CNx nanofibers converted from polypyrrole nanowires as platinum support for methanol oxidation†
Yanwen
Ma
ab,
Shujuan
Jiang
a,
Guoqiang
Jian
a,
Haisheng
Tao
a,
Leshu
Yu
a,
Xuebin
Wang
a,
Xizhang
Wang
a,
Jianmin
Zhu
c,
Zheng
Hu
*a and
Yi
Chen
a
aKey Laboratory of Mesoscopic Chemistry of MOE and Jiangsu Provincial Lab for Nanotechnology, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, China. E-mail: zhenghu@nju.edu.cn; Fax: +86 25-83686251; Tel: +86 25-83686015
bJiangsu Key Lab for Organic Electronics and Information Displays, Institute of Advanced Materials, Nanjing University of Posts and Telecommunications, Nanjing, 210003, China
cNational Laboratory of Solid State Microstructures, Nanjing University, Nanjing, 210093, China
First published on 18th December 2008
Abstract
A new kind of carbon nitride (CNx) nanofiber has been prepared by the calcination of polypyrrole nanowires at 800 °C. The product maintained a wire-like morphology during calcination, and the pyrrolic nitrogen in the polypyrrole nanowires gradually changed to pyridinic and graphitic nitrogen as annealing temperature increased. These CNx nanofibers, prepared at 800 °C, have a nitrogen concentration of about 10%. Pt nanoparticles with average size of ∼3 nm could therefore be easily immobilized onto the CNx nanofibers because of the inherent chemical activity arising from the nitrogen incorporation. The Pt/CNx composite catalyst thus obtained has a large electrochemically active area and gives good electrocatalytic performance for methanol oxidation, both in activity and stability, suggesting it has potential application in fuel cells.
Introduction
Direct methanol fuel cells (DMFCs) are attracting considerable interest as alternative power sources for automotive and portable applications due to their high energy conversion efficiency and environmental benefits.1 The current state-of-the-art electrocatalysts adopted for direct methanol oxidation are carbon supported Pt-based metal nanoparticles.2,3 Great efforts have been devoted to improving these electrocatalysts for increasing the DMFCs' performance while decreasing their cost. Recent studies have revealed that carbon nanotubes (CNTs) and carbon nanofibers (CNFs) are of great potential for using as supports in the Pt-based electrocatalyst due to their unique surface structure, high electric conductivity, corrosion resistance and large surface areas.3 For the effective dispersion of the ultrafine Pt-based catalysts onto CNTs or CNFs, usually it is necessary to chemically modify the CNTs or CNFs before they are used as supports to immobilize the catalysts.3 The modification processes commonly used, e.g., acid oxidation and side wall functionalization, are obviously complex and/or may result in the introduction of some pollutants, and may even decrease the mechanical properties of the supports. A promising strategy to overcome these disadvantages is to introduce chemically active sites into the CNTs or CNFs during their growth, hereby the latter modification process could be simplified. This thought has been demonstrated in our recent study on methanol oxidation catalyzed by Pt/CNx nanotube hybrids.4 Briefly, according to the six-membered-ring-based growth mechanism,5 CNx nanotubes dominated with pyridinic nitrogen were obtained by using a pyridine precursor.6 Then, Pt nanoparticles could be homogeneously distributed and strongly anchored on the so-prepared CNx nanotubes without needing further modification due to the N-participation.7 The resulting Pt/CNx nanotube hybrid has obvious electrocatalytic activity for methanol oxidation. This progress stimulates our interests to explore the electrocatalyst supports of some other carbon-nitride nanomaterials. In this study, a new kind of CNx nanofiber with nitrogen concentration about 10 at% has been fabricated by the calcination of PPy (polypyrrole) nanowires, which could be then conveniently used for direct immobilization of Pt nanoparticles due to the inherent chemical activity arisen from the nitrogen incorporation. The electrocatalytic properties of the so-constructed Pt-loaded CNx nanofibers for methanol oxidation have been examined by cyclic voltammetry and obvious catalytic activity and good stability obtained as expected, indicating their potential important application in fuel cells.Experimental
Analytical grade chemicals of cetyltrimethylammonium bromide (CTAB), pyrrole monomer and ammonium persulfate (APS) were purchased from Beijing Chemical Reagents Company. Commercial carbon black powder of Vulcan XC-72 was obtained from Cabot International.PPy nanowires were synthesized by the solution chemistry method.8 In short, 12.4 g CTAB was dissolved in 750 mL deionized water, and 1.5 mL pyrrole monomer was dropped into the CTAB solution and stirred for 3 h. Then 45 mL aqueous solution containing 5.1 g APS was added as oxidizing agent to initiate the polymerization for 24 h. All the experiments were performed at 0–5 °C. After reaction, the precipitate was filtrated and thoroughly washed with deionized water and ethanol alternately, then dried at 70 °C overnight. PPy nanowires thus obtained were used for further study.
