structure show photocatalytic activities for water splitting under UV irradiation. In addition, the modified photocatalysts of these layered perovskite compounds have been also reported.22,35 On the other hand, there are few reports of oxide photocatalysts with the (111) plane-type layered perovskite structure.21,24 Moreover, a titanate photocatalyst with the (111) plane-type layered perovskite structure is not reported.
 | ||
| Fig. 1 Layered perovskite structures with different face defects. | ||
A polymerizable complex method (PC method) is one of the preparation methods of functional material powders.43 Various complex metal oxides can be prepared under mild condition by the PC method. We can prepare complex oxides with different crystallinity, particle size, and morphology by changing calcination temperature in a wide range. Investigation using these samples with a series of different conditions is meaningful for clarification of the factors affecting photocatalytic activities. It has been reported that the particles of K2La2Ti3O108 and KTiNbO513 prepared by a PC method have higher crystallinity and finer crystal size than those prepared by a solid-state reaction, resulting in that these photocatalytic activities remarkably increase. We have preliminary reports that the particles of Ba5Nb4O15 with (111) plane-type layered perovskite structure prepared by the PC method were plate-like crystals, and showed high activity for water splitting under UV irradiation.26 On the other hand, A5Ta4O15 (A = Sr and Ba) with (111) plane-type layered perovskite structure prepared by a PC method also shows high activity for water splitting under UV irradiation.21,24 Thus, (111) plane-type layered perovskite oxides with high crystallinity and fine particle size can be prepared by a PC method.
In the present paper, we investigated photophysical properties and photocatalytic activities for water splitting of (111) plane–type A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12 prepared by a PC method. The main characteristic point of the materials reported in the present study is the new-type layered perovskite structure in which alkaline earth metal and lanthanum cations are ordered or not at interlayer and A-site. Diffuse reflectance spectroscopy (DRS), photoluminescence, scanning electron microscope (SEM) measurements, and photodeposition of some metal and metal oxides were carried out as characterizations in order to investigate the properties based on the anisotropy of the layered structure. The relationship between photocatalytic activities, and crystal and energy structures are discussed based on the different ordering of alkaline earth metal and lanthanum cations.
2. Experimental
CaCO3 (Kanto Chemical; 99%), SrCO3 (Kanto Chemical; 99.9%), BaCO3 (Kanto Chemical; 99%), La(NO3)3·6H2O (Wako Pure Chemical; 99.9%), Ti(OC4H9)4 (Kanto Chemical; 97%), Nb(OC2H5)5 (Kojundo Chemical; 99.99%), PrCl3·6H2O (Rare Metallic; 99.99%), citric acid (Sigma Aldrich Japan; 99.5%), ethylene glycol (Kanto Chemical; 99.5%), and propylene glycol (Kanto Chemical; 99.0%) were employed as starting materials for PC method according to a previous report.43 Metal compounds and a citric acid were dissolved in a mixed solvent of methanol and ethylene glycol or propylene glycol. The mixed solution was aged at 353 K for 2 h. At this stage, a solution containing citric complexes was formed. The solution was dehydrated to form gel by polymerization at 393 K for 12 h. A precursor was obtained by calcination of the gel containing a citric acid using a mantle heater. Various titanates and niobates were prepared by calcination of these precursors at 1073–1473 K for 10 h. Phase purity of the obtained powders was confirmed by X-ray diffraction (Rigaku; MiniFlex). Diffuse reflection spectra were measured using a UV-vis-NIR spectrometer (Jasco; UbestV-570) and were converted from reflectance to absorbance by the Kubellka-Munk method. Photoluminescence spectra of these complex metal oxides with and without Pr-doping as a luminescent probe were measured using a fluorospectrometer (HORIBA JOBIN YVON; SPEX Fluorolog-3). Temperature control of the photoluminescence measurement was carried out using a cryostat (Oxford; ITC 503S). The photocatalyst particles were observed by a scanning electron microscope (SEM, JEOL JSM-6700F). In order to investigate the surface reactivity of photocatalysts, Au, Pt, Ni, and PbO2 particles photodeposited on photocatalysts from aqueous HAuCl4, K2PtCl6, [Pt(NH3)4]Cl2, Ni(NO3)2, and Pb(NO3)2 solutions were observed by SEM.NiO and RuO2 cocatalysts were loaded by an impregnation method from aqueous Ni(NO3)2 and RuCl3 solutions. The powder impregnated with Ni(NO3)2 was calcined at 543 K for 1 h in air. Pretreatment of reduction with 200 torr of H2 at 773 K for 2 h followed by oxidation with 100 torr of O2 at 373–573 K for 1 h was carried out for NiO-loaded photocatalysts, if necessary.44 The powder impregnated with RuCl3 was calcined at 673 K for 2 h in air. Water splitting reactions were carried out in a gas-closed circulation system. The photocatalyst powder (0.5 g) was dispersed in pure water (380 mL) by a magnetic stirrer in an inner irradiation reaction cell made of quartz equipped with a 400 W high-pressure mercury lamp (SEN: HL400EH-5). The amounts of evolved H2 and O2 were determined using on-line gas chromatography (Shimadzu; MS-5A column, TCD, Ar carrier). Apparent quantum yields at 270 nm were measured using a reaction cell with a top window made of quartz and a 300 W Xe illuminator (ILC technology; CERMAX-LX300) attached with a band-pass filter (Kenko; BPUV-270, WHM: 16 nm). The number of incident photon was determined using a photodiode (OPHIRA: PD300-UV of a head and NOVA of a power monitor). The photocatalyst powder (0.05–0.3 g) was dispersed in pure water (300 mL) for the measurement of the apparent quantum yield. The apparent quantum yields were determined by eqn (1).
