Solar energy conversion using photochemical molecular devices: photocatalytic hydrogen production from water using mixed-metal supramolecular complexes
Krishnan
Rangan
,
Shamindri M.
Arachchige
,
Jared R.
Brown
and
Karen J.
Brewer
*
Department of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061-0212, USA. E-mail: kbrewer@vt.edu; Fax: +1 540-231-3255; Tel: +1 540-231-6579
First published on 6th March 2009
Abstract
Photocatalytic generation of hydrogen from water is an integral part of the next generation clean fuel technologies. The conversion of solar energy into useful chemical energy is of great interest in contemporary investigations. The splitting of water is a multi-electron process involving the breaking and making of chemical bonds which requires multi-component photocatalytic systems. Supramolecular complexes [{(TL)2Ru(BL)}2RhX2](Y)5 (where TL = terminal ligand, BL = bridging ligand, X = Cl− or Br−, and Y = PF6− or Br−) have been synthesized and studied for their light absorbing, electrochemical and photocatalytic properties. The supramolecular complexes in this investigation are multi-component systems comprised of two ruthenium based light absorbers connected through bridging ligands to a central rhodium, which acts as an electron collecting center upon excitation. These complexes absorb light throughout the ultraviolet and visible regions of the solar spectrum. The supramolecular complexes possess ruthenium based highest occupied molecular orbitals (HOMO) and a rhodium based lowest unoccupied molecular orbital (LUMO). These molecular devices have been investigated and shown to function as photoinitiated electron collectors at the reactive rhodium metal center, and explored as photocatalysts to generate hydrogen from water in an aqueous solution in the presence of an electron donor.
Broader contextThe work described herein provides basic chemical studies of the process of light to energy conversion via the collection of reducing equivalents in a molecular system. This system further delivers the reducing equivalents to a water substrate to produce hydrogen. The harvesting of solar energy efficiently by molecular systems is largely restricted by the lack of a fundamental understanding of multielectron chemistry. It is further severely restricted by the lack of systems that undergo multielectron photochemistry. Systems that harvest visible light efficiently typically undergo single electron reactions which is energetically prohibitive for solar energy water splitting. The first visible light induced photoinitiated electron collection and hydrogen generation from water by polyazine derived Ru, Rh, Ru heteronuclear water soluble system is described. This article places this unique chemistry in the context of the very few, typically organic soluble systems even known to under photoinitiated electron collection. The reported system is unusual in its ability to use low energy visible light to drive multielectron photochemistry, ability to function in water, and further ability to deliver the photocollected electrons to a substrate leading to hydrogen production from water. |
Introduction
The need for renewable energy sources has driven an exploration for alternative energy. The present consumption rate of fossil fuels makes it necessary to explore alternative energy sources. One appealing alternative energy source is solar energy. The harvesting of solar energy can occur through the conversion of light energy into heat energy in thermal conversion schemes, or electrical potentials in solar cells, or fuels through light to chemical energy conversion schemes. The conversion of light energy into a fuel focuses on the conversion of a widely available chemical feedstock into a transportable fuel. The chemical feedstocks commonly suggested are carbon dioxide (via artificial photosynthesis) and water (via solar water splitting). Water is an attactive chemical feedstock for solar energy conversion as the fuel produced is hydrogen. Molecular hydrogen is an attractive nonpolluting clean burning fuel with a combustion energy of −282 kJ mol−1 and with potential as a long-term replacement for natural gas.1The solar spectrum at the earth's surface consists of the infrared, visible and ultraviolet regions of the electromagnetic spectrum. Solar water splitting is a process by which light energy from the sun is harvested to convert water into hydrogen and oxygen.2–4 The splitting of water is an energetically uphill process which requires 1.23 V of energy.4 Much of the visible and ultraviolet regions of the solar spectrum, that reaches the earth from the sun, has sufficient energy to drive the splitting of water into hydrogen and oxygen. Yet, this process does not happen by direct irradiation with sunlight because of the transparent nature of the water. One molecule of water yields, in a balanced chemical equation, one equivalent of hydrogen gas and a half equivalent of oxygen gas. The overall reaction for water splitting and the related energetics of the half reactions are represented below, where E is the redox potential vs. NHE at pH = 7.
| 2H+(aq) + 2e− → H2(g) E = −0.41 V |
| H2O(l) → ½O2(g) + 2e− + 2H+(aq) E = −0.82 V |
| H2O(l) → H2(g) + ½O2(g) E = −1.23 V |
The energy requirements for water splitting through a multielectron process is much less (1.23 V)4–6 relative to water splitting through a single electron process (∼ 5 V). Fujishima and Honda showed the light activated water splitting reaction over UV light irradiated TiO2 semiconductors.7
The discovery of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) has inspired photochemical and photophysical studies of similar polyazine complexes for light to energy conversion research.8–11Ru(II)-polyazine chromophores absorb light with a high efficiency throughout the UV and much of the visible regions, and possess long-lived metal-to-ligand charge-transfer (MLCT) excited states. The photochemical and photophysical properties of ruthenium polyazine complexes are easily tuned by the selection of the ligand set bound to the metal center. The prototypical [Ru(bpy)3]2+ possesses intense intra-ligand (IL) transitions in the UV with MLCT transitions in the visible, Fig. 1.
