Metabolic engineering of cyanobacteria for ethanol production†
Jason
Dexter
a and
Pengcheng
Fu
*b
aDepartment of Molecular Biosciences & Bioengineering, University of Hawaii, 1955 East-West Road, Honolulu, Hawaii 96822-2321, USA
bFaculty of Chemical Sciences and Engineering, China University of Petroleum, 18 Fuxue Road, Changping District, Beijing, 102249, P. R. China. E-mail: pengcheng@cup.edu.cn; Fax: + 86-10-6074-3667; Tel: +86-10-8973-1283
First published on 10th June 2009
Abstract
Development of renewable energy is rapidly being embraced by our society and industry to achieve the nation's economic growth goals and to help address the world's energy and global warming crises. Currently most of the bioethanol production is from the fermentation of agricultural crops and residues. There is much debate concerning the cost effectiveness and energy efficiency of such biomass based ethanol production processes. Here, we report the creation of a Synechocystis sp. PCC 6803 strain that can photoautotrophically convert CO2 to bioethanol. Transformation was performed using a double homologous recombination system to integrate the pyruvate decarboxylase (pdc) and alcohol dehydrogenase II (adh) genes from obligately ethanol producing Zymomonas mobilis into the Synechocystis PCC 6803 chromosome under the control of the strong, light driven psbAII promoter. PCR based assay and ethanol production assay were used to screen for stable transformants. A computerized photobioreactor system was established for the experimental design and data acquisition for the analysis of the cyanobacterial cell cultures and ethanol production. The system described here shows an average yield of 5.2 mmol OD730 unit−1 litre−1 day−1 with no required antibiotic/selective agent.
Broader contextThe development of renewable energy is rapidly being embraced by our society and industry to meet the energy growth and emission reduction goals. Ethanol has recently surged to the forefront of biofuels technology. Most of the current ethanol production is from the biomass fermentation of agricultural crops and residues. It remains a challenge for ethanol distilleries to be environmentally friendly and economically competitive using biomass as the raw materials. The technology reported in this paper involves genetic engineering of cyanobacterial strains for direct conversion of sunlight and the greenhouse gas CO2 into ethanol. It will integrate photosynthesis, emission reduction and biofuels production in one single process. Another important advantage for our approach is that by directly using CO2 and solar energy for cyanobacteria growth and ethanol synthesis, agricultural space can be preserved for food production. This novel ethanol production approach thus has great potential for industrial applications for many interrelated sociological/political/environmental/economical reasons. |
Introduction
Biologically produced ethanol as a fuel is an attractive renewable energy concept. Bioethanol can be readily integrated with existing fuel processing/utilization platforms while offering the benefit of control over both production and utilization reactions (i.e. control over the entire carbon cycling of ethanol, as opposed to fossil fuels which we cannot effectively synthesize). Ethanol–gasoline hybrid fuel can be used by most internal combustion engines at levels up to 15% ethanol and with modification they could operate on a mixture of 85% ethanol–15% gasoline.1 Currently most bioethanol production is from the fermentation of agricultural crops and residues. Statistics indicate that the world ethanol production is 60% from sugar crops, 33% from other crops and 7% from chemical synthesis (http://attra.ncat.org/guide/a_m/biomass.html). Concerns over agricultural land/grain use for fuel, along with the high energy input associated with traditional corn/starch and other biomass based fermentation systems have been significant hurdles for the ethanol community. Ethanol production from biomass as a primary feedstock is somewhat controversial since some experts argue that such bioprocesses are neither cost-effective nor energy-efficient.2,3Alternative bioethanol production methods need to be developed so that the costs associated with the land, labor, and time of traditionally fermented crops can be circumvented. Genetic engineering of photosynthetic prokaryotes is one possibility for efficient bioethanol production which would combine the conversion of solar energy, carbon sequestration and ethanol production in a single organism. For example, Deng and Coleman4 demonstrated that through genetic engineering, ethanol could be produced from the uptaken CO2 by a photosynthetic organism via photoautotrophic metabolism. In that experiment, cyanobacterium species Synechococcus sp. strain PCC 7942 was transformed with pCB4-based shuttle vectors bearing the Z. mobilispdc/adh gene cassette, which was first described by Ingram et al.5,6 The genes were expressed under the control of the rbcLS operon promoter, both alone and in combination with the E. coli lac promoter. Their results showed ethanol yields on an order of magnitude of 54 nmol OD730 unit−1 litre−1 day−1. Also in the same study, an ethanol concentration of approximately 5 mM was achieved after 4 weeks of growth. More recently, the Coleman group filed a patent which indicates a yield of 1.7 µmol of ethanol per mg of chlorophyll per hour using the CI-PL temperature inducible promoter. (Woods et al., US patent 6699696, 2004).7
Synechocystis sp. strain PCC 6803 (Synechocystis), a freshwater, non-filamentous, non-nitrogen fixing cyanobacteria capable of heterotrophic growth, was the first photosynthetic autotrophic organism that was completely sequenced and annotated.8 In addition, the natural transformability of this strain has allowed for the establishment of techniques for precise genome manipulation.9–11Synechocystis has thus served as a model organism for understanding fundamental physiological and metabolic processes involved in O2 evolving photosynthesis. The standard method for Synechocystis mutation induction involves transformation with a plasmid vector that is targeted to the genome via double homologous recombination and selection under appropriate conditions.11 For gene disruption (knockout), the vector may contain an antibiotic resistance cassette that has been inserted into the cloned open reading frame (ORF). Simultaneous genomic insertion of both an antibiotic resistance gene (positive selection) and a negative selection component, such as the sacB gene that is lethal in the presence of sucrose, will then allow for introduction of any desired genetic material into the genome replacing the above positive/negative selection cassette with the genes of interest through negative selection.12
The primary objective of this work is to create a strain of the freshwater cyanobacterium Synechocystis sp. PCC 6803 that will form the foundation for an efficient process of ethanol production. Here, we report the creation of a Synechocystis sp. PCC 6803 strain that can photoautotrophically convert CO2 to bioethanol. Transformation was performed using a double homologous recombination system to integrate the pyruvate decarboxylase (pdc) and alcohol dehydrogenase II (adh) genes from obligately ethanol producing Zymomonas mobilis into the Synechocystis 6803 chromosome under the control of the strong, light driven psbAII promoter. A PCR based assay and ethanol production assay were used to screen for stable transformants. A computerized photobioreactor system was established for the experimental design and data acquisition for the analysis of the cyanobacterial cell cultures and ethanol production. The system described here shows an average yield of 5.2 mmol OD730 unit −1 litre−1 day −1 with no required antibiotic/selective agent.