 | (1) |
3. Results and discussion
3-1. Synthesis and characterization
A single phase of A5Nb4O15 (A = Sr and Ba) was obtained by a solid-state reaction (SSR method) at 1373 K. In construct, single phases of Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12 were not obtained even at 1673 K of the calcination temperature. It is probably caused by low reactivity of lanthanum oxide with alkali earth metal oxides. Therefore, synthesis of these compounds was attempted by a polymerizable complex method (PC method). These pure complex metal oxides were successfully prepared by calcination of the precursors at 973–1473 K. The synthetic processes by the PC method for Ba5Nb4O15 and BaLa4Ti4O15 that showed high photocatalytic activities as mentioned later were examined in detail.Fig. 2 shows XRD patterns of Ba5Nb4O15 and BaLa4Ti4O15 prepared by a PC method. Broad diffraction patterns of both precursors were assigned to a simple perovskite structure. Almost pure phases of Ba5Nb4O15 and BaLa4Ti4O15 were successfully obtained by calcination at 973 and 1323 K, respectively. The diffraction peaks of (103) and (203) of BaLa4Ti4O15 and Ba5Nb4O15 became larger than that of (110), as the calcination temperature became high. Fig. 3 shows SEM images of Ba5Nb4O15 prepared at different calcination temperatures. The shape of particles was plate-like derived from the layered perovskite structure. As the calcination temperature was high, the aspect ratio of particles was decreased by crystal growth to the direction in a plane parallel to the (−110) plane. This is the reason why the (110) diffraction peak became relatively small with increasing the calcination temperature. La4Ti3O12, Ba3LaNb3O12, Sr5Nb4O15, and ALa4Ti4O15 (A = Sr and Ca) were also obtained as plate-like particles.
 | ||
| Fig. 2 XRD patterns of (I) Ba5Nb4O15 and (II) BaLa4Ti4O15 prepared by a PC method. (a) and (b) precursor, powders calcined at (c) 873 K, (d) 973 K, (e) 1073 K, (f) 1273 K, (g) 1173 K, (h) 1273 K, and (i) 1323 K. Calcination time was 10 h. | ||
 | ||
| Fig. 3 SEM images of Ba5Nb4O15 powders synthesized at (a) 1073 K and (b) 1273 K. | ||
3-2. Crystal and energy structures
Factors affecting the band gaps of layered perovskite oxides are considered as follows.Factor (i): the bond angle between metal and oxygen ions of octahedra units (M–O–M). As the bond angle is apart from 180°, the band gap becomes wide and the excited energy state is localized.5,14,45,46
Factor (ii): the thickness of the perovskite layer. As the thickness of the perovskite layer decreases, the two dimensionality of crystal structure becomes high, resulting in the band gap becoming wide and the excited energy state more localized.
Factor (iii): the interaction between perovskite layers. As the ionic radius of cations at the interlayer becomes large, the distance between perovskite layers becomes long, accordingly an excited energy state becomes localized by decreasing interaction between the layers.
Factor (iv): the polarization ability of cations at the interlayer towards the oxygen ions of octahedra faced at the interlayer. For example, H+-exchange of layered oxides sometimes causes red shift in absorption spectra due to high polarization ability of proton showing band gap narrowing.47,48 The relationships between these factors and band gaps are summarized in Table 1.