CNx nanofibers were prepared by the decomposition of PPy nanowires. Briefly, 0.1 g PPy nanowires were placed in a quartz tube furnace and then heated under Ar protection from room temperature to 400, 600, and 800 °C respectively at a rate of 1 °C min−1 and kept there for 2 h. After the furnace was cooled down to room temperature, a black powder was obtained.
Platinum nanoparticles were supported on CNx nanofibers by the microwave-polyol method.9 20 mg CNx nanofibers converted from PPy nanowires at 800 °C were placed in 50 mL ethylene glycol (EG) and sonicated for 20 min to obtain a liquid suspension. 1 mL EG solution of hexachloroplatinic acid (7.5 mg Pt mL−1 EG) was added to the suspension dropwise and magnetically stirred for 3 h, and 2 mL NaOH/EG solution (0.1 mol L−1) was then added into the above mixture. After stirring for 10 min, the mixture was put into a microwave oven (700 W) for 90 s irradiation. The solid sample with about 27.3 wt% Pt loading was collected after multi-wash and filtration with ethanol and finally vacuum-dried at room temperature, denoted as Pt/CNx. For comparison, Pt/PPy and Pt/XC-72 catalysts were also constructed by replacing CNx nanofibers by PPy nanowires and Vulcan XC-72 with the same procedure, respectively.
The product was characterized by transmission electron microscopy (TEM, JEOL-JEM-1005 at 100 kV), high-resolution transmission electron microscopy (HRTEM, JEM2010 at 200 kV), Fourier transform infrared spectroscopy (FTIR, BRUKER VECTOR22) with a KBr pressed pellet, X-ray diffraction (XRD, Philips X'pert Pro X-ray diffractometer with a Cu Kα radiation of 1.5418 Å) and X-ray photoelectron spectroscopy (XPS, VG ESCALAB MKII).
The electrochemical performance of the catalysts of Pt/CNx, Pt/PPy and Pt/XC-72 were evaluated at 25 °C in a three-electrode cell connected to a CHI 660C workstation (CH Instrument, Inc.). A glass carbon disk with an area of 0.071 cm2 was used as a working electrode. Ag/AgCl and platinum wire served as reference and counter electrodes, respectively. Thin film electrode was prepared similarly to those reported in the literature.10 Briefly, a suspension of Pt/CNx catalyst with a concentration of 1.0 mg mL−1 was made by ultrasonically dispersing 2 mg Pt/CNx in 2 mL ethanol/water solution (1 : 1, v/v). Then 10 µL Pt/CNx suspension was dropped onto the glassy carbon disk, giving about 2.73 µg Pt, i.e. 38.5 µg cm−2 Pt loading. When the glassy carbon disk was dried in air, 10 µL diluted Nafion solution (5 wt%, Alfa Aesar) was dropped onto the catalyst to act as binder. Thin film electrodes for Pt/PPy and Pt/XC-72 were prepared by the same procedure with the same Pt loading.
Hydrogen electrosorptions of the catalysts were measured by cyclic voltammetry (CV) in nitrogen saturated 1 M H2SO4 solution between −0.25 and 1.4 V at a scan rate of 50 mV s−1. CO stripping experiments were conducted in 1 M H2SO4 solution which was purged with nitrogen for 30 min and then bubbled with CO gas (99.99%) for 15 min for CO adsorption on catalysts. The residual CO in the solution was subsequently removed by nitrogen purging for 40 min. The CO stripping CV curves were recorded within the potential between 0 and 1.25 V at a scan rate of 10 mV s−1. The activities of the catalysts for methanol oxidation were examined by the stable CV curves in 1 M H2SO4 solution containing 1 M methanol in the potential range of 0 to 1.0 V at a scan rate of 50 mV s−1. Electrochemical stabilities of the catalysts were evaluated by immersing the thin film electrodes into the electrolyte for one to four days, then measuring the CV curves of methanol oxidation at the freshly-made electrolyte.
Results and discussion
Fig. 1 shows typical TEM images of PPy nanowires and the annealed samples. The PPy nanowires (Fig. 1a) have a diameter of about 80 nm and length up to a micron. After annealing at 400 to 800 °C, the samples still maintained their wire-like morphology without an obvious change in diameter. The evolution of the FTIR spectra (Fig. 2) shows the gradual diminishing of the characteristic peaks for PPy with increasing annealing temperature, indicating the increasing carbonization. Specifically, the FTIR spectrum of PPy nanowires (line a) presents the peaks at 1552 and 1473 cm−1 for pyrrole ring vibration and at 1310 and 1090 cm−1 for![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H in-plane deformation.8 These peaks become weaker and weaker with increasing annealing temperature and almost vanish at 800 °C (line b–d). This means that PPy nanowires have been converted to another kind of carbon-based nanowire at 800 °C.
C–H in-plane deformation.8 These peaks become weaker and weaker with increasing annealing temperature and almost vanish at 800 °C (line b–d). This means that PPy nanowires have been converted to another kind of carbon-based nanowire at 800 °C.
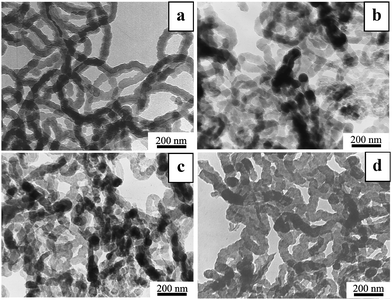 | ||
| Fig. 1 Typical TEM images of PPy nanowires (a) and the samples after 2 h annealing at 400 °C (b), 600 °C (c) and 800 °C (d), respectively. | ||
 | ||
| Fig. 2 FTIR spectra of PPy nanowires (a) and the samples after 2 h annealing at 400 °C (b), 600 °C (c) and 800 °C (d), respectively. | ||
XRD curves of the PPy nanowires and the annealed samples are shown in Fig. 3. The diffraction peaks at 23.6, 24.1, 24.6 and 25.5° correspond to d spacings of 0.38, 0.37, 0.36 and 0.35 nm respectively. The peak for PPy nanowires at 23.6° should arise from the π–π interaction of partial PPy chains similar to that of aromatic groups.8 With increasing annealing temperature, the peak shifts gradually to the higher diffraction-angle side accompanied by an increase of its intensity, finally approaching a d spacing close to 0.34 nm, i.e., d002 in graphite. The XRD curve for the sample annealed at 800 °C also displays a weak peak at 43.4° corresponding to graphite (100), which might be a sign of the complete transformation of PPy to another kind of carbon-based nanowire, in agreement with the FTIR results.
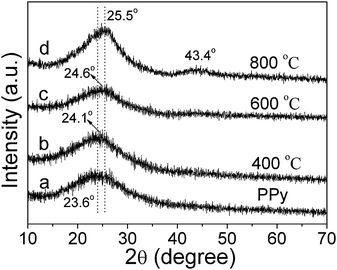 | ||
| Fig. 3 XRD curves of the PPy nanowires and the samples after 2 h annealing at 400 °C (b), 600 °C (c) and 800 °C (d), respectively. | ||
The PPy nanowires before and after annealing were further analyzed by XPS as shown in Fig. 4, with the data listed in Table 1. It is seen that nitrogen in PPy nanowires exists as pyrrolic nitrogen at 400.1 eV.11 With increasing annealing temperature, the pyrrolic nitrogen gradually transforms to pyridinic nitrogen at 398.5 eV and graphitic nitrogen at 401.0 eV. After annealing at 800 °C, the nitrogen exists as pyridinic and graphitic nitrogen with the total concentration of about 10 at% (Table 1). The above experimental results point to the fact that CNx nanofibers have been obtained by annealing PPy nanowires at 800 °C.
| Samples | Atom concentration (at%) | ||
|---|---|---|---|
| C1s | N1s | O1s | |
| PPy | 72.8 | 17.5 | 9.7 |
| 400 °C | 81.4 | 12.6 | 6.0 |
| 600 °C | 85.1 | 12.4 | 2.5 |
| 800 °C | 88.1 | 10.2 | 1.7 |
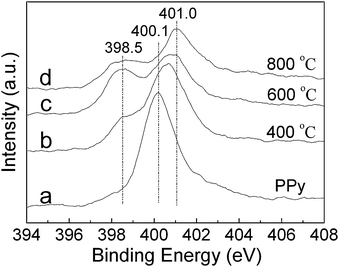 | ||
| Fig. 4 N1s XPS spectra of PPy nanowires (a) and the samples after 2 h annealing at 400 °C (b), 600 °C (c) and 800 °C (d), respectively. | ||
As revealed in our recent study, replacing some of the carbon atoms of CNTs with nitrogen could create active sites for anchoring Pt nanoparticles as a potential electrocatalyst for DMFC.4 Hence, we would like to examine the performance of this new kind of carbon nitride material, i.e., CNx nanofibers, for similar application in DMFC. Pt nanoparticles have been immobilized onto the CNx nanofibers, also onto the PPy nanowires and Vulcan XC-72 for comparison, by the microwave-polyol method. Fig. 5 displays the XRD curves of Pt/CNx, Pt/PPy and Pt/XC-72. It is seen that new peaks at 39.6, 46.1 and 67.6° appeared in addition to the background diffraction from CNx nanofibers, PPy nanowires and Vulcan XC-72, which could be attributed to the (111), (200) and (220) planes of face-centered cubic Pt (JCPDS, No. 04-0802), respectively. The XRD result indicates that Pt species exists in the metallic state. Fig. 6 shows typical TEM and HRTEM images of the Pt/CNx, Pt/PPy and Pt/XC-72 catalysts. It is clearly seen that Pt nanoparticles are homogeneously deposited onto the CNx nanofibers, PPy nanowires and Vulcan XC-72 carbon black powder with high dispersion. From the size-distribution histograms inserted in Fig. 6a, c, and e, it can bee seen that the average size d of Pt nanoparticles for these three catalysts could be distinguished in the relative order d (Pt/PPy) < d (Pt/CNx) < d (Pt/XC-72) with the mean particle size of 2.5, 3 and 4 nm, respectively. This is understandable since, for the same Pt loading of 27.3 wt%, CNx nanofibers have less nitrogen concentration (10.2 at%) as active sites for nucleation and growth of Pt nanocrystals than PPy nanowires (17.5 at%) (Table 1), while Vulcan carbon has the least active sites only from native defects. The chemical stabilities of Pt/CNx and Pt/PPy were intuitively reflected by their morphologies' changes after four-days' immersion in 1 M H2SO4 solution, as shown in Fig. 7. It's seen that Pt/CNx is quite stable and the CNx nanofibers keep the one-dimensional morphology (Fig. 7a), while Pt/PPy is unstable and the PPy nanowires collapse in H2SO4 solution (Fig. 7b),12 which indicates the much higher chemical stability of Pt/CNx than Pt/PPy. Apparently, the so-constructed Pt/CNx catalyst is attractive for use in electrocatalysis due to the highly uniform dispersion of Pt nanoparticles with excellent stability.

 | ||
| Fig. 6 Typical TEM and HRTEM images of Pt/CNx, Pt/PPy and Pt/XC-72 catalysts. (a) and (b) for Pt/CNx, (c) and (d) for Pt/PPy, (e) and (f) for Pt/XC-72. Inserts in (a), (c) and (e) are the corresponding size-distribution histograms of Pt nanoparticles for each catalyst based on observation of 300 particles, in which P. D. means particle diameter and F. frequency. Inserts in (b), (d) and (f) are the enlarged images. | ||
 | ||
| Fig. 7 Typical TEM images of Pt/CNx (a) and Pt/PPy (b) catalysts after 4 days' immersion in 1 M H2SO4 solution. | ||
The electrocatalytic performances of the Pt/CNx, Pt/PPy and Pt/XC-72 catalysts for methanol oxidation have been systematically examined in a parallel manner for comparison. Firstly, the electrochemically active areas (EAA) of the three catalysts have been evaluated by both hydrogen electrosorptions13 and CO stripping voltammetries13a,14 as shown in Fig. 8. EAAH of Pt/CNx, Pt/PPy and Pt/XC-72 catalysts are 104, 117 and 84 m2 g−1 Pt, and EAACO 92, 101 and 78 m2 g−1 Pt, respectively. It is seen that both experimental results of hydrogen electrosorptions and CO stripping voltammetries show the relative order of EAA (Pt/PPy) > EAA (Pt/CNx) > EAA (Pt/XC-72). This relative order means that the average size d of Pt nanoparticles has the relative order of d (Pt/PPy) < d (Pt/CNx) < d (Pt/XC-72), which is in good agreement with the TEM observation in Fig. 6.
 | ||
| Fig. 8 Hydrogen electrosorption (a) and CO stripping (b) CV curves for Pt/CNx (green), Pt/PPy (red) and Pt/XC-72 (black) catalysts in 1 M H2SO4 with a scan rate of 50 mV s−1 and 10 mV s−1 respectively. Note: Current density = current/area of the working electrode, i.e. 0.071 cm2. | ||
The stable CV curves of methanol oxidation as the anodic half-cell reaction in DMFCs for the three catalysts are shown in Fig. 9. It is seen that the peak potentials of Pt/CNx, Pt/PPy and Pt/XC-72 catalysts are all around 0.66 V. The onset potential of ∼0.4 V for Pt/CNx and Pt/PPy are slightly lower than that of ∼0.5 V for Pt/XC-72. The maximum stable peak current density (Jmax) for Pt/CNx and Pt/PPy catalysts are 7.0 and 14.1 mA cm−2 respectively, and both of them are higher than that for Pt/XC-72 catalyst (4.6 mA cm−2). Hence the catalytic activities of the catalysts follow the order Pt/PPy > Pt/CNx > Pt/XC-72, which matches well with the relative order of EAA as expected, i.e. EAA (Pt/PPy) > EAA (Pt/CNx) > EAA (Pt/XC-72) obtained from the experiments of hydrogen electrosorptions and CO stripping voltammetries (Fig. 8). In the reverse scan curve for each catalyst in Fig. 9, another oxidation peak appears around 0.4 V, which is attributed to the oxidation of CO or CO-like species.3c,15 The corresponding peak current density is denoted as JCO. A high ration of Jmax/JCO suggests a better CO-poisoning tolerance.3c,15 In this case, the Jmax/JCO for Pt/CNx, Pt/PPy and Pt/XC-72 catalysts are 1.5, 1.2 and 2.2 respectively, indicating the CO-poisoning tolerance of the catalysts follow the order Pt/XC-72 > Pt/CNx > Pt/PPy. This order is consistent with the relative order of the average sizes of the Pt nanoparticles for the three catalysts. This suggests that the different CO-poisoning tolerances for the three catalysts resulted from the different Pt nanoparticle sizes in them, i.e., the larger Pt nanoparticles have higher tolerance to CO-poisoning, in accordance with the literature study.16
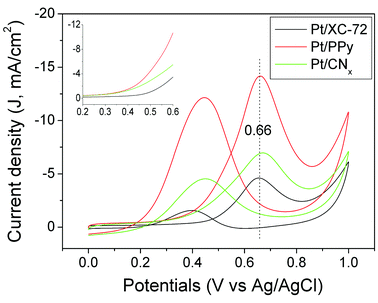 | ||
| Fig. 9 Cyclic voltammetries of Pt/CNx (green), Pt/PPy (red) and Pt/XC-72 (black) catalysts in 1 M H2SO4 aqueous solution with 1 M CH3OH. Inset is the forward parts of the CV curves. Note: Current density = current/area of the working electrode, i.e. 0.071 cm2. | ||
By dividing the maximum current by EAA, the current density J′ for each catalyst has been obtained to evaluate the catalytic activity per active site (see ESI†). It is shown that J′(Pt/PPy) is much larger than J′(Pt/CNx), hence the much bigger peak current in the stable CV for Pt/PPy (Fig. 9) not only comes from the larger EAA due to the smaller Pt size, but also from the modified chemical state of the Pt species by PPy probably due to the influence of abundant pyrrolic nitrogen.17 Of note, the stability of these catalysts are rather different as shown in Fig. 10. Both Pt/CNx and Pt/XC-72 are quite stable and keep the stable Jmax of ∼6.0 mA cm−2 and ∼4.5 mA cm−2, while Pt/PPy degrades seriously within two days and reach the stable state with Jmax of ∼5.0 mA cm−2. The deterioration of Pt/PPy could be ascribed to the degradation of PPy nanowires in acid solution (Fig. 7) rather than the CO poisoning (see ESI†). Therefore, the Pt/CNx catalyst is much more stable than the Pt/PPy catalyst, and has higher catalytic activity than the Pt/XC-72 catalyst, which indicates the potential applications of the Pt/CNx catalyst in DMFC.
 | ||
| Fig. 10 The maximum peak current densities of Pt/CNx (green), Pt/PPy (red) and Pt/XC-72 (black) catalysts change with the immersion time in electrolyte. | ||
Conclusions
In summary, a new kind of carbon nitride nanofiber has been prepared via the carbonization of polypyrrole nanowires at high temperature while keeping the wire-like morphology. During the annealing process, the pyrrolic nitrogen in polypyrrole nanowires was gradually changed to pyridinic and graphitic nitrogen, and the CNx nanofibers obtained by 800 °C annealing contains the nitrogen concentration of about 10 at%. Due to the inherent chemical activity arising from the nitrogen incorporation, Pt nanoparticles with average size of ∼3 nm could be easily anchored onto the CNx nanofibers. The so-constructed Pt/CNx catalyst presents a high electrochemically active area, good stability and obvious electrocatalytic activity for methanol oxidation, suggesting its potential application in fuel cells. The anticipated results in this study indicate that our strategy, i.e. by creating chemically active sites into the CNTs or CNFs during their growth to realize convenient surface modification and construction of composite nanocatalyst, is quite promising.Acknowledgements
This work was financially supported by NSFC (20833002, 20525312), “973” program (2007CB936300) and the Scientific Research Foundation of Graduate School of Nanjing University.References
- (a) S. Srinivasan, Fuel cells: from fundamentals to applications, Springer, New York, 2006 Search PubMed; (b) J. D. Morse, Int. J. Energy Res., 2007, 31, 576 CrossRef CAS.
- (a) H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95 CAS; (b) E. Antolini, J. Power Sources, 2007, 170, 1 CrossRef CAS.
- (a) W. Z. Li, C. H. Liang, W. J. Zhou, J. S. Qiu, Z. H. Zhou, G. Q. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292 CrossRef CAS; (b) T. Matsumoto, T. Komatsu, K. Arai, T. Yamazaki, M. Kijima, H. Shimizu, Y. Takasawa and J. Nakamura, Chem. Commun., 2004, 840 RSC; (c) Y. L. Hsin, K. C. Hwang and C. T. Yeh, J. Am. Chem. Soc., 2007, 129, 9999 CrossRef CAS; (d) K. M. Metz, D. Goel and R. J. Hamers, J. Phys. Chem. C, 2007, 111, 7260 CrossRef CAS; (e) K. Lee, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Appl. Electrochem., 2006, 36, 507 CrossRef CAS.
