Carbon dioxide (C16O2 and C18O2) adsorption in zeolite Y materials: effect of cation, adsorbed water and particle size
Pragati
Galhotra
a,
Juan G.
Navea
a,
Sarah C.
Larsen
a and
Vicki H.
Grassian
*ab
aDepartment of Chemistry, University of Iowa, Iowa City, IA 52246, USA. E-mail: vicki-grassian@uiowa.edu; Fax: +1 319 335 1270; Tel: +1 319 335 1392
bDepartment of Chemical and Biochemical Engineering, University of Iowa, Iowa City, IA 52246, USA
First published on 25th February 2009
Abstract
In this study, CO2 adsorption in the presence and absence of co-adsorbed H2O was investigated in zeolite Y. Several different zeolite Y materials were investigated including commercial NaY, commercial NaY ion-exchanged with Ba2+ and nanocrystalline NaY; herein referred to as NaY, BaY and nano-NaY. Following heating of these zeolites to 573 K and cooling to room temperature, CO2 was adsorbed as a function of pressure. FTIR spectra show that a majority of CO2 adsorbs in the pores of these three zeolites (NaY, BaY and nano-NaY) in a linear complex with the exchangeable cation, as indicated by the intense absorption band near 2350 cm−1, assigned to the ν3 asymmetric stretch of adsorbed CO2. Most interestingly is the formation of carbonate and bicarbonate on the external surface of nano-NaY zeolites as indicated by the presence of several broad absorption bands in the 1200–1800 cm−1 region, suggesting unique sites for CO2 adsorption on the surface of the nanomaterial. For the other two zeolite materials investigated, bicarbonate formation is only evident in BaY zeolite in the presence of co-adsorbed water. Adsorption of 18O-labeled carbon dioxide and theoretical quantum chemical calculations confirm these assignments and conclusions.
Broader contextAs carbon dioxide plays an important role in global warming, it has become necessary to think about new approaches and novel ideas for carbon dioxide management. Therefore, carbon dioxide storage and conversion are topics of great importance to the scientific community and the public because of the global implications related to climate change, sustainability and energy. There is a great deal of interest in using microporous materials for the capturing and storage of carbon dioxide. The research described herein focuses on fundamental interactions of carbon dioxide in zeolite materials, including nanocrystalline NaY, as investigated by FTIR spectroscopy, isotope studies and theory. Differences are observed between the different zeolite materials investigated, with the formation of carbonates and bicarbonates depending upon the experimental conditions, dry versus wet, the exchangeable cation and, very interestingly, the zeolite particle size. For nanocrysalline zeolite materials, i.e. materials with zeolite crystals less than 100 nm, it is shown here that the external surface sites play a role in carbon dioxide uptake and these sites can be utilized to enhance the uptake of carbon dioxide. Furthermore, these external sites may have the potential to be useful in the conversion of carbon dioxide to more useful compounds such as methanol. |
Introduction
Carbon dioxide (CO2) is the fourth most abundant gas in the atmosphere uniformly distributed over the earth's surface with a concentration of about 385 ppm.1 Globally, CO2 concentration has increased by more than 40% since the Industrial Revolution and is continuously increasing.2 As CO2 plays an important role in global warming, it has become necessary to think about new approaches and novel ideas for CO2 management. Some of the strategies to reduce its concentration include reducing emission from vehicles and industries, using alternative sources of energy (wind energy, tidal energy, and solar energy), gas capture and adsorption, sequestration and recycling similar to radioactive waste, and adsorbing it on reactive substrates such as metal oxides. Recent studies indicate that zeolites have the potential to adsorb, store and convert CO2 into more useful products.3–7Zeolites can perhaps be ideal for the mass scale sequestering and/or conversion of CO2 to more useful products. Zeolites are crystalline, porous aluminosilicates that are extensively used in catalysis, chemical separations and as adsorbents.8,9 Nanocrystalline zeolites are a class of zeolite materials with particle sizes below 100 nm. Due to their small size, nanocrystalline zeolites have unique properties such as both high internal and external surface areas, high concentration of reactive sites on the external surface and enhanced diffusional properties.8,9 Recent studies have shown that nanocrystalline zeolites have potential use in a number of environmental applications.8–12Faujasite zeolites (e.g.NaY) are considered promising environmental catalysts because of their cation exchange capacity and acid–base properties, and nanocrystalline NaY zeolites have shown enhanced performance for the selective catalytic reduction of NOx (NOx = NO + NO2) with urea.13
In this study, adsorption of C16O2 and C18O2 was investigated on synthesized nanocrystalline NaY zeolite (nano-NaY) as well as commercially available zeolite catalysts (NaY, BaY) under both dry conditions and in the presence of co-adsorbed water. FTIR spectroscopy was used to probe adsorbed CO2. In particular, we present in this study: (i) experimental data for CO2 adsorption in three zeolite Y materials including isotope data to support vibrational mode assignments; (ii) CO2 adsorption in a new type of zeolite: nanocrystalline zeolites with particle size below 100 nm, where unique carbon dioxide adsorption sites are observed due to the high external surface area and; (iii) experimental data which probes the impact of adsorbed water on the reactivity of the three zeolites investigated (NaY, BaY and nano-NaY). Besides the isotope data, quantum chemical calculations are used to aid in the mode assignments. It is shown here that zeolite Y adsorbs CO2 both molecularly, in a linear bonded complex, as well as through the formation of carbonates and bicarbonates depending upon the zeolite and experimental conditions, i.e. in the presence and absence of co-adsorbed water, the exchangeable cation and the zeolite particle size.