| Factors | Niobate | Titanate | ||
|---|---|---|---|---|
| Sr5Nb4O15 | Ba5Nb4O15 | CaLa4Ti4O15 | BaLa4Ti4O15 | |
| Cation of interlayer | Sr2+ | Ba2+ | La3+, Ca2+ | La3+, Ba2+ |
| Ionic radius/Å | 1.44 | 1.61 | 1.36, 1.34 | 1.36, 1.61 |
| Cation of perovskite slab | Sr2+ | Ba2+ | La3+, Ca2+ | La3+ |
| Ionic radius/Å | 1.44 | 1.61 | 1.36, 1.34 | 1.36 |
| Bond angle between metal and oxygen ions | 168 | 180 | Similar | |
| Effect on band gap | Wider | Narrower | Similar | |
| Thickness of perovskite layer | Four | Four | Four | Four |
| Effect on band gap | Similar | Similar | ||
| Interaction between perovskite layers | Large | Small | Large | Small |
| Effect on band gap | Narrower | Wider | Narrower | Wider |
| Polarization ability of cation | Large | Small | Large | Small |
| Effect on band gap | Narrower | Wider | Narrower | Wider |
| Band gaps/eV | 4.04 | 3.91 | 3.79 | 3.85 |
Fig. 4 shows diffuse reflection spectra of A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12. The band gaps of Sr5Nb4O15 and Ba5Nb4O15 with four-octahedra thickness of perovskite layer were estimated to be 4.0 eV and 3.9 eV from the onsets of absorption, respectively (Fig. 4(I)). The bond angles of Nb–O–Nb in the frameworks of perovskite structures of Ba5Nb4O15 and Sr5Nb4O15 are 180 and 168°, respectively.49,50 Because the bond angle of Ba5Nb4O15 is closer to 180° than that of Sr5Nb4O15, the band gap of Ba5Nb4O15 is narrower than that of Sr5Nb4O15 according to the factor (i). The difference between bond angles of Ba5Nb4O15 and Sr5Nb4O15 is almost the same as those of KTaO3 and NaTaO3 with bulk-type perovskite structures, respectively. However, the difference in the band gap between Ba5Nb4O15 and Sr5Nb4O15 (0.1 eV) was smaller than that between KTaO3 and NaTaO3 (0.4 eV).5 This result implies that the factors (iii) and (iv) also affect the band gaps. The interaction between perovskite layers of Ba5Nb4O15 would be smaller than that of Sr5Nb4O15 because Ba2+ at the interlayer is larger than Sr2+. In addition, the energy structures of the perovskite layers are changed by different polarization abilities between Sr2+ and Ba2+ at the interlayer. The balance of these factors seems to cause the small difference in band gaps between Ba5Nb4O15 and Sr5Nb4O15.
 | ||
| Fig. 4 Diffuse reflection spectra of (I) niobates and (II) titanates; (a) Ba3LaNb3O12, (b) Sr5Nb4O15, (c) Ba5Nb4O15, (d) CaLa4Ti4O15, (e) SrLa4Ti4O15, (f) BaLa4Ti4O15, and (g) La4Ti3O12. | ||
The band gaps of CaLa4Ti4O15, SrLa4Ti4O15, and BaLa4Ti4O15 were 3.78 eV, 3.82 eV, and 3.85 eV, respectively. This order in the band gap was reverse to that for ATiO3 (A = Ca, Sr, and Ba) with bulk-type perovskite structure. The positions of La3+, Ca2+, and Ba2+ in crystal structures of ALa4Ti4O15 (A = Ca and Ba) give some information to consider this difference in the order of band gaps. The ratio of Ba2+ to La3+ at the interlayer is 1 : 1 in BaLa4Ti4O15 while only La3+ exists in the perovskite layer as shown in Fig. 5.51 On the other hand, Ca2+ and La3+ randomly exist at the interlayer and perovskite layer in CaLa4Ti4O15. The distortion of the perovskite framework structure of BaLa4Ti4O15 would be close to that of CaLa4Ti4O15 because the ionic radius of La3+ (1.36 Å) is nearly equal to that of Ca2+ (1.34 Å). Therefore, the factor (i) would be negligible. The band gap of BaLa4Ti4O15 is wider than that of CaLa4Ti4O15 due to an increase in the distance between layers (factor (iii)) and the different polarization ability of Ca2+ and Ba2+ (factor (iv)) because the ionic radius of Ba2+ is larger than that of Ca2+. The band gap of Ba3LaNb3O12 with layered perovskite structure with three-octahedra thickness was estimated to be 4.1 eV. The perovskite layer of Ba3LaNb3O12 is thinner than that of Ba5Nb4O15. Therefore, the band gap of Ba3LaNb3O12 was wider than that of Ba5Nb4O15 according to the factor (ii). The band gap of La4Ti3O12 with layered perovskite structure with three-octahedra thickness was also wider than that of ALa4Ti4O15 with layered perovskite structure with four-octahedra thickness.
 | ||
| Fig. 5 Crystal structures of ALa4Ti4O15 (A = Ca and Ba) with layered perovskite structure.50 | ||
Ba5Nb4O15 shows a broad yellow emission with maximum at 580 nm.52 The photoluminescent properties of A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12 were measured as shown in Fig. 6. These materials did not show any emission at room temperature, but did so at 77 K. The onsets of excitation spectra of these materials agreed with those of absorption spectra, indicating that these emissions were due to the band gap excitation.