Results and discussions
Comparison with the results of previous research
In the patent filed by Woods et al. (US patent 6306639, 2001),13 the most critical alteration to their ethanol producing system described in 1999 was the substitution of the rbcLS operon endogenous promoter (from Synechococcus PCC 7942) with the temperature inducible CI-PL promoter, EMBL Accessive No. L05669, SEQ. ID. No.7. Obviously, the first point of difference lies in the different organisms chosen to express the pdc/adh cassette. Deng and Coleman chose to use Synechococcus PCC 7942, whose genome was completely sequenced in 2004, as their model organism. We have chosen Synechocystis PCC 6803, which was the first cyanobacteria to be sequenced in 1996.8 Use of Synechocystis thus has the advantage of having a larger body of methodological work associated with the genetics and genetic engineering strategies.In their work Deng and Coleman have used plasmids pCB4-LRpa, pCB4-LR(TF),4 and pCB4-CPpa,13 based on pCB4 as the replicating plasmid vector for expression of the pdc/adh cassette, and plasmid selection via ampicillin. In our work, we used the non-replicating vector for direct integration of genes into the genome at the psbA2 genetic locus, under control of the strong, light-driven psbA2 promoter. The original pPSBAIIKS was based on pSL1180 and was designed for double homologous recombination with the Synechocystis 6803 psbA2 locus, thus the strains did not need to be maintained on antibiotics or other selection factors after isolation. Given the number of variables involved, it is difficult to attribute the difference in ethanol production observed between the Deng and Coleman effort and this work to any single factor. The combined results from both studies thus suggest that there would be much benefit in screening multiple strains of cyanobacteria using various promoter systems to determine the best combination for ethanol production. Yet, one must also consider the additional key advantages of not requiring antibiotics for plasmid maintenance, despite the increase in effort for genetic transformation and screening. This is because during extended photobioreactor experiments, one does not need to be concerned with the antibiotic concentration within the culture media and its sufficiency for maintaining the plasmid.
Creation of pMota for the expression of pdc/adh in Synechocystis
A pararosaniline indicator plate assay was used to confirm the successful creation of E. coli transformant pMota. The indicator plates, as described by Ingram et al.,14 contain the Schiff reagent, allowing for the detection of the alcohol dehydrogenase (adh). Therefore, only the E. coli that contained a plasmid with the adh gene (pLOI295 and pMota) would generate the intensely red Schiff base on the indicator plates. Fig. 1 shows the plasmid constructs (2B) tested positive (in red) with the control (2A, 2C and 2D). Here 2A is a negative control plate with non-ethanogenic pPSBAIIKS. 2B is for the plates with isolated colonies expressing the pdc/adh cassette within pMota. 2C is the positive control plate with pLOI295. Plates in 2A, 2B, and 2C were incubated for 24 hours at 37 °C as given in the standard protocol for the indicator plate assay. And 2D is for a pMota plate which was incubated for ∼2 hours. The indication reaction has not had time to diffuse throughout the LB agar. This method thus allows for rapid screening of isolated colonies of possible interest.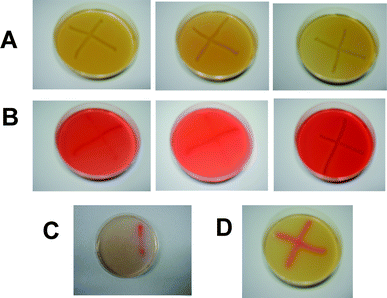 | ||
| Fig. 1 Results from the pararosaniline indicator plate assay on E. coli containing various plasmid constructs. (A) Negative control plates with non-ethanogenic pPSBAIIKS. (B) Plates with isolated colonies expressing the pdc/adh cassette within pMota. (C) Positive control plate with pLOI295. (D) pMota plate. | ||
The plasmid constructs tested positive using the above indicator plate assay were then subjected to restriction mapping for more stringent analysis of the plasmid construct, as shown in Fig. 2. Here the ∼1 kb band located in lane 2 is some contaminant. This is not a problem, as plasmid from this later purification was not used for the initial transformation of PCC 6803 or for the construction of pMota. This batch of pPSBAIIKS will be used solely for novel plasmid construction in the future, eliminating the possible interference of the 1 kb contaminant. Lane 3 shows the expected fragment sizes resulting from a SacI digest of pMota. Expected fragment sizes are observed at 5010 bp and 2276 bp. After successful restriction mapping, the construct was sequenced for final verification of pMota. Primers used for this sequencing reaction are listed in Appendix I, ESI.† The complete pMota sequence is given in Appendix II, ESI.†
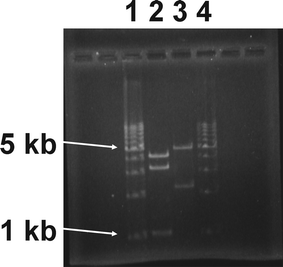 | ||
| Fig. 2 Restriction digest mapping of pMota and pPSBAIIKS (control). Lanes 1 and 4 are Promega™ 1 kb step ladders. Lane two is NdeI/BamHI double digest of pPSBAIIKS with fragment sizes as expected. Lane 3 shows the expected fragment sizes resulting from a SacI digest of pMota. | ||
