Deposition of an oxomanganese water oxidation catalyst on TiO2 nanoparticles: computational modeling, assembly and characterization†
Gonghu
Li
,
Eduardo M.
Sproviero
,
Robert C.
Snoeberger III
,
Nobuhito
Iguchi
,
James D.
Blakemore
,
Robert H.
Crabtree
*,
Gary W.
Brudvig
* and
Victor S.
Batista
*
Department of Chemistry, Yale University, P.O. Box 208107, New Haven, CT 06520-8107, USA. E-mail: robert.crabtree@yale.edu; gary.brudvig@yale.edu; victor.batista@yale.edu
First published on 12th January 2009
Abstract
Inexpensive water oxidation catalysts are needed to develop photocatalytic solar cells that mimic photosynthesis and produce fuel from sunlight and water. This paper reports the successful attachment of a dinuclear di-µ-oxo manganese water oxidation catalyst [H2O(terpy)MnIII(µ-O)2 MnIV(terpy)H2O](NO3)3 (1, terpy = 2,2′:6′2″-terpyridine) onto TiO2 nanoparticles (NPs) via direct adsorption, or in situ synthesis. The resulting surface complexes are characterized by EPR and UV-visible spectroscopy, electrochemical measurements and computational modeling. We conclude that the mixed-valence (III,IV) state of 1 attaches to near-amorphous TiO2 NPs by substituting one of its water ligands by the TiO2 NP, as suggested by low-temperature (7 K) EPR data. In contrast, the analogous attachment onto well-crystallized TiO2 NPs leads to dimerization of 1 forming Mn(IV) tetramers on the TiO2 surface as suggested by EPR spectroscopy and electrochemical studies.
Broader contextIn Nature, the production of O2 by oxidizing water is catalyzed by an Mn4Ca inorganic unit in the oxygen-evolving complex of photosystem II. Heterogeneous arrays consisting of a light harvester, a water oxidation catalyst, and an electron acceptor have been considered a promising approach for artificial photosynthesis. In this study, we deposited a di-nuclear manganese water oxidation catalyst on the surfaces of TiO2 nanoparticles, which have shown to be efficient electron acceptors. The synthesized materials were thoroughly characterized with a variety of techniques and computational modeling. Both experimental and theoretical studies indicate the successful surface immobilization of an oxomanganese water oxidation catalyst. This approach could be utilized to build an artificial photosynthetic system, in which the oxomanganese catalyst facilitates water oxidation and the TiO2 surface collects photogenerated electrons for solar hydrogen production. |
1. Introduction
Solar energy is a vast potential source of renewable energy but we still lack cheap, efficient and robust solar cells. For large-scale applications, it is important to avoid rare elements, such as ruthenium needed in Grätzel cells. Beyond electrical energy production, it should be possible to mimic photosynthesis and produce fuel from sunlight and water by incorporating a water oxidation catalyst into a solar cell.1 In this paper, we report the attachment of a dimanganese water oxidation catalyst on TiO2 nanoparticles (NPs). We describe the synthesis and characterization based on computational modeling, UV-visible and electron paramagnetic resonance (EPR) spectroscopy, and electrochemical measurements.Oxide semiconductors have been widely explored as catalysts for H2 production, based on solar water splitting.2 In particular, TiO2-based materials have been particularly attractive as photocatalysts,3 largely because the TiO2 band edges bracket the redox potential for water splitting.4 However, TiO2 has a relatively large band-gap and its direct photoactivation requires UV light (λ < 400 nm), accounting for only less than 4% of the natural sunlight spectrum.5 While it is possible to extend the absorption edges of TiO2 into the visible-light region,5–7 multiphoton activation would still be required to accumulate the energy needed for water splitting. Several other composite semiconductors are also visible light catalysts for water splitting,8,9 but their efficiencies are still too low. Common challenges include the recombination of the photogenerated electron-hole pairs, and the usual weak photocatalytic activity of semiconductor surface sites.