 | ||
| Fig. 6 Excitation and emission spectra of (I) niobates and (II) titanates; (a) Sr5Nb4O15, (b) Ba5Nb4O15, (c) CaLa4Ti4O15, (d) SrLa4Ti4O15, and (e) BaLa4Ti4O15 at 77 K. | ||
Information on the migration of excited energy in the layered oxide photocatalysts can be obtained by monitoring the emission of some doped lanthanide cations as guests.53–55 Photoluminescent properties of ALa4Ti4O15 (A = Ca and Ba) doped with 1% of Pr3+ were examined as shown in Fig. 7. Pr3+ would be doped in the site of La3+ at an A-site of the perovskite layer and interlayer, judging from the ionic radius and electric charge of Pr3+. Luminescence of the doped Pr3+ was observed for ALa4Ti4O15:Pr(1%) (A = Ca and Ba) by the host excitation at room temperature. This result indicates that the photo-generated carrier or excited energy in the host material can migrate to the Pr3+ guest. The ratio of an excitation band of a host around 280 nm to that of a Pr3+ guest at 449.7 nm for BaLa4Ti4O15 was larger than that for CaLa4Ti4O15. It suggests that excited energy in the perovskite framework migrates to the Pr3+ guest more easily for BaLa4Ti4O15 than CaLa4Ti4O15. It may also be due to the amount of Pr3+ in the perovskite framework of BaLa4Ti4O15 being greater than that in CaLa4Ti4O15. Reported niobate and titanate photocatalysts for water splitting show photoluminescence at 77 K.20,53–55 Moreover, the photo-generated carrier or excited energy can migrate in the perovskite layer. Therefore, A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12 were expected to work as photocatalysts for water splitting.
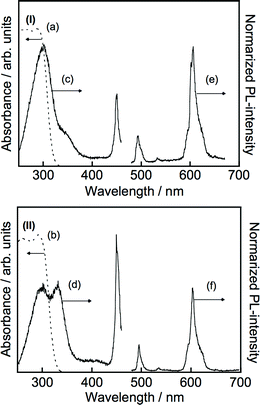 | ||
| Fig. 7 Diffuse reflection spectra and photoluminescence spectra of (I) BaLa4Ti4O15:Pr(0.1%) and (II) CaLa4Ti4O15:Pr(0.1%) at room temperature. (a) and (b): the diffuse reflection spectra, (c) and (d): the excitation spectra were obtained by monitoring luminescence of Pr3+ at 606 nm, (e) and (f): the emission spectra were obtained by host excitation at 280 nm. | ||
3-3. Photocatalytic water splitting
Table 2 shows photocatalytic activities of A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15(A = Ca, Sr and Ba), and La4Ti3O12. The amounts of NiO-loaded and pretreatment conditions were optimized for the photocatalytic water splitting over these photocatalysts. The activities of these photocatalysts without cocatalysts were low. However, when NiO cocatalysts were loaded, the activities were improved more than two orders of magnitude.| Photocatalyst | Preparation condition | Surface area | Band gap | NiO loaded | Pretreatmentb | Activity/µmol h−1 | |
|---|---|---|---|---|---|---|---|
| /m2 g−1 | /eV | (wt%) | H2 | O2 | |||
| a Catalyst: 0.5 g, pure water: 380 mL, inner irradiation cell made of quartz, 400 W high-pressure mercury lamp. b R and O indicate temperatures in K for H2reduction for 2 h and O2oxidation for 1 h, respectively. | |||||||
| La4Ti3O12 | 1473 K, 10 h | 3.4 | 3.95 | None | No | 9 | 0 |
| La4Ti3O12 | 1473 K, 10 h | 3.4 | 3.95 | 0.6 | No | 357 | 179 |
| La4Ti3O12 | 1473 K, 10 h | 3.4 | 3.95 | 0.6 | R673–O373 | 330 | 150 |
| CaLa4Ti4O15 | 1373 K, 10 h | 6.2 | 3.79 | None | No | 3 | 0 |
| CaLa4Ti4O15 | 1373 K, 10 h | 6.2 | 3.79 | 0.2 | No | 11 | 4 |
| CaLa4Ti4O15 | 1373 K, 10 h | 6.2 | 3.79 | 0.2 | R673–O373 | 593 | 276 |
| SrLa4Ti4O15 | 1373 K, 10 h | 6.2 | 3.82 | None | No | 2 | 0 |
| SrLa4Ti4O15 | 1373 K, 10 h | 6.2 | 3.82 | 0.7 | No | 23 | 11 |
| SrLa4Ti4O15 | 1373 K, 10 h | 6.2 | 3.82 | 0.7 | R673–O373 | 1171 | 546 |
| BaLa4Ti4O15 | 1323 K, 10 h | 6.1 | 3.85 | None | No | 5 | 2 |
| BaLa4Ti4O15 | 1323 K, 10 h | 6.1 | 3.85 | 0.5 | No | 118 | 62 |
| BaLa4Ti4O15 | 1323 K, 10 h | 6.1 | 3.85 | 0.5 | R673–O373 | 2300 | 1154 |
| Ba3LaNb3O12 | 1073 K, 10 h | 5.8 | 4.07 | None | No | 2 | 0 |
| Ba3LaNb3O12 | 1073 K, 10 h | 5.8 | 4.07 | 0.5 | R773–O473 | 1185 | 588 |
| Sr5Nb4O15 | 1073 K, 10 h | 7.9 | 4.04 | None | No | 3 | 0 |
| Sr5Nb4O15 | 1073 K, 10 h | 7.9 | 4.04 | 0.7 | R773–O473 | 2200 | 1100 |
| Ba5Nb4O15 | 1073 K, 10 h | 7.2 | 3.91 | None | No | 3 | 0 |