![Electronic absorption (solid line) and emission spectra (dotted line) of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) in acetonitrile at room temperature.](/image/article/2009/EE/b812049h/b812049h-f1.gif) | ||
| Fig. 1 Electronic absorption (solid line) and emission spectra (dotted line) of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) in acetonitrile at room temperature. | ||
The intense Ru(dπ) → bpy(π*) MLCT transition occurs at 450 nm in acetonitrile solution for [Ru(bpy)3]2+. Excitation of a Ru polyazine type of light absorber (LA) leads to intersystem crossing and internal conversion, which results in population of the lowest lying triplet metal-to-ligand charge-transfer (3MLCT) excited state with unit efficiency, Fig. 2. This 3MLCT excited state displays a strong emission in deoxygenated acetonitrile solution centered at 630 nm with an excited-state lifetime of 860 ns at room temperature.12 The 3MLCT state of ruthenium polyazine complexes is typically long-lived and can undergo excited state energy and electron transfer reactions. As the excited state of [Ru(bpy)3]2+ is both a more powerful oxidizing and reducing agent than its ground state, it is capable of undergoing either oxidative or reductive electron transfer quenching.10,12–16
![Energy state diagram for [Ru(bpy)3]2+, (bpy = 2,2′-bipyridine, GS = ground state, 1MLCT = singlet metal-to-ligand charge-transfer state, 3MLCT = triplet metal-to-ligand charge-transfer state, kisc = rate constant for intersystem crossing, kr = rate constant for radiative decay, knr = rate constant for non-radiative decay, krxn = rate constant for reaction).](/image/article/2009/EE/b812049h/b812049h-f2.gif) | ||
| Fig. 2 Energy state diagram for [Ru(bpy)3]2+, (bpy = 2,2′-bipyridine, GS = ground state, 1MLCT = singlet metal-to-ligand charge-transfer state, 3MLCT = triplet metal-to-ligand charge-transfer state, kisc = rate constant for intersystem crossing, kr = rate constant for radiative decay, knr = rate constant for non-radiative decay, krxn = rate constant for reaction). | ||
The complex [Ru(bpy)3]2+ and other light absorbing analogs possess 3MLCT states of sufficient energy to split water into hydrogen and oxygen, yet direct photocatalysis has not been observed. Water splitting is a multistep and multielectron process involving breaking and making of chemical bonds. Water splitting requires multicomponent systems containing proper catalytically active sites in addition to adequate energy input. Complicated multicomponent systems incorporating light absorbing units, electron relays, and redox catalysts have been extensively utilized in solar energy conversion schemes.
Nocera et al. have made a significant contribution in the area of photochemical hydrogen production. They have utilized metal–metal bonded dirhodium photocatalysts, such as [Rh2(dfpma)3(PPh3)CO] (dfpma = MeN(PF2)2), which undergo photochemical transformations and catalytically produce hydrogen from hydrohalic acids in the presence of a halogen trap.1,2,17–20
Multicomponent systems using polyazine light absorbers have been studied and shown to photocatalytically produce H2. Lehn et al.5,6 and Sutin et al.21,22 have studied a four component system for hydrogen production using the well studied [Ru(bpy)3]2+ light absorber (LA), a [Rh(bpy)3]3+ electron relay, triethanolamine (TEOA) as a sacrificial electron donor (ED), and a colloidal Pt catalyst system. Photolysis results in the transfer of electrons from the excited state of *[Ru(bpy)3]2+ to [Rh(bpy)3]3+. The newly generated [RhII(bpy)3]2+ is proposed to transfer electrons to colloidal Pt, which produces hydrogen in the presence of water. Oishi23 and Bauer and Werner24 studied the Wilkinson's catalyst analog [RhICl(dpm)3]3− (dpm = diphenylphosphino-benzene-m-sulfonate), which when photolyzed in the presence of [Ru(bpy)3]2+ and ascorbic acid produces hydrogen. Eisenberg et al. reported the photocatalytic hydrogen production activity of a system containing a series of luminescent platinum(II) terpyridyl acetylide complexes in the presence of dialkylated bipyridinium cations, an electron donor (TEOA), and colloidal Pt catalyst.25,26
Developing suitable molecules that can absorb the solar spectrum and utilize the absorbed light energy for a productive purpose is a challenge and involves the art of molecular design and development. Photocatalytic systems for fuel production typically combine (i) a chromophore or LA, (ii) an electron collector (EC), and (iii) a catalytic site (CAT).9 Molecular devices applied in this forum should be able to perform multiple tasks including: the absorption of light over a broad region of the electromagnetic spectrum, storage of the absorbed energy for an appropriate length of time, transferring electrons to an active center, and catalyzing a suitable reaction to carry out the chemical transformation. A schematic orbital energy diagram for a photochemical molecular device for photointiated electron collection is represented in Fig. 3. Supramolecular assemblies which couple an ED to a LA and EC represent photochemical molecular devices for electron collection.
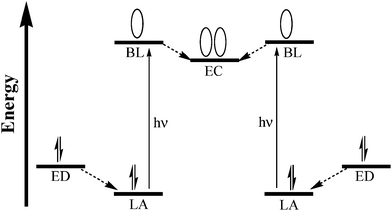 | ||
| Fig. 3 Schematic representation of an orbital energy diagram showing the functioning of a molecular device for photoinitiated electron collection (LA = light absorber, BL = bridging ligand, EC = electron collector, ED = electron donor). | ||
Photochemical electron collection in molecular complexes employ chromophores connected, through proper bridging ligands (BL), to an electron collector. Utilization of solar energy typically proceeds through MLCT excitation of the chromophore. The excited electron is transferred and held by a suitable EC. This process is repeated, providing two electrons collected at the EC site. Bridging ligands provide a point of attachment between the light absorbing chromophores and the electron collector. A variety of TL and BL can be designed to tune the properties and functioning of the molecular devices. Some of the representative TL and BL are given in Fig. 4.
![Chemical structures of some of the representative terminal ligands (TL) and bridging ligands (BL), where bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline, Ph2phen = 4,7-diphenylphenanthroline, tpy = 2,2′:6′,2″-terpyridine, dpp = 2,3-bis(2-pyridyl)pyrazine, dpq = 2,3-bis(2-pyridyl)quinoxaline, bpm = 2,2′-bipyrimidine, tppz = 2,3,5,6-tetrakis(2-pyridyl)pyrazine, and tatpp = 9,11,20,22-tetraazatetrapyrido[3,2-a:2′,3′-c:3″,2″-1:2‴,3‴-n]pentacene.](/image/article/2009/EE/b812049h/b812049h-f4.gif) | ||
| Fig. 4 Chemical structures of some of the representative terminal ligands (TL) and bridging ligands (BL), where bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline, Ph2phen = 4,7-diphenylphenanthroline, tpy = 2,2′:6′,2″-terpyridine, dpp = 2,3-bis(2-pyridyl)pyrazine, dpq = 2,3-bis(2-pyridyl)quinoxaline, bpm = 2,2′-bipyrimidine, tppz = 2,3,5,6-tetrakis(2-pyridyl)pyrazine, and tatpp = 9,11,20,22-tetraazatetrapyrido[3,2-a:2′,3′-c:3″,2″-1:2‴,3‴-n]pentacene. | ||
Despite the increasing interest in photochemical molecular devices for photoinitiated electron collection, working systems that store multiple electrons remain a challenge. The first functioning photoinitiated electron collector in a molecular system was reported by Brewer et al.27 The supramolecular assembly couples two ruthenium LA units to a central iridium that functions to electronically isolate the two LA subunits, Fig. 5. The [{(bpy)2Ru(dpb)}2IrCl2](PF6)5 (dpb = 2,3-bis(2-pyridyl)benzoquinoxaline) system, when excited with visible light in the presence of the electron donor, N,N-dimethylaniline (DMA), photochemically reduces by two electrons to form [{(bpy)2Ru(dpb−)}2IrCl2]3+. MacDonnell et al.28,30,31 studied the Ru–Ru bimetallic systems containing interesting extended π-conjugated bridging ligands which can collect up to four electrons during photolysis in the presence of an electron donor triethylamine (TEA), Fig. 5. Bocarsly et al.29,32 studied a series of Pt(IV) centered trimetallic complexes of the form [(NC)5MII(CN)PtIV(NH3)4(NC)MII(CN)5]4− (M = Fe, Ru, or Os) which undergo photoinitiated electron collection at the platinum metal center Fig. 5. The complexes, however, are not stable and disassemble to monometallic components with formation of the two electron reduced Pt(II) complex.