Experimental and theoretical methods
Zeolite materials
Commercial zeolite, NaY (Aldrich) and synthesized nano-NaY11 were used for these studies. The crystal sizes were ∼1 µm and 38 nm for NaY (Aldrich) and nano-NaY, respectively. Fig. 1A and 1B shows scanning electron micrographs of these two different zeolite samples. The most important difference between the two samples is the particle size and the difference in external surface area. The external surface area for the two sizes has been determined to be ca. 1 m2 g−1 and 100 m2 g−1, respectively.14 As discussed previously, the external surface area for the 38 nm nano-NaY is 100 times greater than that of the commercial sample. The external surface provides additional reaction sites and functionality in these nanocrystalline zeolite materials. The Si/Al ratio determined for NaY (Aldrich) and nano-NaY is 2.0 and 1.8, respectively, as measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES).
BaY was prepared from NaY (Aldrich) using standard ion-exchange procedures as described previously.15 Briefly, aqueous 0.5 M BaCl2 solution was added to NaY and stirred with heating to 363 K for 24 h. The resulting solution was washed with deionized water and filtered until the filtrate was free of chloride ions. The Ba/Al ratio was determined by ICP/OES to be 0.65.
Transmission FTIR spectroscopy
In situ transmission FTIR spectroscopy was employed to investigate the adsorption of CO2 in zeolite materials. The infrared sample cell used here has been described previously.12 The Mattson Galaxy 6000 infrared spectrometer is equipped with a narrowband MCT detector. It consists of a stainless steel IR cell equipped with two BaF2 windows for infrared measurements and is connected to a vacuum/gas handling system. Inside the IR cell is the sample holder that consists of a 3 cm × 2 cm photoetched tungsten grid held in place by nickel jaws. The nickel jaws are attached to copper leads so that the sample can be resistively heated. A thermocouple wire attached directly to the tungsten grid is used to measure the temperature of the sample. Approximately 12 mg of zeolite sample was mixed with a few drops of methanol and sonicated for 20 minutes. This preparation resulted in the formation of a suspension. The suspension was then coated onto half of the tungsten grid and allowed to dry. The IR cell was then placed inside the stainless steel cube that sits on a linear translator inside the FTIR sample compartment. The linear translator allows each half of the sample grid to be translated into the infrared beam and permits the detection of gas phase and adsorbed species under identical reaction conditions. The samples were heated overnight under vacuum at 573 K or higher temperature to remove adsorbed water. Reactant gases were introduced into the reaction cell through the gas handling system. Two absolute pressure transducers were used to monitor the pressures. In the dry experiments, i.e. in the absence of co-adsorbed water, the zeolite was equilibrated for around 30 minutes with carbon dioxide prior to a spectrum being recorded. Carbon dioxide and water were also co-adsorbed into the zeolite. In these experiments, water vapor (1% RH) was introduced into the reaction cell and allowed to equilibrate followed by evacuating the cell for an hour to remove any gas-phase water in the reaction cell. Carbon dioxide was then adsorbed at different pressures at a temperature of 298 K. The exact pressures are given in the figure captions. Each spectrum was obtained by averaging 64 scans at an instrument resolution of 4 cm−1. Each absorbance spectrum shown represents a single beam scan referenced to the appropriate single beam scan of the clean zeolites or the blank grid, unless otherwise noted. It should be noted that the zeolite absorbs strongly below 1300 cm−1 and can result in a sloping background in this region as a result of poor spectral subtraction.Quantum chemical calculation of carbon dioxide, bicarbonate and carbonate complexes
In order to better understand the influence of adsorption on the structure and vibrational frequencies of carbon dioxide, several quantum chemical calculations were performed. All the electronic structures minimizations and vibrational calculations were performed using a density-functional method, with the hybrid Becke (B3) exchange and the Lee, Yang, and Parr (LYP) correlation (B3LYP) functionals in combination with the Los Alamos ECP and double-ζ level of theory (LanL2DZ) as implemented in the GAUSSIAN 03 package.16As it has been previously shown that the primary interaction between carbon dioxide and zeolites is with the exchangeable cation,17–21 calculations were done using different gas-phase cluster structures with two different cations, Na+ and Ba2+. In order to have a formally neutral cluster, the hydroxyl complexes of the corresponding cations have been used. The electrostatic contribution from the hydroxyl counter ion (OH) serves as a model to resemble the interaction between the cation and the zeolite oxygen atoms. Fig. 2 shows the minimized structures for the different models: (I) and (II) show the CO2 complex of the corresponding metal hydroxide, Na(OH) and Ba(OH)2. In addition, possible carbonate and bicarbonate products have also been investigated with B3LYP LanL2DZ, and the geometry optimization for carbonate and bicarbonate models are shown in Fig. 2 as the neutral clusters (III) BaCO3; (IV) Na2CO3; (V) BaOH(CO3H) and; (VI) Na(CO3H).