We found that the modification of the pMota plasmid with a desired multiple cloning site should allow for simultaneous expression and analysis of other complementary gene products with the pdc/adh cassette. This may open up possibilities for future metabolic modifications via the following methods: (1) overexpression of pathway components; (2) overexpression of sigma factor/sigma factor binding proteins;15 (3) gene-silencing via antisense based RNAi.16
Transformation and screening for stable ethanol production
After successful creation of the pMota plasmid, Synechocystis strains used in this study were sequentially transformed and isolated for correct construction, first with pPSBAIIKS (creating WT[r]) and then with pMota (creating WT[OH], here the subscript [OH] denotes the plasmid bearing pdc/adh genes). Results from the PCR based assay, shown in Fig. 3, were used to confirm isolation of strains with homologous psbA2 loci. The first lane in gels (a)–(e) is a 1 kb BRL ladder. Gel conditions in gels (a)–(e) were constant, 1% agarose gel in 2X TAE buffer run at 100 V for 45 min. The last lane in each of the gels (a)–(e) is unused. Starting from the first lane after the BRL ladder, samples were run, with each set of three consecutive lanes corresponding to the same sample tested with the WT, [r], and [OH] assays respectively: (a) Sample 1 and 2 were the WT Synechocystis 6803 strain. As shown here, the WT psbA2 locus was intact in each of these strains and the other internal primers show no cross-reactivity with the WT psbA2 gene (i.e. the 1st lane for each sample set bears the expected 749 bp amplicon, while the 2nd and 3rd lanes corresponding to the [r] and [OH] assay show no amplification). (b) Sample 3 and 4 were for WT[r]. Here it can be seen that the WT[r] strain was completely segregated, while there was no cross reaction with the [OH] assay. In (c) it can be seen that the above WT[OH] (ethanol producing) isolates gave consistent results after additional bioreactor runs. As shown in both samples 5 and 6, the ethanol cassette hybridized only with the pdc-specific 3′ primer, as expected. The Synechocystis strains were then grown under photoautotrophic conditions in a bioreactor and shown to be ethanol producing with respect to the WT control strain. During growth in the bioreactor, samples were taken and assayed again, yielding the results for samples 7–11. In gels (d), a 1 kb Hyperladder® (Bioline, Taunton, MA, USA) was used as a molecular weight marker. Gel (d) also shows the isolation of 5 strains (7–11) of WT[OH], screened via the [OH] reaction of the PCR assay. Single bands at the expected 554 amplicon size were present for samples 7–11. These same five isolates were then tested for reactivity with the [r] reaction of the PCR assay on gel (e); showing no response to the [r] primed reaction (lanes labeled 12–16 were the same samples as 7–11).![PCR assay results showing isolation of completely segregated transformants of WT[r] and WT[OH].](/image/article/2009/EE/b811937f/b811937f-f3.gif) | ||
| Fig. 3 PCR assay results showing isolation of completely segregated transformants of WT[r] and WT[OH]. | ||
Colonies showing successful isolation of desired strains were then glycerol stocked. Successful isolates bearing the ethanol cassette were screened using the primary seed reactor based assay, as described above. Many of the ethanol cassette bearing isolates were shown to give reduced ethanol yields with continued culturing (data not shown). Typically, these strains would show significant reduction in ethanol yields with each consecutive serial inoculation, such that by the second or third serial culture we were able to determine the stability of ethanol production within the strain. One particular isolate was shown to give strong and stable ethanol production after repeated culturing. This isolate was designated as WT5.2[OH] which has been successfully and stably transformed with the ethanol production cassette. As shown in Fig. 4, the WT5.2[OH] strain shows repeatable ethanol production during five consecutive cultures.
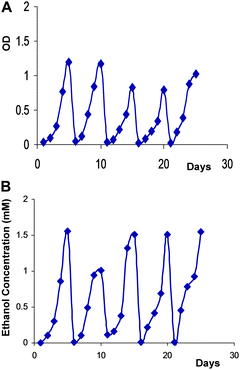 | ||
| Fig. 4 Primary seed reactor assay testing for stability of Ethanol production during repeated culturing. (A) Optical density vs. days for five consecutive batch runs; (B) Ethanol concentration (mM) vs. days for five consecutive batch runs. | ||
Confirmation of stability of the created ethanol producing strains via the seed reactor based assay was critical for strain isolation. It was found through the seed reactor based assay that several isolates with complete segregation for the ethanol production cassette did not show stable ethanol production (data not shown). Typically, this could be deduced by the third serial liquid culturing of an isolate. This process has also been invaluable for the continued monitoring of strains after revival from glycerol stock. Unfortunately, our primary seed reactor system is quite rudimentary, not allowing for pH control, gas flow rate control, and not having condensers on the off-gas outlet, resulting in continuous and unequal evaporative losses from parallel reactors. Also, our current platform can only support the simultaneous growth of 6 such primary seed reactors. Increasing our capacity for parallel processing of primary seed reactors is critical for increasing the rate of obtaining results, ideally with added control regarding the aforementioned parameters.
Creation of novel fermentation pathway in Synechocystis metabolic network
The autotrophic growth of Synechocystis strains encompasses two fundamental processes: (1) cells use light as the energy source to drive the electron-transfer reactions that enable electrons from chlorophyll to move along an electron transport chain that leads to the reduction of NADP to NADPH. During the photosynthetic electron transport, H+ is pumped across the membrane and the resulting proton-motive force drives the formation of ATP. Thus, the light reactions couple with the production of energy and reducing power, as well as the evolution of O2. (2) Carbon fixation reactions in which the ATP and NADPH produced by the photosynthetic electron transport reactions are utilized for the conversion of CO2 to carbohydrate and other metabolites. By our genetic engineering efforts, we have successfully inserted a novel ethanol-producing pathway into the Synechocystis metabolic network. As a result, the carbon flow from pyruvate was directed to this fermentation pathway for formation of the end metabolic product, ethanol. It can be seen that a unique combination of three cellular functions, oxygenic photosynthesis, carbon fixation and ethanol production within the single organism, Synechocystis W5.2[OH], has been achieved. This forms the foundation for the direct conversion of CO2 to ethanol driven by the light energy.Autotrophic photobioreactor fermentation
The WT5.2[OH] strain was then examined under the more stringent control of a commercially based photobioreactor. Fig. 5A (averaging of three runs of growth experiments) shows that the WT5.2[OH] strain has a significantly reduced growth compared to the non-ethanol-producing control. Fig. 5A also shows that the decrease in photon flux into the reactor due to increased optical density of the Synechocystis cultures. Fig. 5B (averaging of three runs of growth experiments) shows that the WT5.2[OH] strain produces detectable amounts of ethanol, whereas the WT control strain does not.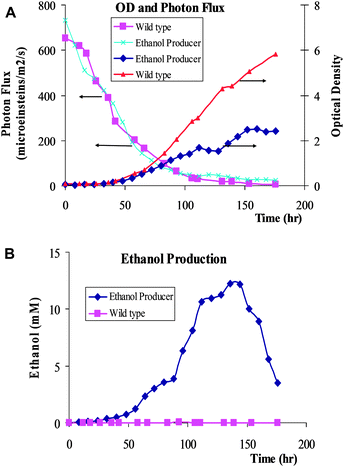 | ||
| Fig. 5 (A) Effect of cell density (optical density = OD730) on photon flux through the photobioreactor. (B) Ethanol concentration vs. time (hour). The results are the average of three individual experimental runs. | ||
The ethanol yields from the BioFlo 110® photobioreactor (New Brunswick Scientific, Edison, NJ, USA) are significantly greater than those obtained from the primary seed reactors. This could be due to a number of factors, but it is significantly due to the increased illumination of the BioFlo 110® and the presence of a condenser at the gas outlet. Given that this system had control of critical features, such as gas flow rate, pH and a condenser on the off-gas, in comparison to the primary seed platform, the results were deemed more representative of actual ethanol yields and growth rate. The pH-stat control strategy also played a critical role. Without pH control, the cells may become starved by insufficient CO2 supply. Multiple discreet pH measurements in the primary seed reactor have shown the pH was always greater than 10.5 at the later phase of the Synechocystis cell cultures. This indicates the cultures lack of CO2 supply—we only sparged compressed air into the primary seed reactors, this did not appear to meet the demand. Also, with access to only a single reactor it is impossible to co-ordinate experiments with parallel variables/negative control. For this reason, analysis using the BioFlo 110® photobioreactor platform was a slow and tedious process. Despite this, the BioFlo 110® platform described here offered the most controlled system for analysis of generated cyanobacterial strains.
Perhaps one of the most important observations from the combined BioFlo 110® runs is the fundamental consideration of limiting factors for a given photobioreactor design. The standard vat type reactor shape was shown to severely limit light flux through the reactor volume; once the OD730 was increased to be around 1, the photon flux at about 1 cm within the reactor was reduced by between 60% and 80%. Thus, light limitation is a critical factor that must be considered for reactor design. An additional complexity is that light availability also effects pdc/adh expression using the psbAII promoter expression system. Gas limiting has also been observed in primary seed cultures (data not shown). Thus, CO2 and light availability have presented themselves as the two major limiting factors in both the culturing of Synechocystis 6803 and in ethanol production. Maintenance of the pH at a given setpoint will allow for the control of CO2 availability, this leaves light availability as the most critical environmental variable for analysis and control.
Some issues for future study
Fig. 5 indicates that due to the ethanol production (which was a metabolic burden for the Synechocystis cells), the WT5.2[OH] strain could reach a final cell density level of about half that of the wild type Synechocystis cells which would direct more carbon resources towards biomass formation. It should be noted that in Fig. 5B, the ethanol concentration peaked around 12 mM. It then started to decrease. Ethanol reutilization may be possible by the Synechocystis cell cultures because Z. mobilis alcohol dehydrogenase II has been shown to be a bidirectional catalyst17 and Synechocystis 6803 contains an endogenous aldehyde dehydrogenase (ORF: slr0091) that may shunt acetaldehyde to acetate biosynthesis. This competing pathway may need to be addressed in future studies.Another question that arises is the possibility that ethanol accumulation in the media may have direct toxic effects on the cell culture, and thus inhibit Synechocystis growth. The toxicity of ethanol on the Synechocystis cell cultures was examined by culturing cells in parallel with the addition of various amount of ethanol to the culture media to make the residual ethanol concentration to vary from 0.5% to 5% (∼115–1150 mM). The experimental results indicate that Synechocystis can tolerate concentrations of up to 1% (∼230 mM) ethanol in the media with negligible impacts on cell growth (data not shown). It can thus be concluded that the ethanol accumulation in the media caused by our Synechocystis mutants has not reached the inhibitory level. Therefore, reduced cell density in the cell cultures with ethanol-producing Synechocystis mutants was not caused by the alcohol inhibition, but by the metabolic reprogramming of the cells with the insertion of new pathway into its metabolic network.
Several microorganisms, including Clostridium sp., are ethanologenic microbes, among them, the yeast Saccharomyces cerevisiae and facultative bacterium Zymomonas mobilis are considered as better candidates for industrial ethanol production. S. cerevisiae is capable of fermenting sugars to produce ethanol anaerobically or microaerobically with high yields (over 15%). Zymomonas mobilis is a prokaryotic organism that produces ethanol. In comparison to S. cerevisiae, Z. mobilis possesses the following advantages: 1. its higher sugar uptake and ethanol yield, 2. its lower biomass production, 3. its higher ethanol tolerance, 4. it does not require controlled addition of oxygen during the fermentation, and 5. its amenability to genetic manipulations.18 Wild type Synechocystis does not possess the pathway for ethanol production. In this paper, we report our genetic engineering efforts of creating ethanol-producing cyanobacteria for the production of biofuels. So far, the ethanol yield in our engineered Synechocystis strains is lower than both S. cerevisiae and Z. mobilis. Since optimization strategies have not yet been implemented, and since the pyruvate decarboxylase (pdc) and alcohol dehydrogenase II (adh) enzymes are not evolved to fully function in the oxygenic photosynthetic organisms, there is no reason to assume that its performance would be optimal. Here we only intend to show that cyanobacteria cells can stably express pdc and adh proteins and produce ethanol from CO2 and light when transformed with heterologous genes. There exists a significant room for the improvement of Synechcystis ethanol production from the aspects of both molecular biology strategies and bioprocess development.
Materials and methods
Strains/culture conditions
E. coli strain C600 was used for the plasmid cloning. Standard LB formulation (10 g NaCl, 10 g tryptone, 5 g yeast extract per litre, pH 7) was used to culture E. coli in both liquid and solid (add 15 g agar per liter) media. Antibiotics ampicillin and kanamycin were supplemented to the LB media at concentrations of 100 µg ml−1 and 50 µg ml−1, respectively. Wild Type (WT) Synechocystis sp. strain PCC 6803 was cultured on standard BG-11 media plates, with or without the addition of: 5 mM glucose, 5% sucrose, and/or either 5 µg ml−1, 25 µg ml−1, or 50 µg ml−1 kanamycin. Plates containing Synechocystis strains were incubated at 30 °C under a light intensity of ∼100 microeinsteins m−2 s−1. All Synechocystis liquid cultures were grown in standard BG-11 medium with the addition of 50 µg ml−1 kanamycin when appropriate. A series of two seed cultures, each requiring about 4 days of growth, were used for the inoculation of a 7.5 L photobioreactor (3 L working volume). Liquid primary seed cultures were grown at 24 °C, with a maximal surface photon flux of ∼200 microeinsteins m−2 s −1 on the seed reactor wall. The seed cultures were agitated via magnetic stir bar, and sparged continuously with compressed air at a rate of approximately 0.5 L min−1. Primary seed cultures consisted of a working volume of 25 mL in a standard 100 mL Pyrex™ media bottle, with a two Pasteur pipettes serving as the sparge and the off-gas tubes. Upstream of the sparge tube was a Whatman® PFTE 0.1 µm filter and the off-gas tube was loosely capped with aluminium foil. Secondary seed cultures used for photobioreactor inoculation was nearly identical to the primary seed bottles except that the working volume was 300 mL in a standard 1 L Pyrex™ media bottle, and that the sparging pipette was replaced with a level C porosity diffuser with pore size of 25–50 µm from Ace Glass Inc. Lights used were 40 W cool white fluorescent tubes. Cell growth in liquid culture was monitored by determination of optical density (OD) of the samples at 730 nm (OD730) using a spectrophotometer (ThermoSpectronic Genesys® 10-S, Beckman Coulter, Inc., Fullerton, CA, USA). UltraVette® 1.5 ml disposable cuvettes from Plastibrand (Vineland, NJ, USA) were used for the OD measurement.Creation of the transformation vector, pMota
All plasmid/PCR product cleanup kits and Taq DNA polymerase were acquired from Qiagen (Valencia, CA, USA). All restriction enzymes, VentR® Polymerase and T4 DNA ligase were obtained from New England Biolabs (Ipswich, MA, USA). The plasmid PSBAIIKS was obtained from Dr Vermaas at Arizona State University. The plasmid LOI295, containing the Z. mobilis pdc and adhII genes, was obtained from Dr Lonnie Ingram at the University of Florida. PCR reaction was used for both the amplification of the pdc/adhII cassette from pLOI295 and for the simultaneous introduction of NdeI and BamHI sites at the 5′ and 3′ ends, respectively. These sites then allowed for subcloning pdc/adhII into the backbone of pPSBAIIKS. It resulted from the removal of aphX/sacB selection cassette via NdeI/BamHI dual digestion, and in the creation of transformation vector, pMota (see Appendix III, ESI).† The following primers were used for the above PCR reaction (restriction sites are italicised, induced mutations are in bold): upstream: 5′-ggAgTAAgCATATgAgTTATACTg-3′ Downstream: 5′-ggATCTCgACTCTAgAggATCC-3′, with a resultant amplicon of 3117 bp. PCR reaction was carried out as follows: total reaction volume of 50 µl, 0.36 µg of pLOI295 as template, 4 units of VentR® polymerase, a final concentration of 0.5 µM for each primer, 300 µM of each dNTP. The PCR was run on the following program on an Eppendorf Mastercycler®: initial denaturation at 94 °C for 2 min, followed by 35 cycles of 10 s denaturation at 94 °C, 1 min annealing at 47 °C, and 3.7 min extension at 68 °C; finally, hold at 4 °C.Screening of E. coli for correct pMota construct
After transformation of competent E. coli C600 cells with the pMota ligation reaction, single colonies were analyzed on aldehyde indicator plates to verify the activity of the Z. mobilis alcohol dehydrogenase II enzyme. These indicator plates were formulated by the addition of 8 ml of pararosaniline (2.5 mg of the dry powder per ml of 95% ethanol; not autoclaved) and 100 mg of sodium bisulfite (unsterilized dry powder) to 400-mL batches of LB agar. Mixtures of pararosaniline and bisulfite are often referred to as Schiff reagent. It has been widely used to detect aldehydes, to detect sugars on glycoproteins after periodic acid oxidation, or in a broth to test for organisms such as Z. mobilis which secrete aldehydes into the culture media.18 These components have been incorporated into a solid medium which is relatively nontoxic and can be used to identify clones expressing enzymes which produce aldehydes. Ethanol diffuses into the E. coli cells, where it can be converted to acetaldehyde by alcohol dehydrogenase. The leuco dye serves as a sink, reacting with acetaldehyde to form a Schiff base which is intensely red.19E. coli colonies that yielded positive results on the indicator plates via presence of intense red coloration were subsequently subjected to restriction mapping. Qiagen® miniprep kits were used for the preparation of plasmid DNA from positive colonies on the indicator plates. Purified plasmids were subjected to NdeI/BamHI double digestion and SacI digestion for verification of fragment sizes. Digests were run on 2X TAE, 1% agarose gels at 100 V for 40 min. Gels were stained with ethidium bromide and visualized on UV light box. After successful verification via restriction mapping, pMota was sequenced using the primer set for sequencing analysis of the pdc/adh cassette within Synechocystis PCC 6803. Sequencing results showed successful creation of the pMota plasmid for insertion of the pdc/adh cassette into the psbA2 locus.
Transformation and screening for stable ethanol production
Following successful creation of pMota, the mutant strains of Synechocystis sp. PCC 6803 were transformed and screened sequentially with pPSBAIIKS and pMota via the protocol described by Williams11 and Lagarde et al.12 After transformation with pPSBAIIKS, a serial replating on increasing kanamycin concentrations were conducted for the isolation of Synechocystis mutant strains WT[r] (here the subscript [r] denotes the plasmid with resistance cassette), which were completely segregated with respect to the aphX/sacB selection cassette, as verified by a PCR based assay. The constructs were in turn grown in liquid seed culture, under the presence of 50 µg mL−1 kanamycin, and subsequently transformed with pMota. The transformants were then screened on BG-11 plates containing 5% sucrose. Screening was performed via a serial streaking of single colonies coupled with a PCR based assay used for probing the psbA2 loci. The PCR assay consisted of three PCR reactions per sample, probing for the presence of the (1) WT psbA2 gene, (2) aphX/sacB selection cassette, and (3) pdc/adhII ethanol pathway cassette. Each of the three reactions shared a common upstream primer that lies outside of the psbA2 gene loci, while each reaction is defined by the downstream primer that is specific for each of the three possible genetic constructs. The upstream primer used in all three reactions was: 5′-gTCAgTTCCAATCTgAACATCGA-3′, with the amplicon beginning 48 bp upstream of the psbA2 start codon. The downstream primer for probing the WT psbA2 gene: 5′-AATTTgTAACCgTAgTTCTgggAT-3′, and the resultant amplicon is 749 bp. For probing the aphX/sacB selection cassette, downstream primer: 5′–TTggTgATTTTgAACTTTTgCTTgC-3′ was used, resulting in a 3.1 kb amplicon. The downstream primer for probing the pdc/adhII cassette: 5′-TTgCAAgCgATTTCgAgATAAA-3′, resulting in a 554 bp amplicon. All PCR reactions were formulated as described in the Qiagen® Taq Polymerase Handbook in the section for long PCR products, modified only by the exclusion of any high fidelity polymerase. The PCR assay utilized the following cycling program: initial denaturation at 94 °C for 3 min, followed by 35 cycles of 10 s denaturation at 94 °C, 1 min annealing at 48 °C, and 3.5 min extension at 68 °C; a final 3 min extension at 68 °C, hold at 4 °C.To perform the PCR assay on a given cyanobacterial sample, FTA® cards (Whatman Inc., Piscataway, NJ, USA) were used for rapid preparation of genomic DNA for use as a template in the above PCR reaction. For testing a liquid culture, 5 µL aliquot was spotted onto the FTA® card. For testing cultures streaked on solid media, multiple colonies were lifted from the plate, streaked on the inside of a 1.5 mL tube and resuspended in 10 µL of BG-11 via vortexing; 5 µL aliquots were then spotted onto the FTA® card, as above. The FTA® protocol for preparation of the archived DNA from a bacterial source was followed for preparation of the template for the PCR assay.
A seed reactor based assay was also used for determination of stability of ethanol generation in consideration of the absence of selective pressure. The assay focused on screening colonies that showed to be completely segregated for the ethanol cassette for stable ethanol production. Seed reactors as described above were inoculated with multiple colonies from a plate of a given isolate. The cells were grown to an OD730 greater than 0.1. They were then centrifuged at 3220 × g for 6 min at room temperature, and resuspended in a fresh seed reactor at an initial OD730 = 0.025. This would constitute the first experimental reactor in a series of five runs. Each seed reactor cell culture was run for five days with continuous feeding of compressed air, at which point the cells were again collected by centrifugation and used to inoculate the second experimental reactor in the series to the above OD730 = 0.025. Only a subset of the total cell biomass was used for this serial inoculation, while the rest was discarded or used to make glycerol stocks. Each day of a particular run, the OD730 were recorded, and a 550 µL aliquot was taken for the ethanol concentration assay (the ‘before’ aliquot). The cells were then washed by collection via centrifugation (as above), discarding the supernatant, resuspended by vortexing of the entire pellet in 25 mL of fresh BG-11, and returned to the seed reactor. The OD730 was again recorded and another aliquot was taken for the ethanol concentration assay (the so called ‘after’ aliquot). After isolation of a stable ethanol producing isolate, the PCR based assay was applied a final time for complete confirmation (data not shown).