![Representative examples of photoinitiated electron collector molecular devices, [{(bpy)2Ru(dpb)}2IrCl2]5+,27[(phen)2Ru(tatpp)Ru(phen)2]4+,28 and [(NC)5Fe(CN)Pt(NH3)4(NC)Fe(CN)5]4−.29](/image/article/2009/EE/b812049h/b812049h-f5.gif) | ||
| Fig. 5 Representative examples of photoinitiated electron collector molecular devices, [{(bpy)2Ru(dpb)}2IrCl2]5+,27[(phen)2Ru(tatpp)Ru(phen)2]4+,28 and [(NC)5Fe(CN)Pt(NH3)4(NC)Fe(CN)5]4−.29 | ||
Mixed-metal polyazine complexes containing ruthenium LA coupled to reactive metal sites have been explored. Sakai et al.33–35 recently investigated a Ru–Pt bimetallic system capable of photochemically producing hydrogen from water in the presence of the electron donor, ethylenediaminetetraacetic acid (EDTA). In this system the platinum component, of the form cis-PtIICl2 was linked to the ruthenium LA through an amide linkage on the phenanthroline ligand coordinated to ruthenium. This system functioned with Φ ≈ 0.01, and 5 turnovers in 10 h. This low turnover has been postulated to reflect decomplexation of the reactive metal to form colloidal platinum, which functions as the hydrogen generation site.36 Rau et al.37 reported a Ru–Pd bimetallic system that photochemically produces hydrogen in the presence of an electron donor, triethylamine (TEA). This system was shown to give 56 turnovers in ∼30 h. Recently, colloidal Pd formation was suggested for Ru–Pd molecular systems.38
Brewer et al.39,40 recently reported the photoinitiated electron collection at a metal center in [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 (dpp = 2,3-bis(2-pyridyl)pyrazine). When excited with visible light, this complex is photochemically reduced converting the Rh(III) to Rh(I), which undergoes subsequent chloride loss to form [{(bpy)2Ru(dpp)}2RhI]5+.39 This complex photochemically reduces water to hydrogen using visible light excitation with a Φ ≈ 0.01 in the presence of the sacrificial electron donor DMA.40 The ability of this system to undergo photoinitiated electron collection on the rhodium center with the molecular architecture remaining intact is unprecedented and allows use in multielectron photochemistry. One supramolecular complex used in our study as a photochemical molecular device is given in Fig. 6.
![Supramolecular complex [(bpy)2Ru(dpp)RhBr2(dpp)Ru(bpy)2]5+ for photoinitiated electron collection and solar water splitting (LA = light absorber, BL = bridging ligand, and EC = electron collector). Hydrogens are omitted for clarity.](/image/article/2009/EE/b812049h/b812049h-f6.gif) | ||
| Fig. 6 Supramolecular complex [(bpy)2Ru(dpp)RhBr2(dpp)Ru(bpy)2]5+ for photoinitiated electron collection and solar water splitting (LA = light absorber, BL = bridging ligand, and EC = electron collector). Hydrogens are omitted for clarity. | ||
Brewer et al. developed a number of mixed-metal supramolecular complexes containing ruthenium or osmium polyazine light absorbers coupled through a variety of bridging ligands to a rhodium metal center with interesting photophysical properties, which have also shown to be efficient as light activated DNA photocleaving agents.41–45
In this paper, we report our study of Ru,Rh,Ru molecular devices for solar energy conversion and generation of hydrogen from water in the presence of a suitable electron donor in aqueous medium.
Experimental
The supramolecular complexes [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 and [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 were prepared following literature procedures.42,44 The water soluble complex, [{(bpy)2Ru(dpp)}2RhBr2]Br5, was prepared from [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 by metathesis with Et4NBr in acetone. All solvents used were spectroscopic grade obtained from VWR Scientific and used without further purification. High purity DMA and TEOA were obtained from Aldrich Chemical Company and used without further purification.Electronic absorption spectra were collected using a Hewlett Packard 8452A or Agilent 8453 diode array spectrophotometer. Emission spectra were recorded using a QuantaMaster Model QM-200-45E Fluorimeter from Photon Technology Inc. Cyclic voltammetry and constant potential electrolysis experiments were conducted with a BioAnalytical Systems (BAS) electrochemical analyzer. Potentials were referenced to a Ag/AgCl reference electrode (0.197 V vs. NHE). The supporting electrolytes were 0.1 M Bu4NPF6 in acetonitrile, or 0.1 M KCl in water.
The photocatalytic hydrogen production experiments were performed under argon atmosphere as follows. For the photolysis reaction in acetonitrile, the septa capped deoxygenated photolysis reaction cells contained 4.5 mL final volume of the reaction solution containing the metal complex (65 µM, [{(bpy)2Ru(dpp)}2RhCl2](PF6)5, or [{(bpy)2Ru(dpp)}2RhBr2](PF6)5), electron donor (1.5 M) and water (0.62 M, acidified to pH = 2 with triflic acid, final [CF3SO3−] = 110 µM). The solutions were photolyzed using 470 nm LED light array constructed locally with flat optical bottom cells.45 For the photolysis reaction in water, the septa capped deoxygenated photolysis reaction cells contained 4.5 mL final volume of the reaction solution containing the metal complex (65 µM, [{(bpy)2Ru(dpp)}2RhBr2]Br5) and electron donor (1.5 M, neat or adjusted to pH = 7.9 with triflic acid, HBr or H3PO4). The solutions were photolyzed using 470 nm LED light. DMA or TEOA were used as the electron donors. After 4 h photolysis time, the hydrogen was quantitated by headspace analysis using gas chromatography. Samples of the headspace (100 µL) were injected into a series 580 GOW-MAC gas chromatograph equipped with a 5 Å molecular sieves column using ultra-high purity argon as the carrier gas, purchased from Airgas Inc. (Radnor, PA), equipped with a rhenium tungsten thermal conductivity detector. The signal was amplified with a Vernier Software instrument amplifier and recorded with Logger Pro 3.4.5 software. The system was calibrated for hydrogen signal sensitivity by hydrogen standard measurements. The total amount of hydrogen produced in a photolysis experiment was obtained by the sum of the hydrogen found in the headspace as well as the hydrogen in the solution. The amount of hydrogen in the solution was calculated according to Henry's Law using the reported solubility of hydrogen in solvents.46,47 Each photocatalysis experiment was conducted three times with the reported µmol hydrogen being the average of the trials.