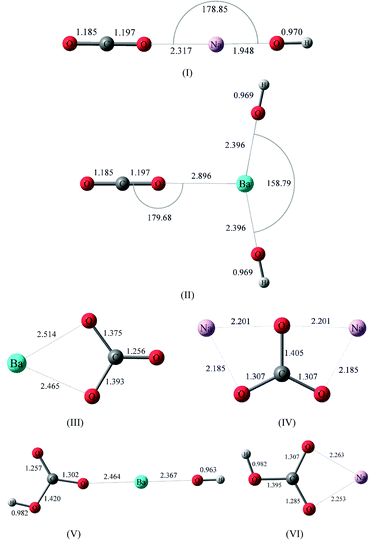 | ||
| Fig. 2 Energy minimized structures for (I) (OH)Na⋯O–C–O and (II) (OH)2Ba⋯O–C–O, and energy minimized structures for carbonate and bicarbonate complexes with Ba2+ and Na+: (III) BaCO3; (IV) Na2CO3; (V) Ba(OH)CO3H; and (VI) (OH)NaCO3H. | ||
To aid in the interpretation of the experimental data, calculations were performed for both C16O2 and C18O2 on different carbon dioxide isotopes. These fundamental vibrational frequencies on gas-phase carbon dioxide have been reported in previous studies using B3LYP/6-31G(d) level of theory.17 Here, in order to determine the effect of the basis set used, fundamental vibrational frequencies were determined for C16O2 and the labeled C18O2 at B3LYP LanL2DZ level of theory. In addition, the frequencies of the clusters were calculated using the basis set 6-311 + G(d,p) for Na+ coordinated to CO2, as well as several other ions coordinated to CO2, to determine the effect of the LAN2DZ basis set on the frequencies. Differences in the frequencies calculated between the two basis sets were determined to be small (≤2%), which is in good agreement with previously reported studies.22
It is important to note that because all the calculated frequencies correspond to the vibrational modes of the molecule in gas phase, the experimentally observed frequencies of the adsorbed species may be slightly different than those in the calculated spectra. Furthermore, the calculated harmonic frequencies tend to differ from experimental values due to a combination of anharmonicity effects, electron density correlation effects and basis set deficiencies. In order to best correct for these, an empirical scaling has been applied based on the harmonic frequencies calculated and the observed experimental data for gas-phase carbon dioxide. The scaling factor used to correct the calculated CO2 frequencies was obtained from averaging the calculated-to-experimental ratio of the different vibrational modes for the isolated CO2 molecule that are infrared active: the asymmetric stretching ν3 and the doubly degenerated ν2 bending mode (the symmetric stretching ν1 is only Raman active).22–25 The averaged scaling factor was determined using also the experimental and calculated values for the isotopic labeled C18O2. Thus, the frequencies of the CO2 vibrational modes have been scaled using an empirical scaling factor of 0.95. The scaling factor facilitates the assignments of the vibrational frequencies measured using FTIR spectroscopy.
Results and discussion
Quantum chemical calculations of carbon dioxide, bicarbonate and carbonate complexes
CO2 interactions with hydroxyl metal clusters were calculated, with the surface being modeled as Mn(OH) (M correspond to the ion exchange, Ba2+ or Na+), as illustrated in Fig. 2. The OH counter ion modified the electron density associated with the carbon dioxide molecule and the cation (Mn+) of interest, thus resembling the influence of a local zeolite structure in a simplified fashion. It can be seen that for the minimized structures shown in I and II, the interaction with the cation causes an increase in the length of the C–O bond adjacent to the cation, subsequently decreasing the length of the other C–O bond. As discussed below, this change in molecular symmetry gives rise to an infrared active symmetric ν1 vibrational mode. The calculated bond lengths for carbon dioxide are identical for both structures containing either Na+ or Ba2+. In particular, the bond lengths calculated for the clusters are calculated as: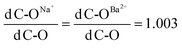 | (1) |
Importantly from the perspective of this study, the ν1 vibrational mode becomes IR active as a result of the interaction with the cation and the asymmetry in the bond lengths. Fundamental vibrational frequencies calculated for unlabeled and 18O labeled gas phase CO2 and CO2clusters (I and II) are given in Tables 1 and 2, respectively, along with their relative intensities. The reported frequencies are calculated at the same level of theory and scaled to a proper scaling factor, as discussed in the Experimental. Compared with the asymmetric stretch of carbon dioxide (Table 1), ν3, and the Raman active ν1, both vibrational frequencies show a surface influence. In addition, Table 1 shows the calculated frequencies for the equivalent-energy in-plane and out-of-plane CO2 bending vibrational modes (ν2). Upon interaction with the surface, this doubly degenerate CO2 bending mode (ν2) of the isolated molecule is separated into two different vibrations attributed to the change of symmetry of the molecule: an in-plane bending mode, ν2 (O–C–O)a, and an out-of-plane bending mode, ν2 (O–C–O)b. For the models shown in Fig. 2, the symmetry is approximately kept in most cases, although the contrast between these two bending modes becomes more pronounced. This energy difference in the ν2 bending modes is more obvious in the (OH)2Ba⋯O–C–O cluster, where an in-plane bending mode could be further distinguished from an out-of-plane bending mode due to cluster geometry. On a zeolite frame, this symmetry distortion will be resembled by the ion-exchange solvating zeolite ring. The CO2 ν3 vibrational frequency difference upon interaction with Na(OH) and Ba(OH)2 is about 2 cm−1. In addition, there is a transition dipole moment now for ν1 symmetric frequency mode, otherwise inactive for gas-phase CO2.