- B. Yue, Y. W. Ma, H. S. Tao, L. S. Yu, G. Q. Jian, X. Z. Wang, X. S. Wang, Y. N. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1747 RSC.
- Y. J. Tian, Z. Hu, Y. Yang, X. Z. Wang, X. Chen, H. Xu and Q. Wu, J. Am. Chem. Soc., 2004, 126, 1180 CrossRef CAS.
- H. Chen, Y. Yang, Z. Hu, K. F. Huo, Y. W. Ma, Y. Chen, X. S. Wang and Y. N. Lu, J. Phys. Chem. B, 2006, 110, 16422 CrossRef CAS.
- (a) A. Zamudio, A. L. Elías, J. A. Rodríguez-Manzo, F. López-Urías, G. Rodríguez-Gattorno, F. Lupo, M. Rühie, D. J. Smith, H. Terrones, D. Díaz and M. Terrones, Small, 2006, 2, 346 CrossRef CAS; (b) S. H. Yang, W. H. Shin, J. W. Lee, H. S. Kim and J. K. Kang, Appl. Phys. Lett., 2007, 90 Search PubMed 013103; (c) X. Lepró, Y. Vega-Cantú, F. J. Rodríguez-Macías, Y. Bando, D. Golberg and M. Terrones, Nano Lett., 2007, 7, 2220 CrossRef CAS; (d) X. L. Li, Y. Q. Liu, L. Fu, L. C. Cao, D. C. Wei, G. Yu and D. B. Zhu, Carbon, 2006, 44, 3113 CrossRef; (e) Y. Y. Shao, J. H. Sui, G. P. Yin and Y. Z. Gao, Appl. Catal., B, 2008, 79, 89 CrossRef CAS.
- X. T. Zhang, J. Zhang, W. H. Song and Z. F. Liu, J. Phys. Chem. B, 2006, 110, 1158 CrossRef CAS.
- M. Tsuji, M. Kubokawa, R. Yano, N. Miyamae, T. Tsuji, M. S. Jun, S. Hong, S. Lim, S. H. Yoon and I. Mochida, Langmuir, 2007, 23, 387 CrossRef CAS.
- L. Li and Y. Xing, J. Phys. Chem. C, 2007, 111, 2803 CrossRef CAS.
- J. Casanovas, J. M. Ricart, J. Rubio, F. Illas and J. M. Jiménez-Mateos, J. Am. Chem. Soc., 1996, 118, 8071 CrossRef CAS.
- K. Cheah, M. Forsyth and V. T. Truong, Synth. Met., 1999, 101, 19 CrossRef CAS.
- (a) A. Pozio, M. D. Francesco, A. Cemmi, F. Cardellini and L. Giorgi, J. Power Sources, 2002, 105, 13 CrossRef CAS; (b) Z. Q. Tian, S. P. Jiang, Y. M. Liang and P. K. Shen, J. Phys. Chem. B, 2006, 110, 5343 CrossRef CAS.
- C. Bock, M. A. Blakely and B. MacDougall, Electrochim. Acta, 2005, 50, 2401 CrossRef CAS.
- Z. Liu, X. Y. Ling, X. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234 CrossRef CAS.
- Z. F. Liu, M. Shamsuzzoha, E. T. Ada, W. M. Reichert and D. E. Nikles, J. Power Sources, 2007, 164, 472 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Current densities (J′) of Pt/CNx, Pt/PPy and Pt/XC-72 catalysts representing the catalytic activity per active site; analysis on the degradation of Pt/PPy catalyst after immersion in electrolyte. See DOI: 10.1039/b807213m |
| This journal is © The Royal Society of Chemistry 2009 |