Surface functionalization of TiO2 NPs has been extensively investigated within the context of Grätzel cells as the most successful strategy for maximizing light-harvesting with wide band-gap semiconductors.10,11 Dye-molecules attached to TiO2 surfaces shift the absorption threshold to the visible light region and efficiently inject electrons into the semiconductor conduction band upon visible photoexcitation. However, the photo-generated holes (h+) are typically localized in the oxidized adsorbate molecules with a potential that is insufficient to oxidize water. Therefore, while dye-sensitized solar cells may be efficient for light harvesting and photon-to-electricity conversion, they do not necessarily lead to solar water splitting.
Water oxidation into O2, protons and electrons is a four electron oxidation reaction,12
| 2H2O → O2 + 4H+ + 4e−, |
Complex 1 is thought to be biomimetic since some of its structural and mechanistic features are similar to those of the photosynthetic reaction center.18,24–27 While the catalytic mechanism of water oxidation based on complex 1 is not yet established,26 it is thought to involve the formation of a high-valent oxyl radical species MnIV–O˙ species according to the following reactions:
| [H2O(terpy)MnIII(µ-O)2MnIV(terpy)H2O]3+ → [H2O(terpy)MnIV(µ-O)2MnIV(terpy)OH]3+ + H+ + e− |
| [H2O(terpy)MnIV(µ-O)2MnIV(terpy)OH]3+ → [H2O(terpy)MnIV(µ-O)2MnIV–O˙(terpy)]3+ + H+ + e− |
| [H2O(terpy)MnIV(µ-O)2MnIV–O˙(terpy)]3+ + H2O → [MnII⋯MnIII] + O2 + 2H+ |
A few studies have already investigated the catalytic and photochemical properties of oxomanganese water oxidation catalysts on solid supports. Yagi and co-workers deposited complex 1 on clay materials and saw catalytic oxygen evolution from Ce4+ as the single-electron oxidant.28,29 They suggested that complex 1 was autoxidized to the (IV,IV) state on kaolin, which is then capable of catalyzing single-electron water oxidation.28 They later found that adsorption on clay suppressed the decomposition of 1 to MnO4− and suggested that water oxidation is facilitated by the cooperation of two equivalents of 1.29 Recently, Weare et al.30 reported a nanostructured assembly in which a related oxomanganese complex, [(bpy)2MnIII(µ-O)2MnIV(bpy)2](NO3)3 (bpy = 2,2′-bipyridine), was coupled to single CrVI charge-transfer chromophore in the channels of nanoporous silica. FT-Raman and EXAFS studies demonstrated that visible light induced electron transfer from the MnIII(µ-O)2MnIV core to CrVI forming MnIV(µ-O)2MnIV and CrV.30 In this study, we build upon earlier work31,32 and we focus on the attachment of complex 1 onto TiO2 NPs and on the characterization of the resulting surface complexes by computational studies, UV-visible and EPR spectroscopy, and electrochemical measurements.
2. Experimental procedures
All reagents and solvents were purchased from Aldrich and used without further purification. MilliQ water was used to make all of the aqueous solutions.2.1. Materials synthesis
2.2. Materials characterization
2.3. Oxygen evolution assay
Using Ce4+ as the oxidant, oxygen evolution from aqueous solutions of 1–TiO2 dispersed in water was measured at 298 K by an oxygen probe (Clark electrode) connected to a YSI 5300A oxygen monitor with digital readout, interfaced to a computer for data logging.35 The sample chamber was maintained at 25 °C with a circulating water bath. The instrument readout was calibrated against air-saturated distilled water kept stirred in the air-tight sample chamber under the Clark electrode. In a typical run, 3 µmol 1 and 50 mg TiO2 were mixed in water and stirred at room temperature for 90 min. The resulting 1–TiO2 was then washed with water to remove excess 1 and was dispersed in 4 ml water in the sample chamber. After allowing time for equilibration and ensuring a constant baseline reading, 50 µL of an aqueous solution of 2 M ceric ammonium nitrate was introduced into the sample chamber.3. Computational analysis
This section describes the computational methodology, including the preparation of structural models, the description of the Quantum Mechanics/Molecular Mechanics (QM/MM) hybrid methods, the methodology applied for simulations of UV-visible spectra, and the calculation of binding enthalpies. The results and discussion, with an emphasis on mechanistic implications, are presented in Section 4.3.1. Molecular models
The reduced models included 1 adsorbed onto 15 [TiO2] units and capped with terminal OH fragments, where the positions of the O atoms in the capping fragments were constrained to their corresponding positions in the 32 [TiO2] unit precursor structure. To dock the complex to the surface, we assumed that one of the two water ligands is exchanged by the TiO2 NP, leaving the Mn dimer immobilized on the surface by a single oxo bridge and hydrogen bonding interactions. The exchanged water molecule is stabilized by hydrogen bonding to the other water molecule in the complex. The orientation of the resulting complex adsorbate (Fig. 1), relative to the surface, was obtained by energy minimization at the DFT QM/MM level of theory.