| Vibrational mode | C16O2 | C18O2 |
|---|---|---|
| ν1 | 1335 | 1258 |
| ν2 | 647 | 637 |
| ν3 | 2408 | 2363 |
| Vibrational modes | (OH)Na⋯CO2 | (OH)2Ba⋯CO2 |
|---|---|---|
| a Calculated scaled vibrational frequencies in cm−1 for C16O2 complexes. b Values in parentheses represent the calculated scaled vibrational frequenciesa for the analogous C18O2 complexes. | ||
| ν1 | 1356a (1268)b | 1354 (1276) |
| ν2 (O–C–O)a | 643 (634) | 645 (636) |
| ν2 (O–C–O)b | 643 (632) | 641 (631) |
| ν3 | 2442 (2393) | 2439 (2402) |
| Vibrational modes | Na2CO3 | BaCO3 |
|---|---|---|
| ν1 | 945 | 892 |
| ν3 (O–C–O)a | 1263 | 1010 |
| ν3 (O–C–O)b | 1583 | 1625 |
| Vibrational modes | Na(HCO3) | Ba(OH)(HCO3) |
|---|---|---|
| ν1 | 1001 | 1241 |
| ν3 (O–C–O)a | 1409 | 1395 |
| ν3 (O–C–O)b | 1790 | 1668 |
Fig. 2 also shows the minimized structures for possible carbonate and bicarbonate adsorbed products associated with both ion-exchanges examined and using the same level of theory. The vibrational frequency calculations for carbonate/bicarbonate adsorbed to the surface show three bands, where the two higher frequencies correspond to the split in the ν3 degenerate mode upon interaction with the surface and subsequent loss of symmetry; ν3 is, therefore, split into ν3 (O–C–O)a and ν3 (O–C–O)b bands. Table 2 gives the calculated ν1 and ν3 vibrational frequencies for carbonate. Frequencies were scaled using the 0.95 scaling factor, as discussed in the Experimental. In addition, it is important to point out that the presence of water solvating molecules decrease the interaction between the carbonate/bicarbonate species with the surface, which affects the geometry of the molecule. Consequently, the frequency difference for the ν3 (O–C–O)a and ν3 (O–C–O)b modes decreases as the carbonate/bicarbonate interaction with the ions decrease.20 This is reflected in the frequencies for the ν3 (O–C–O)a and ν3 (O–C–O)b stretching modes. Vibrational frequency calculations of the isolated bihydrated barium carbonate cluster, Ba(CO3)2·(H2O)2, show that the ν3 (O–C–O)a and ν3 (O–C–O)b bands are 1124 cm−1 and 1630 cm−1 respectively. This represents a reduction in the magnitude of the band split (Δν3) from 615 cm−1 under dry conditions, to 506 cm−1 in the presence of two solvating water molecules; this difference between the isolated Ba(CO3)2 and the Ba(CO3)2·(H2O)2 is attributed to the carbonate ion relaxing to a “less coordinated” state, and confirms previous observations on different surfaces.17,26 If carbonate/bicarbonate species are produced, analogous ν3 (O–C–O)a and ν3 (O–C–O)b bands are expected to be observed in the experimental infrared data.
Fig. 3 summarizes and provides in stick format the vibrational frequencies calculated from several of the cluster models. These data are used as a guide for the interpretation of the experimental data. It is clear from Fig. 3 that bicarbonate products have an intense band below 1500 cm−1. In addition, Fig. 3C shows calculated frequencies for vibrational modes of carbonate and bicarbonate coordinated to aluminium oxide binuclear clusters. These frequencies are taken from Baltrusaitis et al. and is used to interpret the data for nanocrystalline zeolites spectra (vide infra).17,26
 | ||
| Fig. 3 B3LYP LanL2DZ Calculated frequencies for both (OH)nM⋯16O–C–16O cluster and (OH)nM⋯18O–C–18O cluster. 3A represents the calculated spectra of (OH)Na clustered with CO2 unlabeled (solid blue line) and labeled (dashed blue line); 3B represents the calculated spectra of (OH)2Ba clustered with CO2 unlabeled (solid blue line) and labeled (dashed blue line) and the calculated spectra of (OH)Ba(HCO3) cluster (green solid line); 3C represents the vibrational modes for aluminium oxide carbonate complexes (red line) and aluminium oxide bicarbonate complexes (green line) as reported in ref. 26. | ||
Transmission FTIR spectroscopy of carbon dioxide adsorbed in NaY, BaY and nano-NaY under dry conditions
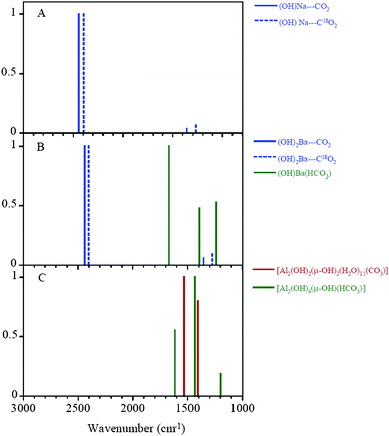