Results and discussion
Assembling supramolecular light harvesting devices
The supramolecular solar energy harvesting photocatalysts under study are multicomponent systems consisting of ruthenium based LAs, an EC rhodium reactive center, and BLs connecting the LA and EC. The multimetallic complexes [{(bpy)2Ru(dpp)}2RhCl2](PF6)5, [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 and [{(bpy)2Ru(dpp)}2RhBr2]Br5 were prepared in good yields using our building-block approach40,44 and counter anion metathesis. The synthetic strategies are depicted in Fig. 7. Initially, the terminal ligand bpy was reacted with ruthenium(III) chloride to form the mononuclear complex [(bpy)2RuCl2].48 The bridging ligand dpp was coordinated to the ruthenium light absorbing center by combining with [(bpy)2RuCl2] followed by addition of excess NH4PF6 salt to form [(bpy)2Ru(dpp)](PF6)2.49 The [(bpy)2Ru(dpp)](PF6)2 was assembled into the mixed-metal trimetallic complexes [{(bpy)2Ru(dpp)}2RhX2](PF6)5 (where X = Cl− or Br−) by reaction with RhCl3·3H2O or RhBr3·3H2O with careful control of stoichiometry.42,44 The water soluble mixed-metal complex [{(bpy)2Ru(dpp)}2RhBr2]Br5 was prepared from [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 by anion metathesis with Et4NBr in acetone. The metal complexes were assayed by electronic absorption spectroscopy and cyclic voltammetry.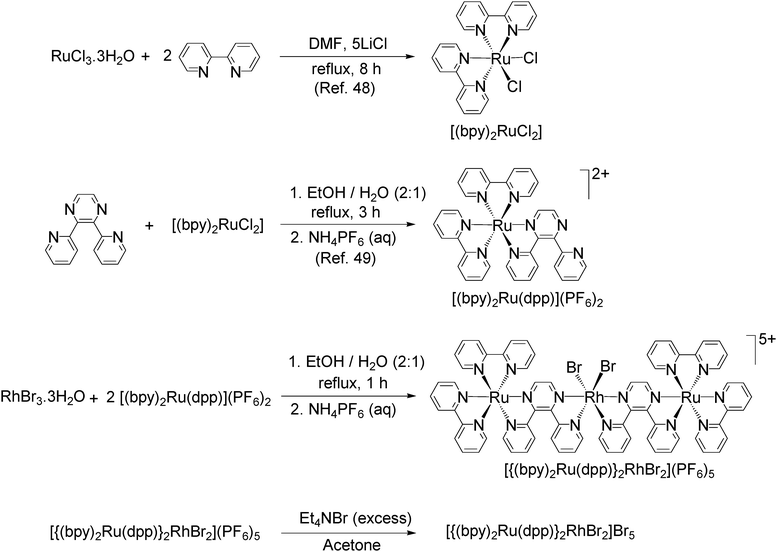 | ||
| Fig. 7 Building block approach synthesis of dpp bridged mixed-metal supramolecular complexes. | ||
5 (bpy = 2,2′-bipyridine, and dpp = 2,3-bis(2-pyridyl)pyrazine) in 0.1 M Bu4NPF6 in CH3CN using a platinum working electrode and a Ag/AgCl reference electrode.](/image/article/2009/EE/b812049h/b812049h-f8.gif) | ||
| Fig. 8 Cyclic voltammogram of [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 (bpy = 2,2′-bipyridine, and dpp = 2,3-bis(2-pyridyl)pyrazine) in 0.1 M Bu4NPF6 in CH3CN using a platinum working electrode and a Ag/AgCl reference electrode. | ||
The cyclic voltammograms can be generalized to display ruthenium based oxidations and Rh, BL and TL based reductions. Overlapping RuII/III based oxidation processes for both Ru centers is seen for [{(bpy)2Ru(dpp)}2RhBr2]5+ at +1.6 V vs. Ag/AgCl illustrative of the electronically isolated nature of the two ruthenium sites.44 The reductive side consists of a first reduction at −0.33 V due to two sequential one electron irreversible reductions of the rhodium center from RhIII to RhII to RhI.39,42,50 At more negative potentials, two reversible reduction events at −0.72 V and −1.02 V occur due to the two dpp0/−reduction processes. Reduction of the bpy ligands occur at more negative potentials.
DeArmond et al.51,52 and Brewer et al.39,50 described the electrochemical properties of cis-dihalide rhodium(III) complexes. [Rh(bpy)2Br2]+ and [Rh(bpy)2Cl2]+ undergo ECECEE reductive electrochemical processes, Fig 9. Similar behavior is observed for the dpp ligand bridged trimetallic complex, [{(bpy)2Ru(dpp)}2RhBr2]5+.39 This is consistent with the observed electrochemical properties of the [{(bpy)2Ru(dpp)}2RhBr2]Br5. The electrochemical features clearly show that the supramolecular complexes possess Ru(dπ) centered highest occupied molecular orbital (HOMO) and the Rh(dσ*) centered lowest unoccupied molecular orbital (LUMO), Table 1. The water soluble complex [{(bpy)2Ru(dpp)}2RhBr2]Br5 shows an irreversible RhIII/II/Ireduction at −0.42 V in aqueous medium, which is slightly more negative than the corresponding couple of the [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 in acetonitrile. The electrochemical properties predict a low energy Ru → dpp CT transition in the trimetallic complexes with a lowest lying Ru → Rh metal to metal charge transfer (MMCT) state.