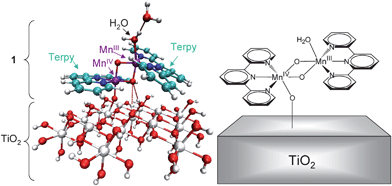 | ||
| Fig. 1 Left panel: DFT/QM-QMM model of the Mn complex 1 anchored to a TiO2–NP. A water ligand is exchanged by the NP and remains hydrogen-bonded to the other water ligand to allow for calculations of binding enthalpies (see text for details). Color scheme: C (light-blue), H (white), Mn (purple), N (blue), O (red), Ti (gray). Right panel: Schematic representation of the 1–TiO2. | ||
3.2. QM/MM hybrid approach
QM/MM computations are based on the two-layer ONIOM electronic-embedding (EE) link-hydrogen atom approach37 as implemented in Gaussian 03.38 The ONIOM-EE method is applied by partitioning the system, according to a reduced high-level molecular domain (X layer) that includes complex 1, its terpy ligands and water molecules (the model system). The rest of the system (Y layer) includes the TiO2 surface and is described at the molecular mechanics level. The ONIOM QM/MM methodology could be efficiently applied, as in previous studies of the multiple nuclear transition metal compounds, only after obtaining high-quality initial-guess states for the cluster of Mn ions (the model system) according to ligand field theory39 as implemented in Jaguar 5.5.40 The resulting combined approach exploits important capabilities of ONIOM, including both the link-hydrogen atom scheme for efficient and flexible definitions of QM layers and the possibility of modeling open-shell systems by performing unrestricted DFT (i.e. UB3LYP) calculations.The total energy E of the system is obtained at the ONIOM-EE level from three independent calculations as follows,
| E = EMM,X + Y + EQM,X − EMM,X |
The efficiency of the QM/MM calculations is optimized by using a combination of basis sets for the QM layer, including the lacvp basis set for Mn ions in order to consider non-relativistic electron core potentials, the 6-31G(d) basis set for O in order to include polarization functions on bridging oxides, and the 6-31G basis set for the N and 3-21G basis set for C and H atoms in the QM layer. Such a choice of basis set has been validated through extensive benchmark calculations on high-valent manganese complexes.36 The molecular structure beyond the QM layer is described by the UFF MM force field.41 Charges of the molecular mechanics layer were computed by using the Charge Equilibration (QEq) method for molecular dynamics simulations,42 which is consistent with the UFF force field.
The model TiO2 slab, with dimensions 1.5 nm × 1.5 nm × 0.4 nm, includes 113 atoms and corresponds to the smallest possible cluster that provides a converged value of the binding affinity of the complex. Its fully relaxed configuration is obtained by energy minimization at the ONIOM-EE (UHF B3LYP/lacvp (Mn),6-31G* (O),6-31G(N),3-21G (C and H) UFF level of theory, subject to the constraint of fixed OH capping fragments. The electronic state of the surface adsorbate complex involves high-spin anti-ferromagnetic couplings between Mn centers. The resulting optimized structures are analyzed and evaluated not only on the basis of the total energy of the system but also as compared to structural, electronic, and mechanistic features that should be consistent with experimental data.