Fig. 4 shows the IR spectra collected upon adsorption of C16O2 in NaY zeolite as a function of pressure in the spectral region between 2000 and 2600 cm−1. As expected, an intense absorption band at 2351 cm−1 appears which is assigned to the asymmetric stretching mode (ν3) of CO2. The ν3 peak is shifted from the gas-phase value of 2347 cm−1 as suggested by the calculations due to the interaction with the exchangeable cation. This band increases in intensity as a function of C16O2 pressure. As the equilibrium pressure (Peq) of C16O2 increases, two smaller bands grow in on the low and high frequency side of the ν3 peak. These bands are most distinct at the highest CO2 pressures. Comparison with previous reports of carbon dioxide adsorption in zeolite materials suggests that absorptions at 2405 cm−1 and 2288 cm−1 are due to the combination modes of the ν3 mode with the framework M⋯O stretching modes of the zeolite as observed for NaZSM.27,28 | ||
| Fig. 4 FTIR spectra of adsorbed C16O2 in dry commercial NaY zeolite as a function of pressure (P = 1.0, 5.2, 10.6, 14.2 and 20 Torr). The spectral region shows the ν3 mode of adsorbed carbon dioxide. | ||
Fig. 5 shows an expanded spectral region, from 1200 to 4000 cm−1, for C16O2 adsorbed in NaY at a pressure of 20 Torr. As seen in Fig. 5, several more bands are present in the spectrum. A sharp peak is observed near 1382 cm−1 due to the symmetric stretch of CO2. As already noted, in the gas-phase this peak is IR inactive. However, this peak becomes IR active when CO2 is adsorbed in the zeolite due to interaction with the exchangeable cation, and the symmetry is lowered upon interaction because of two unequal bond lengths of C–O bond, as also indicated by theoretical calculation, giving rise to an IR active mode. Additional bands are observed in the spectrum and are assigned to other vibrational modes of adsorbed carbon dioxide, some of which are overtones and some are combination bands. One of them is a small peak observed at around 1276 cm−1 for these zeolites, which is a combination band arising from Fermi resonance between the Raman-active ν1 symmetric vibration and the overtone of the bending mode ν2 (2ν2). As already discussed, due to the reduction in symmetry of carbon dioxide on interaction with the zeolite surface this mode becomes IR active as well. The combination bands of ν3 + ν1 and ν3 + 2ν2 at 3713 and 3605 cm−1 are also present and have been reported earlier in the literature.28 These experimental frequencies are summarized in Table 3.
| Vibrational Modes | Gas phase28 | NaY | NaY/H2Oc | BaY | BaY/H2Oc | Nano-NaY | Nano-NaY/H2O c |
|---|---|---|---|---|---|---|---|
| a Experimental frequencies in cm−1. b C18O2 (1 Torr) frequencies in parentheses. c In the presence of co-adsorbed water. d This absorption band is broad and underneath the sharper feature in the spectrum (see Fig. 8 and text for details). | |||||||
| CO2 linear complexes | |||||||
| ν1 | 1388.3 | 1382 (1336) | 1382 | 1379 (1330) | 1382 (1377) | 1381 (1333) | 1381 (1361) |
| ν2 | 667.3 | — | — | — | — | — | — |
| ν3 | 2347.3 | 2351 (2318) | 2351 | 2349 (2318) | 2349 (2286) | 2351 (2319) | 2351 (2316) |
| ν3 + ν1 | — | 3713 (3632) | 3712 | 3707 (3626) | 3710 | 3713 | 3714 |
| ν3 + 2ν2 | — | 3605 (3527) | 3605 | 3597 (3520) | 3604 | 3604 | 3604 |
| 2ν2 | — | 1276 (1227) | 1276 | 1271 (1222) | 1268 | 1276 | — |
| ν3 + ν(M–O) | — | 2405 (2289) | 2403 | 2401 (2367) | 2400 (2349) | 2405 (2382) | 2402 (2352) |
| ν3 − ν(M–O) | — | 2288 (2253) | 2288 | 2285 (2253) | 2285 (2249) | 2287 (2253) | 2287 (2252) |
| Carbonate and Bicarbonate | |||||||
| ν1 | — | — | — | — | — | 1640 (1631) | 1639 |
| ν3 (O–C–O)a | — | — | — | — | 1448 (1439) | 1461 (1368) | 1408 |
| ν3 (O–C–O)b | — | — | — | — | 1382 (1377) | 1381d (1333) | 1381d |
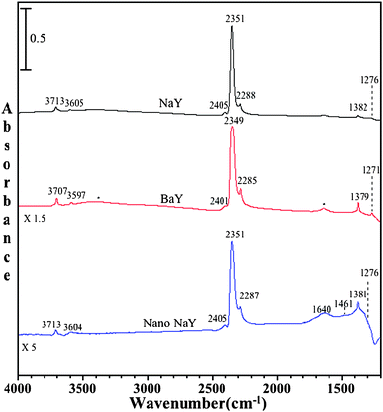 | ||
| Fig. 5 FTIR spectra of adsorbed C16O2 on dry NaY, BaY and nano-NaY zeolite at a CO2 pressure of 20 Torr and temperature of 296 K. The asterisks in the BaY spectrum denote absorptions due to the stretching and bending modes of a small amount of adsorbed water. | ||
In addition to C16O2 adsorption in NaY, Fig. 5 shows C16O2 adsorption in BaY and nano-NaY zeolites. Interestingly, the ν3 vibrational mode for the commercially available NaY, as well as for nanocrystalline NaY zeolite, is observed at exactly the same wavenumber, but for BaY it appears at a slightly different frequency. As can be seen, the experimental ν3 frequency appears at 2351 cm−1, 2349 cm−1 and 2351 cm−1 for NaY, BaY and nano-NaY, respectively. This relatively small shift in the ν3 mode for the BaY zeolite as compared to the NaY zeolites is in agreement with the calculations.
Besides the intense ν3 vibrational band, a number of other vibrational bands are observed in the BaY and nano-NaY spectra that are associated with C16O2 adsorbed inside the zeolite cage. These include the ν1 symmetric vibration and several combination and overtone vibrations.27 The vibrational frequencies for these modes are given in Table 3. Of particular interest are the differences observed in these spectra for the different zeolite materials. In comparing the 1200 to 1800 cm−1 spectral region for the NaY and nano-NaY zeolites, there is evidence for additional bands observed in nano-NaY spectra. Fig. 6 shows the IR spectra collected upon adsorption of C16O2 in nano-NaY zeolite as a function of pressure in the spectral region between 1200 and 4000 cm−1. In particular, for nano-NaY there are several broad features near 1640, 1461 and 1381 cm−1. As the carbon dioxide equilibrium pressure increases, one can, with clarity, distinguish these broad features. The FWHM (full width half maximum) for these peaks are greater than the other absorption bands in the spectrum by more than 20 cm−1. Although the peak at 1381 cm−1 has some contribution to the symmetric stretching mode as already discussed, there is a broad band underlying the sharper feature.
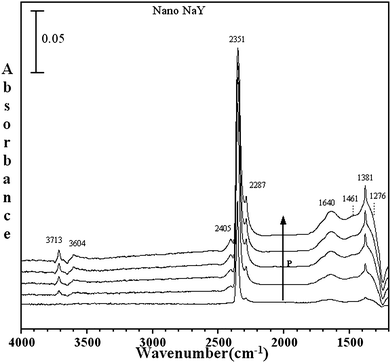 | ||
| Fig. 6 FTIR spectra for of adsorbed C16O2 as a function of P on nano-NaY (P = 1.0, 5.2, 10.6, 14.4 and 19.8 Torr). | ||
Based on earlier studies, these new absorption bands are proposed to be due to the formation of bicarbonate and carbonate species on extra framework aluminium (EFAL) sites8,13 known to be present on the external surface of nanocrystalline zeolites. As has been shown previously, nanocrystalline zeolites not only possess high surface area but in fact have a high concentration of reactive surface sites because of the increased external surface area due to the smaller particle size. EFAL sites are the most notably active catalytic sites in nanocrystalline zeolites that cause these zeolites to demonstrate higher activity as compared to commercial zeolites with large particle size. For example, it has been previously shown that molecules, e.g. NO2, can adsorb to these sites yielding surface complexes unique to nanocrystalline NaY zeolite.12 We propose here that these EFAL sites play a similar role in the adsorption of CO2 in the nanocrystalline NaY zeolite and result in the formation of carbonate and bicarbonate species.
Baltrusaitis et al.17,26 have calculated the vibrational frequencies of bicarbonate and carbonate in binuclear aluminium oxide cluster complexes and have shown these to be in good agreement with the experimental IR frequencies measured for these same species adsorbed on aluminium oxide surfaces. The results of these calculations are summarized in Fig. 3C. It is proposed that the absorption bands seen in the spectra region from 1200 to 1800 cm−1 are a result of carbon dioxide uptake on the EFAL sites, and the formation of carbonate and bicarbonate species on these sites can be explained as being similar to that observed on aluminium oxide surfaces. Bicarbonate species formation can be explained by the reaction mechanism (Scheme 1) already proposed.17 According to this mechanism, CO2 is pre-adsorbed on the surface of the zeolite and is followed by intermolecular proton transfer by the neighboring hydroxyl groups of the extra framework aluminium sites (EFAL), represented by M, resulting in bicarbonate formation. Similar reactions are likely to occur on the EFAL sites on the nanocrystalline external surface.17
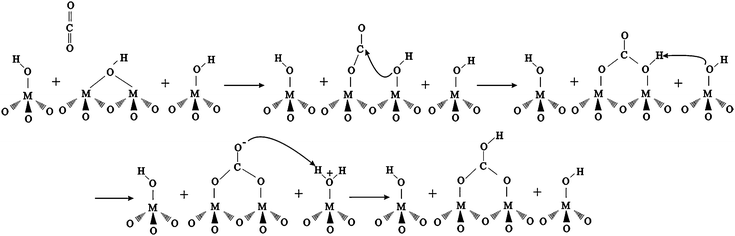 | ||
| Scheme 1 | ||
As isotope-labeling experiments can provide additional information about the adsorbed species, adsorption of 18O-labeled CO2 on all zeolites was also investigated. It is important to note that the pressure of carbon dioxide used in these isotope studies is 1 Torr for both C16O2 and C18O2. An example of the spectra collected for the isotope data is shown in Fig. 7. The spectra shown are for carbon dioxide adsorption in BaY zeolite with C18O2 and C16O2. It is seen that there are some shifts in the peak positions to lower wavenumbers for C18O2, as expected. These shifts are consistent with the quantum chemical calculations for CO2 interaction with the cation, as shown in complex (I) and (II), as well as in the calculated frequencies.