| Complex | Solvent | E 1/2/V | Assignment | Ref. |
|---|---|---|---|---|
| a Potential reported vs. Ag/AgCl. Epc and Epa respectively indicates the value of cathodic and anodic current peak maxima. b In acetonitrile with 0.1 M Bu4NPF6 supporting electrolyte and Pt working electrode. c In water with 0.1 M KCl supporting electrolyte and glassy carbon working electrode. | ||||
| [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 | CH3CNb | +1.63 | 2RuII/III | 41 |
| −0.37 (Epc) | RhIII/II/I | |||
| −0.76 | dpp,dpp/dpp−,dpp | |||
| −1.00 | dpp−,dpp/dpp−,dpp− | |||
| [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 | CH3CNb | +1.60 | 2RuII/III | 44 |
| −0.33 (Epc) | RhIII/II/I | |||
| −0.72 | dpp,dpp/dpp−,dpp | |||
| −1.02 | dpp−,dpp/dpp−,dpp− | |||
| [{(bpy)2Ru(dpp)}2RhBr2]Br5 | CH3CNb | +1.60 | 2RuII/III | |
| −0.39 (Epc) | RhIII/II/I | |||
| −0.66 | dpp,dpp/dpp−,dpp | |||
| −0.90 | dpp−,dpp/dpp−,dpp− | |||
| [{(bpy)2Ru(dpp)}2RhBr2]Br5 | H2Oc | +1.66 (Epa) | 2RuII/III | |
| −0.42 (Epc) | RhIII/II/I | |||
| −0.83 | dpp,dpp/dpp−,dpp | |||
| −1.08 | dpp−,dpp/dpp−,dpp− |
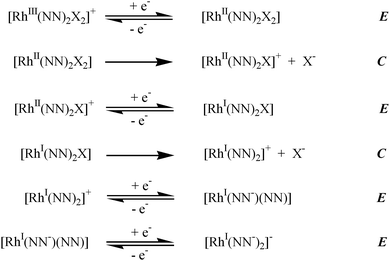 | ||
| Fig. 9 ECECEE mechanism for the electrochemical reduction of cis-dihalide rhodium(III) complexes. | ||
The supramolecular ruthenium(II) polyazine complexes display rich photochemical and electrochemical properties. The electronic absorption spectrum of [(bpy)2Ru(dpp)](PF6)2 is characterized by intense bpy (290 nm) and dpp (320 nm) ligand based π → π* transitions in the UV region, and overlapping absorption bands at 430 nm and 470 nm in the visible region due to the MLCT transitions containing Ru(dπ) → bpy(π*) and Ru(dπ) → dpp(π*) CT transitions respectively. The lower energy band is the Ru(dπ) → dpp(π*) CT transition due to the lower energy π* orbital of dpp in comparison to that of bpy.
The dpp bridged trimetallic complexes are efficient light absorbers throughout the visible and ultraviolet region of the spectrum. The dpp bridged trimetallic complexes [{(bpy)2Ru(dpp)}2RhCl2](PF6)5, [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 and [{(bpy)2Ru(dpp)}2RhBr2]Br5 show nearly identical electronic absorption spectral features.40,44 A representative electronic absorbance spectrum of [{(bpy)2Ru(dpp)}2RhBr2]Br5 in water is shown in Fig. 10. The absorbance at 408 nm (ε = 1.3 × 104 M−1 cm−1) is assigned as a Ru(dπ) → bpy(π*) MLCT transition, and a Ru(dπ) → dpp(π*) MLCT transition appearing at 525 nm (2.2 × 104 M−1 cm−1). In [{(bpy)2Ru(dpp)}2RhBr2]Br5, the bridging ligand dpp π* orbitals are stabilized and the Ru(dπ) → dpp(π*) MLCT transition red shifts relative to that of the monometallic complex [(bpy)2Ru(dpp)](PF6)2. The important lower energy electronic transitions of the dpp ligated ruthenium(II) complexes are summarized in Table 2. Incorporation of the polyazine bridging ligand into the [Ru(bpy)3]2+ chromophoric unit, and further formation of the dpp ligand bridged trimetallic complexes, leads to a modification of the photophysical properties. For example, [(bpy)2Ru(dpp)]2+, and [{(bpy)2Ru(dpp)}2RhX2]5+ display red-shifted absorptions and emissions, and shortened 3MLCT excited-lifetimes (τ) relative to the [Ru(bpy)3]2+, Table 2.12,53 The trimetallic complexes [{(bpy)2Ru(dpp)}2RhX2]5+ (X = Cl− or Br−) display light-absorbing properties dominated by the two Ru light absorbing units and redox properties that illustrate the Rh(dσ*) nature of the lowest unoccupied molecular orbital (LUMO), Fig. 11.