Autotrophic photobioreactor fermentation
Exponentially growing seed cultures in the primary seed reactors were used to inoculate a secondary seed reactor, which was then grown to an OD730 > 0.31 and immediately used for inoculation of the autotrophic photobioreactor. For inoculation, the OD730 was taken and used for determination of the seed culture volume required for the initial bioreactor OD730 = 0.02 (working fermentation volume was 3.1 L). Cells from this determined volume of seed culture were harvested via centrifugation and resuspended in 50 mL of fresh BG-11 by vortexing. After inoculation of the bioreactor with this 50 mL seed, an additional 50 mL of BG-11 was used for flushing of the inoculation tube. A 7.5 L BioFlo® 110 bioreactor system (New Brunswick Scientific, Edison, NJ, USA) was used for our experiments. It has its own controllers for temperature, pH and DO adjustment. The Synechocystis cultivation process was monitored and controlled automatically by a Pentium II (233 MHz, Windows 98) computer equipped with an interface board PCI-MIO-16E-10 (National Instruments Corp., Austin, TX, USA). The data acquisition program was written in LabVIEW7.1 (National Instruments Corp., Austin, TX, USA). The data from Bioflo 110, including pH, agitation, temperature and dissolved oxygen concentration (DO) was acquired through the computer interface board. Six General Electric® 26 W, F26DBX/SPX41/4P light bulbs were arrayed around the reactor to give a maximal surface flux of ∼1000 microeinsteins m−2 s −1 on the reactor wall. The local irradiance level of the light intensity at a fixed point (1 cm away from the internal bioreactor surface) was measured using a cosine (2π) quantum meter (LI-190 SA, LI-COR, Lincoln, NE, USA) with a spherical (4π collector) quantum micro-sensor (US-SQS/LI Heinz Walz GmbH, Effeltrich, Germany), and expressed as photosynthetic photon flux density (PPFD). This fiber-optic micro sensor has a spherical sensor tip (with a diameter of 3 mm) that can sense light from all directions underwater. For protection, the sensor was inserted into a glass optical well that was fixed to the bioreactor head plate and submerged in the culture broth. The reactor was maintained at 27–29 °C throughout the fermentation via an external fan circulating air around the lighting system. The reactor was sparged continuously with compressed air or N2 at a rate of 1 L min−1. Additionally, pure CO2 injection was conducted to maintain the pH below the set point of 8.5. This kind of pH-based CO2 feeding strategy ensures that the cells will have sufficient carbon source for growth and/or for ethanol production. The agitation speed was set to 300 rpm. The condenser on the off-gas port was chilled to 8 °C via a thermo circulating water bath (C10-K20, Haake, Berlin, Germany). Fermentations were maintained for eight days, with sampling every eight hours. For sampling, ∼15 ml aliquot was drawn from the reactor via the sampling port, discarded to clear the harvesting downtube, and a second ∼15 ml aliquot, the sample, was drawn. From each sample OD730 was recorded and an aliquot was taken for the ethanol concentration assay.Ethanol concentration assay
For determination of ethanol concentration of a liquid culture, a 550 µl aliquot was taken, spun down at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 × g for 5 min, and 500 µL (or other appropriate volume) of the supernatant was placed in a fresh 1.5 mL tube and stored at −20 °C until assay was performed. Given the linear range of the spectrophotometer and the sensitivity of the ethanol assay, dilution of the sample (up to 20 fold) was required during the later phase of Synechocystis cell cultures. In this case, an appropriate volume of BG-11 was first added to the fresh 1.5 mL tube, to which the required volume of clarified supernatant was added. This solution was used directly in the ethanol assay. Upon removal from −20 °C and immediately before performing the assay, the samples were spun down a second time at 12100 × g for 5 minutes, also assisting in sample thawing. The Boehringer Mannheim/r-biopharm® (Darmstadt, Germany) enzymatic ethanol detection kit was used for ethanol concentration determination. For the ethanol assays, BG-11 medium was used as the blank.
100 × g for 5 min, and 500 µL (or other appropriate volume) of the supernatant was placed in a fresh 1.5 mL tube and stored at −20 °C until assay was performed. Given the linear range of the spectrophotometer and the sensitivity of the ethanol assay, dilution of the sample (up to 20 fold) was required during the later phase of Synechocystis cell cultures. In this case, an appropriate volume of BG-11 was first added to the fresh 1.5 mL tube, to which the required volume of clarified supernatant was added. This solution was used directly in the ethanol assay. Upon removal from −20 °C and immediately before performing the assay, the samples were spun down a second time at 12100 × g for 5 minutes, also assisting in sample thawing. The Boehringer Mannheim/r-biopharm® (Darmstadt, Germany) enzymatic ethanol detection kit was used for ethanol concentration determination. For the ethanol assays, BG-11 medium was used as the blank.