| Ba5Nb4O15 | 1073 K, 10 h | 7.2 | 3.91 | 0.5 | R773–O473 | 4021 | 1972 |
| Ba5Nb4O15 | 1273 K, 5 h | 1.9 | 3.87 | 0.1 | R773–O473 | 939 | 453 |
Fig. 8 shows the band structures of these titanate photocatalysts and a NiO cocatalyst.56 The band positions were estimated by the band gaps and the valance band maxima that were supposed to be +3.0 eV.57 Photogenerated electrons in a conduction band of a host material must transfer to a NiO cocatalyst that is a hydrogen evolution site. Therefore, it is required that the conduction band level of a photocatalyst is higher than that of a NiO cocatalyst. If the band level of photocatalyst does not satisfy the requirement, the activation pretreatment of H2reduction and subsequent O2oxidation is usually needed.44 The activation pretreatment for the NiO cocatalyst was effective for all photocatalysts as well as previously reported titanate and niobate photocatalysts.7,8,12,14,16,20,29,44,58 On the other hand, La4Ti3O12 showed high activity without the activation pretreatment because of the high conduction band level. NiOx(0.5 wt%)/BaLa4Ti4O15 and NiOx(0.5 wt%)/Ba5Nb4O15 showed the highest activities for water splitting among titanates and niobates, respectively. The photocatalytic activity of Ba5Nb4O15 calcined at 1073 K was higher than that at 1273 K. The SEM images indicated that Ba5Nb4O15 calcined at 1073 K had higher aspect ratio and smaller particle size than that at 1273 K as shown in Fig. 3. In the case of layered perovskite photocatalyst of which the interlayer space is not hydrated, the photogenerated e− and h+ have to reach the surface to react with water molecules. Therefore, the photocatalytic activity is improved by the preparation giving high crystallinity and fine particle. Charge separation of photo-generated e− and h+ might be enhanced by high anisotropy of the crystal structure.14 These factors mean that the photocatalytic activity of Ba5Nb4O15 that had been calcined at 1073 K was higher than that treated at 1273 K.
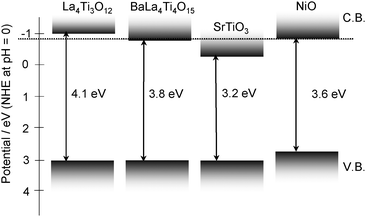 | ||
| Fig. 8 Band structures of various titanate photocatalysts. | ||
Table 3 shows the photocatalytic activities of BaLa4Ti4O15 loaded with various metals and oxides. NiOx/BaLa4Ti4O15 obtained by the impregnation method followed by the activation pretreatment showed the highest activity for water splitting. The activities of Ni/BaLa4Ti4O15 obtained by photo-deposition method and NiOx/BaLa4Ti4O15 prepared by photo-deposition method followed by oxidation pretreatment were low compared with that of NiOx/BaLa4Ti4O15 prepared by the impregnation method. Metallic Au and RuO2 cocatalysts were also effective for BaLa4Ti4O15.
| Cocatalyst (wt%) | Loading method | Pretreatmentb | Activity/µmol h−1 | |
|---|---|---|---|---|
| H2 | O2 | |||
| a Catalyst: 0.5 g, pure water: 380 mL, inner irradiation cell made of quartz, 400 W high-pressure mercury lamp. b R and O indicate temperatures in K for H2reduction for 2 h and O2oxidation for 1 h, respectively. | ||||
| None | — | — | 5 | 2 |
| Ni (0.5) | Photo-deposition | None | 10 | 0.5 |
| NiO (0.5) | Impregnation | O543 | 118 | 62 |
| NiOx (0.5) | Impregnation | R773–O473 | 1333 | 673 |
| NiOx (0.5) | Photo-deposition | R773–O473 | 432 | 203 |
| RuO2 (1.0) | Impregnation | O673 | 575 | 265 |
| Au (1.0) | Photo-deposition | None | 36 | 10 |
| IrO2 (1.0) | Photo-deposition | None | 3 | 0 |
| Pt (1.0) | Photo-deposition | None | 35 | 1 |
| PbO2 (1.0) | Photo-deposition | None | 5 | 0 |
The time course for water splitting on optimized NiOx(0.5 wt%)/BaLa4Ti4O15 photocatalyst is shown in Fig. 9. At the initial stage, NiOx(0.5 wt%)/BaLa4Ti4O15 produced H2 and O2 at the rates of 2.3 and 1.14 mmol h−1 using a 400 W high-pressure mercury lamp, respectively. The turnover number of the amount of reacted electrons/holes to the molar quantity of BaLa4Ti4O15 was 30 at 4 h of the reaction time. It clearly indicated that the reaction proceeded photocatalytically. The apparent quantum yield was 15% at 270 nm.
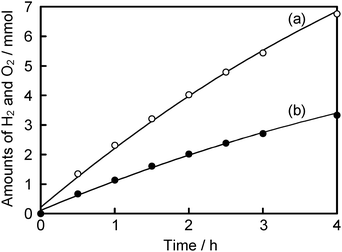 | ||
| Fig. 9 Photocatalytic water splitting over optimized NiOx (0.5 wt%)/BaLa4Ti4O15. (a): H2 and (b): O2. Catalyst: 0.5 g, pure water: 380 mL, inner irradiation cell made of quartz, 400 W high-pressure mercury lamp. | ||
Table 4 summarizes metal oxide photocatalysts with layered perovskite structures for water splitting. Although the detailed experimental condition was not the same for each other, the (111) plane-type layered perovskite oxide photocatalysts found in the present paper showed relatively high activities compared with layered titanate and niobate photocatalysts previously reported.
| Photocatalyst | Interlayer structure type | Cocatalyst | Band gap | Activity/µmol h−1 | Apparent quantum yield | Ref. | |
|---|---|---|---|---|---|---|---|
| /eV | H2 | O2 | (%) | ||||
| a Light source: 400–450 W high-pressure mercury lamp, reaction cell: inner irradiation cell made of quartz, reactant: pure water. NiOx indicates a pretreated NiO cocatalyst. | |||||||
| K2La2Ti3O10 | (100) | NiOx | 3.4 | 2186 | 1131 | — | 8 |
| Sr3Ti2O7 | (100) | NiOx | 3.2 | 144 | 72 | — | 29 |
| Sr4Ti3O10 | (100) | NiOx | 3.3 | 170 | 4.5 | 16 | |
| La2Ti3O9 | (100) | NiOx | 386 | — | 20 | ||
| La2Ti2O7 | (110) | NiOx | 3.8 | 441 | 220 | 12 | 20 |
| La2Ti2O7:Ba | (110) | NiOx | 3.8 | 5000 | 50 | 20 | |
| La4CaTi5O17 | (110) | NiOx | 3.8 | 499 | 20 | 12 | |
| BaLa4Ti4O15 | (111) | NiOx | 3.85 | 2300 | 1154 | 15 | This work |
| KCa2Nb3O10 | (100) | RuO2 | 96 | 47 | — | 22 | |
| Ca2Nb2O7 | (110) | NiOx | 4.3 | 101 | 7 | 13 | |
| Sr2Nb2O7 | (110) | NiOx | 4.0 | 110 | 36 | — | 14 |
| Ba5Nb4O15 | (111) | NiOx | 3.91 | 4021 | 1982 | 17 | This work |
3-4. Difference in photophysical and photocatalytic properties between CaLa4Ti4O15 and BaLa4Ti4O15
The order of photocatalytic activities of ALa4Ti4O15 (A = Ca, Sr, and Ba) was Ba > Sr > Ca. This order seems to depend on the ionic radii of cations at interlayer. Photocatalytic properties would depend on the anisotropy of layered perovskite structure. The difference in the framework distortion of perovskite structure between ALa4Ti4O15 (A = Ca, Sr, and Ba) photocatalysts is not significant for the activity as discussed in section 3-2. The shapes of these particles are similar to each other. Therefore, the difference in photocatalytic activities between ALa4Ti4O15 (A = Ca, Sr, and Ba) is due to the conduction band level and the anisotropy that are caused by A-site cations as shown in Fig. 5. It has been reported that the reactivity of e− and h+ on a TiO2 surface can be examined by observing on which plane Pt and PbO2 are photodeposited reductively and oxidatively, respectively.58 Therefore, the features of photodeposited metals and PbO2 were examined to see the effect of the anisotropy as shown in Fig. 10. Metallic Au was selectively deposited from AuCl4− not on a basal plane but on an edge plane. In contrast, PbO2 was oxidatively photodeposited from Pb2+ on the basal plane. Metallic Ni was reductively photodeposited from Ni2+ only on the basal plane with the characteristic whisker-like shape. Metallic Pt was non-selectively photodeposited from [PtCl6]2− on the whole surface. When metallic Pt was photodeposited from [Pt(NH3)4]2+, the particle size on the basal plane was smaller than that on the edge plane. In contrast, the plane-selective photodeposition was not observed for CaLa4Ti4O15. Moreover, A5Nb4O15 (A = Sr and Ba) in which A-site cations is the same as interlayer cations did not show the selective deposition neither. This unique selective photodeposition is due to the difference in adsorption property for ions between edge and basal planes, and the charge separation promoted by the structural anisotropy due to the different distribution of alkali earth metal cations. This character resulted in that BaLa4Ti4O15 showed higher activity than CaLa4Ti4O15.![SEM images of various metals and PbO2 loaded on BaLa4Ti4O15 photocatalysts by a photo-deposition method; (a) Au, (b) Ni, (c) PbO2, (d) Pt from [PtCl6]2−, (e) Pt from [Pt(NH3)4]2+, and (f) magnification of (e).](/image/article/2009/EE/b818922f/b818922f-f10.gif) | ||
| Fig. 10 SEM images of various metals and PbO2 loaded on BaLa4Ti4O15 photocatalysts by a photo-deposition method; (a) Au, (b) Ni, (c) PbO2, (d) Pt from [PtCl6]2−, (e) Pt from [Pt(NH3)4]2+, and (f) magnification of (e). | ||
The reduction with H2 at 773 K for 2 h followed by oxidation with O2 at 473 K for 1 h was carried out for the Ni/BaLa4Ti4O15 prepared by the photodeposition. The pretreatment gave NiOx loaded only on the basal plane of BaLa4Ti4O15. The shape of this NiOx was similar to the NiOx loaded by an impregnation method. The activity of this NiOx/BaLa4Ti4O15 photocatalyst was lower than that of NiOx/BaLa4Ti4O15 prepared by the impregnation method. It suggests that the NiO cocatalyst loaded on the edge plane was more effective than that on the basal plane.
4. Conclusions
A5Nb4O15 (A = Sr and Ba), Ba3LaNb3O12, ALa4Ti4O15 (A = Ca, Sr, and Ba), and La4Ti3O12 with a (111) plane-type layered perovskite structure were found to be new niobate and titanate photocatalyst materials for water splitting. A PC method was advantageous for the preparation of these photocatalysts at wide-range temperature. These photocatalysts showed photoluminescence at 77 K. Moreover, energy migration in the layered perovskite structure was confirmed by probing luminescence of doped Pr3+. This energy structure revealed by photoluminescent properties is preferable for photocatalysts. BaLa4Ti4O15 showed the highest activity for water splitting among ALa4Ti4O15 (A = Ca, Sr, and Ba) photocatalysts. Ba2+ and La3+ exist at interlayers of BaLa4Ti4O15 while only La3+ exists at an A-site of the perovskite layers. In contrast, Ca2+ and La3+ randomly exist at the interlayers and the perovskite layers of CaLa4Ti4O15. The anisotropic surface reactivity was confirmed by selective photodeposition of Pt, Ni, Au, and PbO2 for BaLa4Ti4O15 but no for CaLa4Ti4O15. These results indicated that the structural anisotropy due to the ordered positions of Ba2+ and La3+ in BaLa4Ti4O15 was one of the major factors for the high activity. The application of plate-like BaLa4Ti4O15 particles prepared by a PC method to other photocatalytic reactions besides water splitting is expected.Acknowledgements
This work was supported by Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Agency (JST) and a Grant-in-Aid (No. 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 050090) for the Priority Area Research (No. 417) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
050090) for the Priority Area Research (No. 417) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- K. Domen, J. N. Kondo, M. Hara and T. Takata, Bull. Chem. Soc. Jpn., 2000, 73, 1307 CrossRef CAS and references therein.
- H. Kato and A. Kudo, Catal. Today, 2003, 78, 561 CrossRef CAS and references therein.
- J. Sato, H. Kobayashi, K. Ikarashi, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2004, 108, 4369 CrossRef CAS and references therein.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC and references therein.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS.
- S. Ikeda, A. Tanaka, K. Shinohara, M. Hara, J. N. Kondo, K. Maruya and K. Domen, Microporous Mesoporous Mater., 1997, 9, 253 CAS.
- S. Ikeda, M. Hara, J. N. Kondo, K. Domen, H. Takahashi, T. Okubo and M. Kakihana, Chem. Mater., 1998, 10, 72 CrossRef CAS.
- H. Kato and A. Kudo, Catal. Lett., 1999, 58, 153 CrossRef CAS.
- A. Kudo, S. Nakagawa and H. Kato, Chem. Lett., 1999, 28, 1197 CrossRef.
- C. Mitsui, H. Nishiguchi, K. Fukamachi, T. Ishihara and Y. Takita, Chem. Lett., 1999, 28, 1327 CrossRef.
- H. G. Kim, D. W. Hwang, J. Kim, Y. G. Kim and J. S. Lee, Chem. Commun., 1999, 1077 RSC.
- H. Takahashi, M. Kakihana, Y. Yamashita, K. Yoshida, S. Ikeda, M. Hara and K. Domen, J. Alloys Compd., 1999, 285, 77 CrossRef CAS.
- A. Kudo, H. Kato and S. Nakagawa, J. Phys. Chem. B, 2000, 104, 571 CrossRef CAS.
- M. Machida, J. Yabunaka and T. Kijima, Chem. Mater., 2000, 12, 812 CrossRef CAS.
- Y. G. Ko and W. Y. Lee, Catal. Lett., 2002, 83, 157 CrossRef CAS.
- N. Sato, H. Kadowaki, H. Kobayashi, K. Ikarashi, H. Nishiyama and Y. Inoue, Chem. Lett., 2004, 33, 1452 CrossRef.
- K. Shimizu, Y. Tsuji, T. Hatamachi, K. Toda, T. Kodama, M. Sato and Y. Kitayama, Phys. Chem. Chem. Phys., 2004, 6, 1064 RSC.
- J. Kim, D. W. Hwang, H. G. Kim, S. W. Bae, J. S. Lee, W. Li and S. H. Oh, Top. Catal., 2005, 35, 295 CrossRef CAS.
- Y. Miseki, H. Kato and A. Kudo, Chem. Lett., 2005, 34, 54 CrossRef CAS.
- K. Yoshioka, V. Petrykin, M. Kakihana, H. Kato and A. Kudo, J. Catal., 2005, 232, 102 CrossRef CAS.
- Y. Ebina, N. Sakai and T. Sasaki, J. Phys. Chem. B, 2005, 109, 17212 CrossRef CAS.
- K. Shimizu, S. Itoh, T. Hatamachi, T. Kodama, M. Sato and K. Toda, Chem. Mater., 2005, 17, 5161 CrossRef CAS.
- H. Otsuka, K. Kim, A. Kouzu, I. Takimoto, H. Fujimori, Y. Sakata, H. Imamura, T. Matsumoto and K. Toda, Chem. Lett., 2005, 34, 822 CrossRef CAS.
- M. Machida, T. Mitsuyama and K. Ikeue, J. Phys. Chem. B, 2005, 109, 7801 CrossRef CAS.
- Y. Miseki, H. Kato and A. Kudo, Chem. Lett., 2006, 35, 1052 CrossRef CAS.
- T. Kurihara, H. Okutomi, Y. Miseki, H. Kato and A. Kudo, Chem. Lett., 2006, 35, 274 CrossRef CAS.
- R. Abe, M. Higashi, K. Sayama, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 2219 CrossRef CAS.
- H. Jeong, T. Kim, D. Kim and K. Kim, Int. J. Hydrogen Energy, 2006, 31, 1142 CrossRef CAS.
- S. Ikeda, M. Fubuki, Y. K. Takahara and M. Matsumura, Appl. Catal., A, 2006, 300, 186 CrossRef CAS.
- H. Kadowaki, N. Saito, H. Nishiyama and Y. Inoue, Chem. Lett., 2007, 36, 440 CrossRef CAS.
- W. Yao and J. Ye, Chem. Phys. Lett., 2007, 435, 96 CrossRef CAS.
- Y. Li, G. Chen, C. Zhou and Z. Li, Catal. Lett., 2008, 123, 80 CrossRef CAS.
- T. Mitsuyama, A. Tsutsui, T. Hara, K. Ikeue and M. Machida, Bull. Chem. Soc. Jpn., 2008, 81, 401 CrossRef CAS.
- O. C. Compton, C. H. Mullet, S. Chiang and F. E. Osterloh, J. Phys. Chem. C, 2008, 112, 6202 CrossRef CAS.
- Y. Lee, H. Terashita, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111, 1042 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416 RSC.
- R. Abe, K. Sayama, K. Domen and H. Arakawa, Chem. Phys. Lett., 2001, 344, 339 CrossRef CAS.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348 CrossRef CAS.
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 3829 RSC.
- M. Higashi, R. Abe, K. Teramura, T. Takata, B. Ohtani and K. Domen, Chem. Phys. Lett., 2008, 452, 120 CrossRef CAS.
- M. Kakihana and K. Domen, MRS Bull., 2000, 25, 27 CAS.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, J. Phys. Chem., 1986, 90, 292 CrossRef CAS.
- M. Wiegel, M. H. J. Emond, E. R. Stobbe and G. J. Blasse, J. Phys. Solids, 1994, 55, 773 CrossRef.
- Z. Zou, J. Ye and H. Arakawa, Chem. Phys. Lett., 2000, 332, 271 CrossRef CAS.
- A. Kudo and T. Sakata, J. Phys. Chem., 1996, 100, 17323 CrossRef CAS.
- A. Kudo and E. Kaneko, Microporous Mesoporous Mater., 1998, 21, 615 CrossRef CAS.
- M. Weiden, A. Grauel, J. Norwig and S. Horn, J. Alloys Compd., 1995, 218, 13 CrossRef CAS.
- T. A. Vanderah, T. R. Collins, W. Won-Ng, R. S. Roth and L. Farber, J. Alloys Compd., 2002, 346, 116 CrossRef CAS.
- Y. Tohdo, K. Kakimoto, H. Ohsato, H. Yamada and T. Okawa, J. Eur. Ceram. Soc., 2006, 26, 2039 CrossRef CAS.
- A. M. Srivastava, J. F. Ackerman and W. W. Beers, J. Solid State Chem., 1997, 134, 187 CrossRef CAS.
- A. Kudo and T. Sakata, J. Phys. Chem. B, 1995, 99, 15963 Search PubMed.
- A. Kudo, Chem. Mater., 1997, 9, 664 CrossRef CAS.
- A. Kudo and E. Kaneko, Microporous Mesoporous Mater., 1998, 21, 615 CrossRef CAS.
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett and N. D. Nicholson, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 643 RSC.
- D. E. Scaife, Sol. Energy, 1980, 25, 41 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167 RSC.
Footnote |
| † Current address: Materials and Structures Laboratory, Tokyo Institute of Technology, 4259 Nagatsuda, Midori-ku, Tokohama, 226-8503, Japan. |
| This journal is © The Royal Society of Chemistry 2009 |