| Complex | Solvent | λ max abs/nm | λ max em/nm | τ/ns |
|---|---|---|---|---|
| 1MLCT | 3MLCT | |||
| a Spectral measurements were carried out in deoxygenated solution at room temperature. bpy = 2,2′-bipyridine and dpp = 2,3-bis(2-pyridyl)pyrazine. b Ref. 12. c Ref. 50. | ||||
| [Ru(bpy)3](PF6)2 | CH3CN | 450 | 630 | 860b |
| [(bpy)2Ru(dpp)](PF6)2 | CH3CN | 470 | 691 | 240c |
| [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 | CH3CN | 520 | 760 | 23 |
| [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 | CH3CN | 520 | 760 | 26 |
| [{(bpy)2Ru(dpp)}2RhBr2]Br5 | H2O | 525 | 770 | — |
![Electronic absorption spectrum of [{(bpy)2Ru(dpp)}2RhBr2]Br5 (bpy = 2,2′-bipyridine, and dpp = 2,3-bis(2-pyridyl)pyrazine) in water at room temperature.](/image/article/2009/EE/b812049h/b812049h-f10.gif) | ||
| Fig. 10 Electronic absorption spectrum of [{(bpy)2Ru(dpp)}2RhBr2]Br5 (bpy = 2,2′-bipyridine, and dpp = 2,3-bis(2-pyridyl)pyrazine) in water at room temperature. | ||
![Energy state diagram for [{(bpy)2Ru(dpp)}2RhX2]5+, (bpy = 2,2′-bipyridine, dpp = 2,3-bis(2-pyridyl)pyrazine, X = Cl− or Br−) incorporating low-lying triplet metal-to-metal charge-transfer state (3MMCT). GS = ground state, 1MLCT = singlet metal-to-ligand charge transfer state, 3MLCT = triplet metal-to-ligand charge transfer state, kisc = rate constant for intersystem crossing, kr = rate constant for radiative decay, knr = rate constant for non-radiative decay, ket = rate constant for electron transfer, knr′ = rate constant for non-radiative decay from 3MMCT state, and krxn = rate constant for reaction.](/image/article/2009/EE/b812049h/b812049h-f11.gif) | ||
| Fig. 11 Energy state diagram for [{(bpy)2Ru(dpp)}2RhX2]5+, (bpy = 2,2′-bipyridine, dpp = 2,3-bis(2-pyridyl)pyrazine, X = Cl− or Br−) incorporating low-lying triplet metal-to-metal charge-transfer state (3MMCT). GS = ground state, 1MLCT = singlet metal-to-ligand charge transfer state, 3MLCT = triplet metal-to-ligand charge transfer state, kisc = rate constant for intersystem crossing, kr = rate constant for radiative decay, knr = rate constant for non-radiative decay, ket = rate constant for electron transfer, knr′ = rate constant for non-radiative decay from 3MMCT state, and krxn = rate constant for reaction. | ||
![Photoinitiated electron collection at the Rh center of the supramolecular complex [{(bpy)2Ru(dpp)}2RhBr2]5+ (bpy = 2,2′-bipyridine, and dpp = 2,3-bis(2-pyridyl)pyrazine) forming the two electron reduced [{(bpy)2Ru(dpp)}2RhI]5+ through halide loss in the presence of an electron donor (ED).](/image/article/2009/EE/b812049h/b812049h-f12.gif) | ||
| Fig. 12 Photoinitiated electron collection at the Rh center of the supramolecular complex [{(bpy)2Ru(dpp)}2RhBr2]5+ (bpy = 2,2′-bipyridine, and dpp = 2,3-bis(2-pyridyl)pyrazine) forming the two electron reduced [{(bpy)2Ru(dpp)}2RhI]5+ through halide loss in the presence of an electron donor (ED). | ||
Brewer et al. have shown that the trimetallic complexes [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 and [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 undergo photoinitiated electron collection at the Rh center.59 Herein we show that the water soluble supramolecular complex [{(bpy)2Ru(dpp)}2RhBr2]Br5 is able to photochemically collect electrons in water. Upon photolysis in the presence of an electron donor, the complex undergoes two electron reduction to produce coordinatively unsaturated RhI based assembly by the loss of two bromide ligands. In the presence of triethanolamine, photolysis of [{(bpy)2Ru(dpp)}2RhBr2]Br5 in water leads to the formation of the RhI form of the complex, [{(bpy)2Ru(dpp)}2RhI]5+. The two electron reduced RhI form can also be produced by electrochemical reduction at −0.4 V. This reduction process occurs through two steps in which the RhIII is first reduced to the RhII state followed by conversion to the RhI state. The RhII species is not stable to isolate and study. The photoinitiated electron collection as well as the electrochemical reduction leading to the same RhI form of the molecular device can be followed spectroscopically, Fig. 13.
![Electronic absorption spectra for the conversion of [{(bpy)2Ru(dpp)}2RhIIIBr2]5+ to the two-electron reduced form [{(bpy)2Ru(dpp)}2RhI]5+ electrochemically by electrolysis at −0.4 V in deoxygenated 0.1 M KCl in water using carbon cloth working electrode (A), and photochemically by photolysis at 520 ± 10 nm in deoxygenated water with 7 mM TEOA (B).](/image/article/2009/EE/b812049h/b812049h-f13.gif) | ||
| Fig. 13 Electronic absorption spectra for the conversion of [{(bpy)2Ru(dpp)}2RhIIIBr2]5+ to the two-electron reduced form [{(bpy)2Ru(dpp)}2RhI]5+ electrochemically by electrolysis at −0.4 V in deoxygenated 0.1 M KCl in water using carbon cloth working electrode (A), and photochemically by photolysis at 520 ± 10 nm in deoxygenated water with 7 mM TEOA (B). | ||
Photoinitiated electron collection is seen by comparing the spectrophotochemistry to spectroelectrochemistry of [{(bpy)2Ru(dpp)}2RhBr2]Br5, Fig. 13. This represents the first molecular system shown to undergo photoinitiated electron collection in water. Upon the reduction of [{(bpy)2Ru(dpp)}2RhBr2]5+ to the two electron reduced RhI species [{(bpy)2Ru(dpp)}2RhI]5+ the Ru(dπ) → dpp(π*) MLCT transition blue shifts from 525 nm to 480 nm.
Rh centered complexes that undergo photoinitiated electron collection have been demonstrated to reduce water to H2. These are the only photoinitiated electron collectors that reduce water to H2. In acetonitrile, in the presence of water, photolysis at 470 nm of [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 (65 µM) in the presence of DMA produced 8.2 µmol of hydrogen in 4 h, Table 3.44 The bromide analog [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 generates a slightly higher amount of hydrogen (10.9 µmol) in comparison to the analogous chloride complex [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 in acetonitrile.44 The quantum yields of these photocatalytic processes in acetonitrile medium using DMA as electron donor are ∼0.01. The hydrogen production is catalytic, with 38 turnovers in 4 h for [{(bpy)2Ru(dpp)}2RhBr2]5+, Table 3.