Conclusions
The above results indicate that it is feasible to create and characterize ethanol producing mutants using the light-driven psbA2 promoter system to express the pdc/adh gene cassette in Synechocystis strains. Under defined circumstances ethanol will be accumulated in the liquid media at a concentration on the order of 10 mM approximately 6 days after inoculation. During the log phase of growth, we observe ethanol yields of 5.2 mmol OD730 unit−1 litre−1 day−1.Production of ethanol for fuel is one of the nation's and the world's major initiatives. Currently most of the bioethanol production is from the fermentation of agricultural crops and residues. It has been argued that the ethanol production approaches which primarily use agricultural crops or residues as a feedstock are resource costly. Novel biological solutions for the production of biofuels with the potential to revolutionize the biology-based energy production need to be developed. We have successfully established a foundation for the ethanol production in cyanobacteria under a photoautotrophic metabolic mode. This bioprocess has the potential to scale up for both bioenergy production and CO2 utilization. The latter would prevent carbon dioxide release into the atmosphere, where it has been implicated as a major cause of global warming.
Acknowledgements
Plasmid PSBAIIKS was obtained from Dr Wim Vermaas at Arizona State University. Plasmid LOI295, containing the Z. mobilis pdc and adhII genes, was obtained from Dr Lonnie Ingram at the University of Florida. The authors are grateful to the two anonymous reviewers for their critical and constructive comments that have improved this manuscript.References
- Y. Sun and J. Cheng, Hydrolysis of lignocellulosic materials for ethanol production: a review, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS.
- D. Pimentel and T. W. Patzek, Ethanol Production Using Corn, Switchgrass and Wood; Biodiesel Production Using Soybean and Sunflower, Natural Resources Research, 2005, 14(1), 65–76 Search PubMed.
- T. W. Patzek, A First-Law Thermodynamic Analysis of the Corn- Ethanol Cycle, Natural Resources Research, 2007, 15(4), 255–270 Search PubMed.
- M. D. Deng and J. R. Coleman, Ethanol synthesis by genetic engineering in cyanobacteria, Appl. Environ. Microbiol., 1999, 65(2), 523–8 CAS.
- L. O. Ingram, T. Conway, D. P. Clark, G. W. Sewell and J. F. Preston, Genetic engineering of ethanol production in Escherichia coli, Appl. Environ. Microbiol., 1987, 53(10), 2420–2425 CAS.
- L. O. Ingram, P. F. Gomez, X. Lai, M. Moniruzzaman, B. E. Wood, L. P. Yomano and S. W. York, Metabolic engineering of bacteria for ethanol production, Biotechnol. Bioeng., 1998, 58(2–3), 204–214 CrossRef CAS.
- R. P. Woods, J. R. Coleman, M. de Deng, Genetically modified cyanobacteria for the production of ethanol, the constructs and method thereof, US Patent 6699696, 2004 Search PubMed.
- M. Ikeuchi, Complete genome sequence of a cyanobacterium Synechocystis sp. PCC 6803, the oxygenic photosynthetic prokaryote, Tanpakushitsu Kakusan Koso, 1996, 41(16), 2579–2583 Search PubMed.
- S. Aoki, T. Kondo and M. Ishiura, A promoter-trap vector for clock-controlled genes in the cyanobacterium Synechocystis sp. PCC 6803, J. Microbiol. Methods, 2002, 49(3), 265–274 CrossRef CAS.
- W. F. J. Vermaas, J. G. K. Williams, A. W. Rutherford, P. Mathis and C. J. Arntzen, Genetically engineered mutant of the cyanobacterium Synechocystis 6803 lacks the photosystem II chlorophyll-binding protein CP-47, Proc. Natl. Acad. Sci. U. S. A., 1986, 83(24), 9474–9477 CrossRef CAS.
- J. G. K. Williams, Construction of Specific Mutations in Photosystem II Phosynthetic Reaction Center by Genetic Engineering Methods in Synechocystis 6803, Methods Enzymol., 1988, 167, 766–778 Search PubMed.
- D. Lagarde, L. Beuf and L. W. F. J. Vermaas, Increased production of zeaxanthin and other pigments by application of genetic engineering techniques to Synechocystis sp. strain PCC 6803, Appl. Environ. Microbiol., 2000, 66(1), 64–72 CrossRef CAS.
- R. P. Woods, J. R. Coleman, M. de Deng, Genetically modified cyanobacteria for the production of ethanol, the constructs and method thereof, US Patent 63066639, 2001 Search PubMed.
- L. O. Ingram and T. Conway, Expression of Different Levels of Ethanologenic Enzymes from Zymomonas mobilis in Recombinant Strains of Escherichia coli, Appl. Environ. Microbiol., 1988, 54(2), 397–404 CAS.
- H. Alper, J. Moxley, E. Nevoigt, G. R. Fink and G. Stephanopoulos, Engineering yeast transcription machinery for improved ethanol tolerance and production, Science, 2006, 314(5805), 1565–1568 CrossRef CAS.
- C. Kemmer and P. Neubauer, Antisense RNA based down-regulation of RNaseE in E.coli, Microb. Cell Fact., 2006, 5, 38–49 CrossRef.
- S. Kinoshita, T. Kakizono, K. Kadota, K. Das and H. Taguchi, Purification of two alcohol dehydrogenases from Zymomonas mobilis and their properties, Appl. Microbiol. Biotechnol., 1985, 22, 249–254 CAS.
- T. Conway, G. W. Sewell, Y. A. Osman and L. O. Ingram, Cloning and sequencing of the alcohol dehydrogenase II gene from Zymomonas mobilis, J. Bacteriol., 1987, 169(6), 2591–2597 CAS.
- P. Gunasekaran and K. Chandra Raj, Ethanol fermentation technology – Zymomonas mobilis, Curr. Sci., 1999, 77(1), 56 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Appendices I, II and III. See DOI: 10.1039/b811937f |
| This journal is © The Royal Society of Chemistry 2009 |