3.3. Binding enthalpies
DFT QM/MM calculations of binding energies are based on the fully optimized configurations of the structural models (a), (b) and (c) shown in Scheme 1, modeled as described in Sections. 3.1 and 3.2. The binding enthalpy ΔH = E(a) − [E(b) + E(c)] is obtained by computing the energy difference between the energy of the whole system E(a) (i.e., the energy of 1 adsorbed on TiO2) and the sum of the energies of 1 [i.e., E(c)] and TiO2 [i.e., E(b)]. Complex 1 adsorbed on the TiO2 surface includes an additional water molecule, as shown in Fig. 1, that serves to conserve the total number of atoms in the QM layers of reactants and products. Upon detachment of the complex from the surface, the additional water molecule forms an OH ligand to MnIV and the capping H of the TiOH fragment on the attachment site of the TiO2 surface (see Scheme 1). | ||
| Scheme 1 Binding energies are calculated as the difference between the extrapolated energy of the whole system (a), which includes the TiO2 surface, the terpy complex 1 and a substrate water molecule; minus an OH adsorbed on the surface (b); minus the complex with an H (c). | ||
3.4. Simulations of UV-visible spectra
The simulations of UV-visible spectra are based on electronic structure calculations of the TiO2 anatase nanoparticles functionalized with 1, as previously reported.31 The calculations are based on a tight-binding model Hamiltonian of the complete supercell 1–TiO2, gained from the Extended-Hückel (EH) semiempirical approach for electronic structure calculations. The EH method requires a relatively small number of transferable parameters and is capable of providing semiquantitative descriptions of energy bands of elemental materials (including transition metals) as well as compound bulk materials in various phases. It is applicable to large extended systems and provides valuable qualitative insight on the role of chemical bonding. The method is, therefore, most suitable to develop a clear chemical picture of the nature of electronic transitions and the interfacial electron-transfer mechanism.The EH Hamiltonian of the bare and functionalized TiO2 is diagonalized in the basis of AOs by solving the generalized EH eigenvalue problem, HQq = EqSQq, where H is the EH matrix, S is the atomic orbital overlap matrix, and Qq are the expansion coefficients of the molecular orbital (MO) with eigenvalue Eq.
The oscillator strength f of the transition with energy Eq′ − Eq is computed by using the expression  , where
, where ![[r with combining circumflex]](https://www.rsc.org/images/entities/i_char_0072_0302.gif) is the electronic position operator,
is the electronic position operator, ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) is the wavenumber of the transition, c is the speed of light, m is the electron mass, h is Planck's constant, and 〈r|ψq〉 is the wavefunction of the MO with eigenvalue Eq in the coordinate representation. The oscillator strengths are used to assign relative weights to the transitions and the total absorbance is globally scaled to facilitate the comparison with the experimental results. The stick-spectrum of discrete transitions is convoluted with a Gaussian function to match the experimental inhomogeneous broadening.
is the wavenumber of the transition, c is the speed of light, m is the electron mass, h is Planck's constant, and 〈r|ψq〉 is the wavefunction of the MO with eigenvalue Eq in the coordinate representation. The oscillator strengths are used to assign relative weights to the transitions and the total absorbance is globally scaled to facilitate the comparison with the experimental results. The stick-spectrum of discrete transitions is convoluted with a Gaussian function to match the experimental inhomogeneous broadening.
4. Results and discussion
Well-crystallized TiO2 materials are usually preferred in photocatalytic applications, over amorphous materials, as photogenerated electron-hole pairs tend to recombine at defect sites on an amorphous TiO2 surface.43 In this study, three TiO2 materials with different degrees of crystallinity were analyzed as substrate supports for complex 1. P25, the gold standard for photocatalysts, was synthesized by a high-temperature (greater than 1200 °C) flame hydrolysis method.44 X-ray diffraction data confirm P25 consists of ∼85% anatase and ∼15% rutile [Fig. 2(a)]. For the synthesized TiO2 nanoparticles without further thermal treatment, a broad (101) diffraction in its XRD pattern [Fig. 2(c)] indicates that D70 nanoparticles are mainly anatase phase with a relatively low degree of crystallinity and have small particle sizes (TEM data are provided in Fig. S2†). Upon sintering at 450 °C, the anatase nanoparticles became better crystallized, as demonstrated by the narrowed (101) diffraction in Fig. 2(b) relative to that in Fig. 2(c). The BET surface areas of D70, D450, and P25 were measured to be 310, 85, and 50 m2 g−1, respectively. | ||
| Fig. 2 Powder XRD patterns of (a) P25, (b) D450, and (c) D70. | ||
4.1. Structural characterization of 1 deposited on TiO2 nanoparticles
 | ||
| Fig. 3 EPR spectra of (a) 1–P25, (b) 1–D450, and (c) 1–D70. The EPR spectrum of complex 1 in a HOAc/NaOAc buffer solution (pH 4.5) is also shown in the figure (d). A trapped electron signal and an organic radical signal are also present in spectra (a) and (b), respectively. The spectrum (c) was scaled down to 1/10 of its original magnitude to allow a better comparison. | ||
The photochemical behavior of 1–TiO2 materials was studied with EPR spectroscopy. For each sample, EPR spectra were taken in dark and under illumination with visible light (>425 nm). A difference spectrum was obtained by subtracting the dark spectrum from the one under illumination. As shown in Fig. 4(a), a resonance corresponding to trapped electrons in the rutile lattice was seen for 1–P25 after illumination with visible light. This signal was not seen for bare P25 under the same experimental conditions. An electron signal identical to the one shown in Fig. 3(a) was also observed for 1–P25 prepared in complete dark conditions. Thus, the electrons may be generated in the dark and transferred to the deep trapping sites in P25. One possible process is the dimerization of 1 to form a Mn(IV) tetramer (2 Mn(III,IV) dimers form 1 Mn(IV) tetramer plus 2e−), as will be discussed later in the text. The lack of such trapping sites in D450 or D70 can explain the absence of an electron signal in the EPR spectra in Fig. 3(b) and 3(c), which were collected under the same conditions as the one in Fig. 3(a). Upon visible light illumination, electrons may also be liberated from the surface Mn complexes or residual, free terpy ligand on P25 and transferred to the trapping sites in P25, leading to the further increase in the rutile electron signal.
 | ||
| Fig. 4 Light-minus-dark difference EPR spectra measured at 7 K of (a) 1–P25, (b) 1–D450, and (c) 1–D70. A resonance at g ∼ 1.97, corresponding to trapped electrons in a rutile lattice, was seen for 1–P25. | ||
The structural and electronic analysis of our QM and DFT-QM/MM models indicate that the attachment of 1 does not significantly affect the spin density of the Mn centers (i.e., the spin densities change less than 5% upon attachment, from 3.83 to 3.88 a.u. for MnIII and from 2.57 to 2.53 a.u. for MnIV). Structural rearrangements, however, are more significant. One of the terpy ligands exhibits a slight distortion upon adsorption of the complex, leading to an increase in the MnIII–N distances. The Jahn–Teller elongation axis of MnIII does not change upon adsorption of the complex, but the Mn–N distances increase (see Table 1, third row). Also, replacing a water molecule by an oxo group, that is a better electron donor, decreases the MnIV–O distance (Table 1, second row) and stabilizes the MnIV center.
 | ||
| Fig. 5 UV-visible spectra of (a) 1–P25, (b) 1–D450, (c) 1–D70, and (d) bare D70 NPs. | ||
![Simulated UV-visible difference spectrum (solid line) compared to the experimental difference spectrum (dashed line). The difference spectrum is the spectrum of 1–D70 [Fig. 5(c)] minus the spectrum of D70 [Fig. 5(d)]. The simulated spectrum was convoluted with gaussians with an FWHM of 60 nm to facilitate comparison.](/image/article/2009/EE/b818708h/b818708h-f6.gif) | ||
| Fig. 6 Simulated UV-visible difference spectrum (solid line) compared to the experimental difference spectrum (dashed line). The difference spectrum is the spectrum of 1–D70 [Fig. 5(c)] minus the spectrum of D70 [Fig. 5(d)]. The simulated spectrum was convoluted with gaussians with an FWHM of 60 nm to facilitate comparison. | ||
Complex 1 exhibits two prominent features in the UV-visible absorption spectrum, including one at 450 nm and the other at 650 nm, as indicated by our computational analysis and the experimental spectrum (Fig. 6 and S4†). Upon adsorption to a TiO2 nanoparticle, the two absorption features are blue shifted to 420 nm and 610 nm, respectively. The transition at 420 nm is assigned to a ligand-to-metal-charge-transfer (LMCT) transition from the terpyridyl ligands to the manganese oxo core, while the transition at 610 nm is assigned to a terpyridyl π–π* transition. Cross transitions from 1 to the TiO2 surface are observed in the range 400–550 nm, although with smaller intensity.
The UV-visible spectrum of 1–D450 [Fig. 5(b)] is not very informative, but an intense and broad absorption around 450 nm is observed for 1–P25 [Fig. 5(a)], indicating the possible existence of an oxidized form of 1 containing a MnIV(µ-O)2MnIV core, a Mn tetramer featuring a MnIV(µ-O)2MnIV–O–MnIV(µ-O)2MnIV unit (see S5†),46 or even solid-state compounds of Mn4+ such as MnO2.
| TiO2 | Experimental | Calculateda | ||
|---|---|---|---|---|
| C | N | N | 1/µmol | |
| a Estimation based on the experimental values of C content, making the assumption that the adsorbates remain in the di-µ-oxo dinuclear form with a Mn : N : C atomic ratio of 1 : 3 : 15 when attached on the surfaces of 1–TiO2 materials. | ||||
| P25 | 0.09 | 0.020 | 0.021 | 0.05 |
| D450 | 0.11 | 0.021 | 0.026 | 0.06 |
| D70 | 0.23 | 0.057 | 0.055 | 0.13 |
 | ||
| Fig. 7 O2 evolution using Ce4+ as a single-electron oxidant. 1 was loaded on TiO2 (50 mg) samples: (a) P25, (b) D450, and (c) D70. A control test was also done using (d) bare P25 NP's as the catalyst. | ||
The loading of complex 2, the Mn(IV) tetramer, onto P25 would be ∼0.025 µmol, assuming complex 1 dimerizes on P25 (see Table 2). It can be inferred from Fig. 7(a) that ∼0.04 µmol O2 was generated after 1000 s using functionalized P25 as the catalyst. This leads to a turnover number greater than 1 for O2 evolution, suggesting that water oxidation on functionalized P25 is catalytic. Furthermore, the initial rate of O2 evolution is estimated to be ∼0.04 nmol s−1, comparable to that for complex 1 supported on clay at a loading of ∼0.3 µmol as reported by Yagi and co-workers.28
4.3. Electrochemical characterization of 1–TiO2
To understand the redox properties of 1–P25 and characterize the electrochemistry of 1 on different TiO2 surfaces, we have performed cyclic voltammetry. TiO2 electrodes were prepared by using standard techniques, doctor-blading TiO2 suspensions on FTO conducting glass slides and subsequently sintering them at 450 °C, before use as the working electrode in a standard three-electrode configuration. The requirement for sintering the samples at elevated temperatures before the electrochemical measurements precluded electrochemical study of 1–D70.The observed redox behavior of 1 on TiO2 electrodes is similar to the redox activity previously reported for homogeneous MnIII(µ-O)2MnIV catalysts of water oxidation.47 Our cyclic voltammograms, shown in Fig. 8, exhibit fairly well-resolved redox behavior, including a quasi-reversible redox couple centered at approximately 1 V, corresponding to MnIII(µ-O)2MnIV ↔ MnIV(µ–O)2MnIV (Pa1 and Pc1).
 | ||
| Fig. 8 Cyclic voltammograms using different working electrodes: (a) P25/FTO, (b) D450/FTO, and (c) bare FTO. The solution contained 0.57 mM 1 in 0.1 M KNO3. Redox processes are labeled including: oxidation of MnIII(µ-O)2MnIV (Pa1), reduction of MnIV(µ-O)2MnIV (Pc1), reduction of the Mn(IV) tetramer (Pc2 and Pc2′). | ||
Subsequent dimerization of MnIV(µ-O)2MnIV can occur, forming the Mn(IV)-terpy tetramer (2).47 Using the bare FTO electrode, the reduction of this tetrameric species gives a broad cathodic wave at ca. 850 mV (Pc2). This cathodic current corresponding to reduction of the Mn tetramer was not observed on the P25/FTO electrode and was barely noticeable on the D450/FTO electrode [see Fig. 8(a) and 8(b)]. However, a large cathodic current is seen at ca. 700 mV for both P25/FTO and D450/FTO, suggesting that the Mn tetramer can be stabilized on the surfaces of crystallized NPs (P25 and D450). Thus, we conclude that the reduction of the (IV,IV,IV,IV) species to the (III,IV,IV,IV) species occurs at a more reducing potential, indicating surface stabilization of the tetramer.
These results are consistent with the observation of O2 evolution for 1–P25 and 1–D450 (see Fig. 7). The acidic conditions of Ce4+ solutions are known to give O2 evolution for the Mn(IV) tetramer species 2.48 Furthermore, the cooperative interaction of two equivalents of 1 has been previously suggested to contribute to the catalytic activity when using 1–mica as the catalyst.29 A similar interaction is likely to be present in the current system, where dimeric oxomanganese catalysts on the surface of the nanoparticles additionally form tetrameric species.
5. Conclusions
We have assembled an oxomanganese water oxidation catalyst (complex 1) on the surfaces of near-amorphous TiO2 nanoparticles. The 1–TiO2 hybrid assemblies were characterized with a variety of techniques including computational modeling, EPR and UV-visible spectroscopies and electrochemistry. Quantum mechanics calculations suggest that 1 is anchored on the TiO2 surface via an oxo bridge (Mn–O–Ti), formed upon substitution of a water ligand by the surface. The mixed-valence Mn(III,IV) state of the complex 1 is not the predominant form of the surface adsorbate complex for well-crystallized TiO2 nanoparticles, probably due to formation of a Mn(IV) tetramer. Using Ce4+ as a primary oxidant, oxygen evolution was observed for 1–P25. However, the chemically-driven water oxidation was not observed for 1 in the mixed-valent form on near-amorphous TiO2.Our EPR measurements suggest that the formation/stabilization of a Mn(IV) tetramer from the mixed-valence Mn(III,IV) species 1 may be facilitated by irreversible electron injection into TiO2. However, further work is required to fully characterize the resulting surface adsorbates. Work in progress includes X-ray studies and immobilization of complex 1 by covalently attaching the complex to the TiO2 surface by using molecular linkers. The utilization of the injected electrons, trapped in 1–P25, and the activation of the surface catalysts with visible light will be reported elsewhere.
Acknowledgements
The authors acknowledge support from the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (DE-FG02-07ER15909) and DOE supercomputer time from NERSC. NSF grant CHE-0215926 provided funds to purchase the ELEXSYS E500 EPR spectrometer and the NSF ECCS # 0404191 grant supported preliminary work. G.L. thanks Drs. Ping-yu Chen, Clyde Cady, Yunlong Gao, Siddhartha Das, Gerald Olack, Barry Piekos, Professor Charles Schmuttenmaer, Professor Gary Haller, Xiaoming Wang, and Gözde Ulaş for their assistance in various aspects of experiments and their helpful discussions.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991–1022 CrossRef CAS.
- K. Rajeshwar, J. Appl. Electrochem., 2007, 37, 765–787 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295–295 CrossRef CAS.
- Z. G. Zou, J. H. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727–1752 CrossRef CAS.
- W. Ruttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef CAS.
- N. D. Morris and T. E. Mallouk, J. Am. Chem. Soc., 2002, 124, 11114–11121 CrossRef CAS.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- M. Borgstrom, N. Shaikh, O. Johansson, M. F. Anderlund, S. Styring, B. Akermark, A. Magnuson and L. Hammarstrom, J. Am. Chem. Soc., 2005, 127, 17504–17515 CrossRef.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 3428–3442 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 6728–6730 CrossRef CAS.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916–6920 CrossRef CAS.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS.
- J. Limburg, J. S. Vrettos, H. Y. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS.
- V. K. Yachandra, K. Sauer and M. P. Klein, Chem. Rev., 1996, 96, 2927–2950 CrossRef CAS.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2008, 47, 1815–1823 CrossRef CAS.
- C. S. Mullins and V. L. Pecoraro, Coord. Chem. Rev., 2008, 252, 416–443 CrossRef CAS.
- M. Yagi and K. Narita, J. Am. Chem. Soc., 2004, 126, 8084–8085 CrossRef CAS.
- K. Narita, T. Kuwabara, K. Sone, K.-i. Shimizu and M. Yagi, J. Phys. Chem. B, 2006, 110, 23107–23114 CrossRef CAS.
- W. W. Weare, Y. Pushkar, V. K. Yachandra and H. Frei, J. Am. Chem. Soc., 2008, 130, 11355–11363 CrossRef CAS.
- S. G. Abuabara, C. W. Cady, J. B. Baxter, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Phys. Chem. C, 2007, 111, 11982–11990 CrossRef CAS.
- W. R. McNamara, R. C. I. Snoeberger, G. Li, J. M. Schleicher, C. W. Cady, M. Poyatos, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 14329–14338 CrossRef CAS.
- H. Y. Chen, R. Tagore, S. Das, C. Incarvito, J. W. Faller, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2005, 44, 7661–7670 CrossRef CAS.
- G. Li and K. A. Gray, Chem. Mater., 2007, 19, 1143–1146 CrossRef CAS.
- R. Tagore, H. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Inorg. Biochem., 2006, 100, 786–800 CrossRef CAS.
- S. Dapprich, I. Komaroni, K. S. Byun, K. Morokuma and M. J. Frisch, THEOCHEM, 1999, 461, 1–21 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian, Inc., Wallingford, CT, 2004.
- (a) G. Vacek, J. K. Perry and J. M. Langlois, Chem. Phys. Lett., 1999, 310, 189–194 CrossRef CAS; (b) E. M. Sproviero, J. A. Gascón, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Chem. Theory Comput., 2006, 2, 1119–1134 CrossRef CAS.
- Jaguar 5.5; L. Schroedinger, Portland, OR, 2003 Search PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. I. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- A. K. Rappe and W. A. I. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- G. Li and K. A. Gray, Chem. Phys., 2007, 339, 173–187 CrossRef CAS.
- A. Mills and S. LeHunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- S. R. Cooper, G. C. Dismukes, M. P. Klein and M. Calvin, J. Am. Chem. Soc., 1978, 100, 7248–7252 CrossRef CAS.
- H. Y. Chen, J. W. Faller, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2004, 126, 7345–7349 CrossRef CAS.
- C. Baffert, S. Romain, A. Richardot, J. Lepretre, B. Lefebvre, A. Deronzier and M. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS.
- Y. Gao, C. W. Cady, R. H. Crabtree, G. W. Brudvig, manuscript in preparation.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structure of complex 1, TEM images of TiO2 NPs, EPR spectra of 1-D70 and isolated 1, and UV-visible spectra of complexes 1 and 2. See DOI: 10.1039/b818708h |
| This journal is © The Royal Society of Chemistry 2009 |