| Run | Complex | Solvent | Electron donor | pH (acid used to adjust pH) | TN | H2/µmol |
|---|---|---|---|---|---|---|
| a Photolysis was carried out using 470 nm LED arrays at room temperature for 4 h. b Calculated pH in CH3CN, assuming that their pKa values are not purturbed relative to aqueous condition. c Ref. 44. | ||||||
| 1 | [{(bpy)2Ru(dpp)}2RhCl2](PF6)5 | CH3CN | DMA | 9.1 (CF3SO3H)b | 28 | 8.2c |
| 2 | [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 | CH3CN | DMA | 9.1 (CF3SO3H)b | 38 | 10.9c |
| 3 | [{(bpy)2Ru(dpp)}2RhBr2](PF6)5 | CH3CN | TEOA | 11.8 (CF3SO3H)b | 1.7 | 0.48 |
| 4 | [{(bpy)2Ru(dpp)}2RhBr2]Br5 | H2O | TEOA | 9.8 | 1.0 | 0.30 |
| 5 | [{(bpy)2Ru(dpp)}2RhBr2]Br5 | H2O | TEOA | 7.9 (CF3SO3H) | 1.3 | 0.38 |
| 6 | [{(bpy)2Ru(dpp)}2RhBr2]Br5 | H2O | TEOA | 7.9 (HBr) | 2.6 | 0.74 |
| 7 | [{(bpy)2Ru(dpp)}2RhBr2]Br5 | H2O | TEOA | 7.9 (H3PO4) | 2.9 | 0.83 |
One challenge which is of economical and environmental significance is replacing the organic solvent system with water as the medium. To that end, we have developed a suitable water soluble complex [{(bpy)2Ru(dpp)}2RhBr2]Br5 for photocatalysis, and further researched alternative water miscible electron donors in the place of DMA. Hence, here we employed TEOA as an electron donor and water as the reaction medium and source of hydrogen. Blue light (470 nm) photolysis of the reaction cells containing TEOA (1.5 M) and [{(bpy)2Ru(dpp)}2RhBr2]Br5 (65 µM) in water are able to produce H2. It is very interesting and noteworthy that the supramolecular complexes can be used in very different reaction conditions, such as employing different electron donors, organic solvent mediums, as well as in water medium, while still retaining photocatalytic activity. The aqueous system produces less H2, 0.3 µmol in 4 h, but the catalyst retains functioning. The photocatalysis in water was further studied at different reaction conditions. Hydrogen production was suppressed in the presence of air consistent with known O2 quenching of 3MLCT excited states. The hydrogen production can be improved by adjusting the pH. At a pH value near the pKa of TEOA, pH = 7.9, a notable increase in hydrogen production is observed. For a 4 h reaction period, TEOA buffered with triflic acid, HBr or H3PO4 to pH = 7.9 respectively produced 0.38, 0.74 or 0.83 µmol of hydrogen from water. The use of acids with coordinating anions such as Br− or CF3SO3− produce lesser amounts of H2 with respect to PO43− supporting halide loss as being important in the catalytic process. The solution at pH = 1, acidified with triflic acid, did not produce a detectable amount of hydrogen. In addition, spectrophotochemical studies of the reaction system containing pH = 1 TEOA and [{(bpy)2Ru(dpp)}2RhBr2]Br5 showed no spectral change. This is consistent with protonated TEOA being a poor electron donor unable to reduce the complex. Local in situ production of colloidal Pd or Pt metals are reported to be responsible active centers for the photocatalytic production of hydrogen by Ru–Pd and Ru–Pt based metal complexes in the presence of an electron donor.36,38 The catalytic activities of the colloidal metals can be inhibited by the addition of Hg. However, in our Rh based systems, addition of Hg did not have any effect on the efficiency of [{(bpy)2Ru(dpp)}2RhBr2]5+ for the hydrogen generation.
Conclusions
Supramolecular mixed-metal complexes have been developed for photoinitiated electron collection and photocatalytic water splitting, which have been shown to display both properties. The photoinitiated electron collecting properties of mixed-metal supramolecular complexes can be tuned by selecting proper bridging ligands and electron collecting metal centers. The dpb ligand bridged Ru, Ir, Ru complex, [{(bpy)2Ru(dpb)}2IrCl2](PF6)5, is the first molecular photoinitiated electron collector, and has been shown to collect up to two reducing equivalents at the bridging ligand sites. The development of the mixed-metal Ru,Rh,Ru complexes bridged by dpp provide rhodium centered molecular devices, such as [{(bpy)2Ru(dpp)}2RhCl2](PF6)5, which collect reducing equivalents at the rhodium active site and photoreduce water. The supramolecular complexes allow for variation of individual sub-units in this multicomponent system by selecting different light absorbers, terminal ligands, bridging ligands, central metal centers, and counter anions. A method for photocatalytic water splitting to hydrogen fuel was developed, which has shown to be an efficient model system functioning under various reaction conditions. The supramolecular complex [{(bpy)2Ru(dpp)}2RhBr2]Br5, which is soluble in water, adds a new direction for our photocatalytic hydrogen production studies using a water medium and readily available cheap electron donors. This complex is the first molecular system shown to undergo photoinitiated electron collection in an aqueous medium. This is one of only a handful of photoinitiated electron collectors that delivers the electrons to a substrate producing a fuel. Functioning can be enhanced with an adjustment in solution pH to the pKa of the electron donor, while very acidic solutions do not function due to protonation of the electron donor. The large excess of amines in this solution may be impeding catalyst functioning. Work in our lab is underway to study a series of these types of complexes in more detail as well as to extend the molecular architecture, as made possible by their supramolecular design, for a new generation hydrogen photocatalysts.Acknowledgements
Acknowledgement is made to the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Sciences, U. S. Department of Energy for their generous support of our research. Acknowledgement is also made to the financial collaboration of Phoenix Canada Oil Company which holds long term license rights to commercialize the technology.References
- M. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef.
- J. L. Dempsey, A. J. Esswein, D. R. Manke, J. Rosenthal, J. D. Soper and D. G. Nocera, Inorg. Chem., 2005, 44, 6879–6892 CrossRef CAS.
- J. Rosenthal, J. Bachman, J. L. Dempsey, A. J. Esswein, T. G. Gray, J. M. Hodgkiss, D. R. Manke, T. D. Luckett, B. J. Pistorio, A. S. Veige and D. G. Nocera, Coord. Chem. Rev., 2005, 249, 1316–1326 CrossRef CAS.
- A. J. Bard and M. A. Mary, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- M. Kirch, J. M. Lehn and J. P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- J. M. Lehn and J. P. Sauvage, New J. Chem., 1977, 1, 449–451 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- V. Balzani, L. Moggi and F. Scandola, Supramolecular Photochemistry, NATO ASI Series 214, Reidel, Dordrecht, The Netherlands, 1987 Search PubMed.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef.
- S. Campagna, S. Serroni, F. Puntoriero, C. D. Pietro and V. Ricevuto, in Electron Transfer in Chemistry, ed. V. Balzani, 2001 Search PubMed.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- G. F. Strouse, J. R. Schoonover, R. Duesing, S. Boyde, W. E. Jones and T. J. Meyer, Inorg. Chem., 1995, 34, 473–487 CrossRef CAS.
- P. A. Anderson, G. F. Strouse, J. A. Treadway, F. R. Keene and T. J. Meyer, Inorg. Chem., 1994, 33, 3863–3864 CrossRef CAS.
- T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193–1206 CrossRef CAS.
- M. H. V. Huynh, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2005, 249, 457–483 CrossRef CAS.
- T. R. Cook, A. J. Esswein and D. G. Nocera, J. Am. Chem. Soc., 2007, 129, 10094–10095 CrossRef CAS.