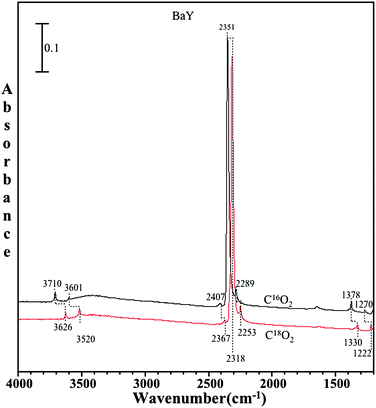 | ||
| Fig. 7 FTIR spectra of adsorbed C16O2 (black) and adsorbed C18O2 (red) on BaY zeolite at a pressure of 1 Torr and a temperature of 296 K. | ||
Transmission FTIR spectroscopy of carbon dioxide adsorbed in NaY, BaY and nano-NaY in the presence of co-adsorbed water
CO2 adsorption was carried out on zeolite surfaces as a function of increasing pressure of CO2 after pre-adsorbing water in the zeolite at 1% relative humidity. Following the introduction of 1% RH at 296 K, the gas phase water was pumped out leaving adsorbed water in the zeolite pores. Evidence for adsorbed water is seen in the spectra shown in Fig. 8, with an absorption band near 1640 cm−1 due to the bending mode of adsorbed water.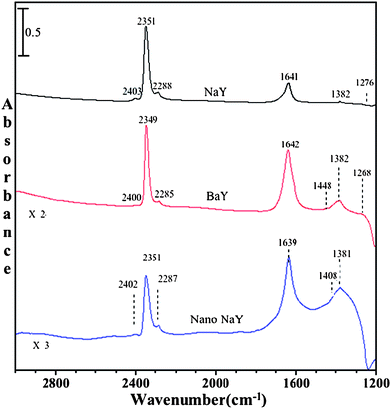 | ||
| Fig. 8 FTIR spectra of adsorbed C16O2 in the presence of co-adsorbed water in NaY, BaY and nano-NaY zeolite at a CO2 pressure of 20 Torr and temperature of 296 K. | ||
Fig. 8 shows the FTIR spectra collected upon adsorption of C16O2 and co-adsorbed water on NaY, BaY and nano-NaY zeolite. In all the three zeolites, peaks due to molecularly adsorbed CO2 were observed, similar to what is observed for CO2 adsorption in the dry zeolite. The frequencies of these absorption bands are given in Table 3. However, there is an overall decrease in the integrated absorbance of these bands for experiments with co-adsorbed water, suggesting that adsorption of water blocks the zeolite pores. For example, at similar pressures, in the presence of water there is ca. 20% decrease in the integrated area of ν3 band of adsorbed carbon dioxide. This decrease in integrated area suggests that co-adsorbed water inhibits carbon dioxide adsorption and blocks sites for adsorption.
As noted previously, we are interested in the differences in carbon dioxide adsorption in the different zeolite materials. For these experiments, in the presence of co-adsorbed water, BaY shows new absorption bands in the region below 1500 cm−1. Beside the absorption band at 1642 cm−1 due to adsorbed water, several other broad bands also become present. A broad band at around 1448 cm−1, with a FWHM of around 45 cm−1, appears upon adsorption of CO2 in the presence of co-adsorbed water; the band at 1382 cm−1 is also quite broad. We suggest that these absorptions, and some of which might be hidden under the intense bending mode of adsorbed water, are due to the presence of bicarbonate. Water most likely activates BaY zeolite for the formation of bicarbonate species. The reason for this difference lies in the exchangeable ion, where the polarizability of Ba2+ (in BaY zeolite) is more than Na+ cation (in NaY zeolite).
Fig. 9 shows the spectra of both C16O2 and C18O2 on BaY zeolite in the presence of co-adsorbed water. The C18O2 and C16O2 pressure is 1 Torr in both cases. Upon absorption of 18O-labeled carbon dioxide on wet zeolite surfaces, a red shift is observed in all peaks with the exception of the peaks from adsorbed water, further providing evidence that carbonate is formed in BaY in the presence of co-adsorbed water.
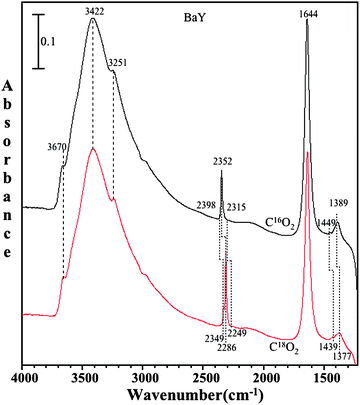 | ||
| Fig. 9 FTIR spectra of adsorbed C16O2 (black) and adsorbed C18O2 (red) in the presence of co-adsorbed water on BaY zeolite at pressure of 1 Torr and temperature of 296 K. | ||
Conclusions
The adsorption of CO2 was carried out on zeolite Y materials (NaY, BaY and nano-NaY) both under dry conditions as well as in presence of co-adsorbed water. Quantum chemical calculations were also performed. Both the theoretical calculations and experimental data provide complementary information and lead to a better understanding of adsorbed CO2 in these zeolite materials. Although CO2 adsorbs molecularly, as in a linear bonded complex, in all three zeolites, differences are observed between the various zeolite materials with the formation of carbonates and bicarbonates depending upon the experimental conditions, dry versus wet, the exchangeable cation and, very interestingly, the zeolite particle size. This carbonate/bicarbonate species formation on the external surface sites of nano-NaY zeolite upon CO2 adsorption, suggests that nanocrystalline zeolite adsorption sites are efficient in conversion of carbon dioxide. These studies show that zeolites can store carbon dioxide in the internal pores. Also, we have seen the bicarbonate formation in BaY zeolite when adsorbed water was also present, which suggests that water activates BaY active sites for the formation of bicarbonate species. Further studies should investigate how zeolites may be used to convert carbon dioxide to more useful products such as methanol. Because of the high external surface area and active sites present on the external surface, nanocrystalline zeolites, in particular, may be useful zeolite materials in CO2 recycling and conversion processes. Studies are underway to investigate these possibilities.Acknowledgements
This material is based upon work partially supported by the National Science Foundation under Grant No. CHE-0503854 (VHG). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.References
- M. Marquis and P. Tans, Science, 2008, 320, 460–461 CrossRef CAS
.
- D. Luthi, M. Le Floch, B. Bereiter, T. Blunier, J. M. Barnola, U. Siegenthaler, D. Raynaud, J. Jouzel, H. Fischer, K. Kawamura and T. F. Stocker, Nature, 2008, 453, 379 CrossRef
- C. L. Angell and M. V. Howell, Can. J. Chem., 1969, 47(20), 3831 CrossRef CAS
- B. Chan and L. Radom, J. Am. Chem. Soc., 2008, 130(30), 9790–9799 CrossRef CAS
- D. Bonenfant, M. Kharoune, P. Niquette, M. Mimeault and R. Hausler, Sci. Tech. Adv. Mater., 2008, 9(1), 013007 Search PubMed
- A. Khelifa, Z. Derriche and A. Bengueddach, Microporous Mesoporous Mater., 1999, 32(1–2), 199–209 CrossRef CAS
- A. Goj, D. S. Sholl, E. D. Akten and D. Kohen, J. Phys. Chem. B, 2002, 106(33), 8367–8375 CrossRef CAS
- S. C. Larsen, J. Phys. Chem. C, 2007, 111, 18464–18474 CrossRef CAS
- A. Corma, J. Catal., 2003, 216(1–2), 298–312 CrossRef CAS
- B. K. Marcus and W. E. Cormier, Chem. Eng. Prog., 1999, 95(6), 47–53 Search PubMed
- W. G. Song, G. H. Li, V. H. Grassian and S. C. Larsen, Environ. Sci. Technol., 2005, 39(5), 1214–1220 CrossRef CAS
- G. H. Li, S. C. Larsen and V. H. Grassian, J. Mol. Catal. A: Chem., 2005, 227(1–2), 25–35 CrossRef CAS
- G. Li, C. A. Jones, V. H. Grassian and S. C. Larsen, J. Catal., 2005, 234, 401 CrossRef CAS
- S. Elzey, A. Mubayi, S. C. Larsen and V. H. Grassian, J. Mol. Catal. A: Chem., 2008, 285, 48–57 CrossRef CAS
- G. Li, M. Xu, S. C. Larsen and V. H. Grassian, J. Mol. Catal. A: Chem., 2003, 194, 169–180 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed
- J. Baltrusaitis, J. D. Schuttlefield, E. Zeitler, J. H. Jensen and V. H. Grassian, J. Phys. Chem. C, 2007, 111, 14870–14880 CrossRef CAS
- M. P. Andersson, J. Blomquist and P. Uvdal, J. Chem. Phys., 2005, 123(22) Search PubMed
- D. Plant, G. Maurin, I. Deroche, L. Gaberova and P. L. Llewellyn, Chem. Phys. Lett., 2006, 426(4–6), 387–392 CrossRef CAS
- A. M. Ferrari, P. Ugliengo and E. Garrone, J. Chem. Phys., 1996, 105(10), 4129–4139 CrossRef CAS
- D. F. Plant, G. Maurin, I. Deroche and P. L. Llewellyn, Microporous Mesoporous Mater., 2007, 99(1–2), 70–78 CrossRef CAS
- V. Krishnakumar and V. Balachandran, Spectrochim. Acta, Part A, 2005, 61(5), 1001–1006 CrossRef CAS
- P. Pulay, G. Fogarasi, G. Pongor, J. E. Boggs and A. Vargha, J. Am. Chem. Soc., 1983, 105(24), 7037–7047 CrossRef CAS
- Y. Yamakita and M. Tasumi, J. Phys. Chem., 1995, 99(21), 8524–8534 CrossRef CAS
- P. Sett, T. Mishra, J. Chowdhury, M. Ghosh, S. Chattopadhyay, S. Kumar Sarkar and P. Kumar Mallick, J. Chem. Phys., 2008, 128(14), 144507 CrossRef
- J. Baltrusaitis, J. H. Jensen and V. H. Grassian, J. Phys. Chem. B, 2006, 110(24), 12005–12016 CrossRef CAS
- E. Garrone, B. Bonelli, C. Lamberti, B. Civalleri, M. Rocchia, P. Roy and C. O. Arean, J. Chem. Phys., 2002, 117(22), 10274–10282 CrossRef CAS
- B. Bonelli, B. Civalleri, B. Fubini, P. Ugliengo, C. O. Arean and E. Garrone, J. Phys. Chem. B, 2000, 104(47), 10978–10988 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2009 |