- A. J. Esswein, J. L. Dempsey and D. G. Nocera, Inorg. Chem., 2007, 46, 2362–2364 CrossRef CAS.
- A. J. Esswein, A. S. Veige and D. G. Nocera, J. Am. Chem. Soc., 2005, 127, 16641–16651 CrossRef CAS.
- A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639–1641 CrossRef CAS.
- S. F. Chan, M. Chou, C. Creutz, T. Matsubara and N. Sutin, J. Am. Chem. Soc., 1981, 103, 369–379 CrossRef CAS.
- G. M. Brown, S. F. Chan, C. Creutz, H. A. Schwarz and N. Sutin, J. Am. Chem. Soc., 1979, 101, 7638–7640 CrossRef CAS.
- S. Oishi, J. Mol. Catal., 1987, 39, 225–232 CAS.
- R. Bauer and H. A. F. Werner, Int. J. Hydrogen Energy, 1994, 19, 497–499 CrossRef CAS.
- P. Du, J. Schneider, P. Jarosz, J. Zhang, W. W. Brennessel and R. Eisenberg, J. Phy. Chem. B, 2007, 111, 6887–6894 Search PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS.
- S. M. Molnar, G. Nallas, J. S. Bridgewater and K. J. Brewer, J. Am. Chem. Soc., 1994, 116, 5206–5210 CrossRef CAS.
- C. Chiorboli, S. Fracasso, F. Scandola, S. Campagna, S. Serroni, R. Konduri and F. M. MacDonnell, Chem. Commun., 2003, 1658–1659 RSC.
- C. C. Chang, B. Pfennig and A. B. Bocarsly, Coord. Chem. Rev., 2000, 208, 33–45 CrossRef CAS.
- C. Chiorboli, S. Fracasso, M. Ravaglia, F. Scandola, S. Campagna, K. L. Wouters, R. Konduri and F. M. MacDonnell, Inorg. Chem., 2005, 44, 8368–8378 CrossRef CAS.
- R. Konduri, H. W. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS.
- D. F. Watson, H. S. Tan, E. Schreiber, C. J. Mordas and A. B. Bocarsly, J. Phys. Chem. A, 2004, 108, 3261–3267 CrossRef CAS.
- H. Ozawa and K. Sakai, Chem. Lett., 2007, 36, 920–921 CrossRef CAS.
- H. Ozawa, Y. Yokoyama, M. Haga and K. Sakai, Dalton Trans., 2007, 1197–1206 RSC.
- H. Ozawa, M. A. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926–4927 CrossRef CAS.
- P. Du, J. Schneider, L. Fan, W. Zhao, U. Patel, F. N. Castellano and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 5056–5058 CrossRef CAS.
- S. Rau, B. Schafer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Gorls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45, 6215–6218 CrossRef CAS.
- P. Lei, M. Hedlund, R. Lomoth, H. Rensmo, O. Johansson and L. Hammarstrom, J. Am. Chem. Soc., 2008, 130, 26–27 CrossRef CAS.
- M. Elvington and K. J. Brewer, Inorg. Chem., 2006, 45, 5242–5244 CrossRef CAS.
- M. Elvington, J. Brown, S. M. Arachchige and K. J. Brewer, J. Am. Chem. Soc., 2007, 129, 10644–10645 CrossRef CAS.
- A. A. Holder, D. F. Zigler, M. T. Tarrago-Trani, B. Storrie and K. J. Brewer, Inorg. Chem., 2007, 46, 4760–4762 CrossRef CAS.
- A. A. Holder, S. Swavey and K. J. Brewer, Inorg. Chem., 2004, 43, 303–308 CrossRef CAS.
- S. Swavey and K. J. Brewer, Inorg. Chem., 2002, 41, 6196–6198 CrossRef CAS.
- S. M. Arachchige, J. Brown and K. J. Brewer, J. Photochem. Photobiol. A: Chem., 2008, 197, 13–17 CrossRef CAS.
- J. R. Brown, M. Elvington, M. T. Mongelli, D. F. Zigler and K. J. Brewer, Proc. SPIE-Int. Soc. Opt. Eng.: Sol. Hydrogen Nanotechnol., 2006, 6340, 634001–634010 Search PubMed.
- Purwanto, R. M. Deshpande, R. V. Chaudhari and H. Delmas, J. Chem. Eng. Data, 1996, 41, 1414–1417 CrossRef.
- T. E. Crozier and S. Yamamota, J. Chem. Eng. Data, 1974, 19, 242–244 CrossRef CAS.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- C. H. Braunstein, A. D. Baker, T. C. Strekas and H. D. Gafney, Inorg. Chem., 1984, 23, 857–864 CrossRef.
- S. C. Rasmussen, M. M. Richter, E. Yi, H. Place and K. J. Brewer, Inorg. Chem., 1990, 29, 3926–3932 CrossRef CAS.
- G. Kew, K. DeArmond and K. Hanck, J. Phys. Chem., 1974, 78, 727–734 CrossRef CAS.
- G. Kew, K. Hanck and K. DeArmond, J. Phys. Chem., 1975, 79, 1828–1835 CrossRef CAS.
- G. Denti, S. Campagna, L. Sabatino, S. Serrni, M. Ciano and V. Balzani, Inorg. Chem., 1990, 29, 4750–4758 CrossRef CAS.
- K. Kalyanasundaram, M. Gratzel and M. K. Nazeeruddin, J. Phys. Chem. A, 1992, 96, 5865–5872 Search PubMed.
- M. T. Indelli, C. Chiorboli, L. Flamigni, L. De Cola and F. Scandola, Inorg. Chem., 2007, 46, 5630–5641 CrossRef CAS.
- M. T. Indelli, F. Scandola, L. Flamigni, J. P. Collin, J. P. Sauvage and A. Sour, Inorg. Chem., 1997, 36, 4247–4250 CrossRef CAS.
- M. T. Indelli, F. Scandola, J. P. Collin, J. P. Sauvage and A. Sour, Inorg. Chem., 1996, 35, 303–312 CrossRef CAS.
- F. Scandola, R. Argazzi, C. A. Bignozzi and M. T. Indelli, J. Photochem. Photobiol. A: Chem., 1994, 82, 191–202 CrossRef CAS.
- S. M. Arachchige, J. R. Brown, E. Chang, A. Jain, D. F. Zigler, K. Rangan and K. J. Brewer, Inorg. Chem., 2009, 48, 1989–2000 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |