Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels†
Eric E. Benson, Clifford P. Kubiak*, Aaron J. Sathrum and Jonathan M. Smieja
Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive MC 0358, La Jolla, CA 92093, USA. E-mail: ckubiak@ucsd.edu; Tel: 1-858-822-2477
First published on 24th October 2008
Abstract
Research in the field of catalytic reduction of carbon dioxide to liquid fuels has grown rapidly in the past few decades. This is due to the increasing amount of carbon dioxide in the atmosphere and a steady climb in global fuel demand. This tutorial review will present much of the significant work that has been done in the field of electrocatalytic and homogeneous reduction of carbon dioxide over the past three decades. It will then extend the discussion to the important conclusions from previous work and recommendations for future directions to develop a catalytic system that will convert carbon dioxide to liquid fuels with high efficiencies.
 Eric Benson, Cliff Kubiak, Aaron Sathrum, Jon Smieja, and Bear | Eric Benson (left) received his BS degree in chemistry from the University of Arizona in 2005. His research is on biomimetic catalysts for the reduction of CO2. |
Cliff Kubiak received the Sc. B. degree with honors in chemistry from Brown University in 1975. He was awarded the PhD (1980) in inorganic chemistry by the University of Rochester, where he worked with Richard Eisenberg. He was a postdoctoral associate with Mark S. Wrighton at M. I. T. Kubiak was a faculty member at Purdue University from 1982–1998. He moved to UCSD in 1998 as Harold C. Urey Professor. His current research interests include ultra-fast electron transfer and molecular electronics, as well as atom transfer of organometallic complexes focusing on the homogeneous reduction of CO2 to liquid fuels. |
Aaron Sathrum (third from left) received a BS in electrical engineering and materials science from UC Davis in 2002. He is currently researching semiconductor photoelectrochemistry. |
Jon Smieja (right) received his BS degree in chemistry from the University of St. Thomas (MN) in 2005. His research is in ligand and catalyst design for the homogeneous reduction of CO2. |
1. Introduction
The catalytic conversion of CO2 to liquid fuels is a critical goal that would positively impact the global carbon balance by recycling CO2 into usable fuels. The challenges presented here are great, but the potential rewards are enormous. CO2 is an extremely stable molecule generally produced by fossil fuel combustion and respiration. Returning CO2 to a useful state by activation/reduction is a scientifically challenging problem, requiring appropriate catalysts and energy input. This poses several fundamental challenges in chemical catalysis, electrochemistry, photochemistry, and semiconductor physics and engineering.1.1 The challenge of CO2 reduction, thermodynamic considerations
With respect to CO2 reduction to liquid fuels or fuel precursors such as CO/H2 (synthesis gas), proton-coupled multi-electron steps are generally more favorable than single electron reductions, as thermodynamically more stable molecules are produced. This is summarized in eqn (1)–(5) (pH 7 in aqueous solution versus NHE, 25 °C, 1 atmosphere gas pressure, and 1 M for the other solutes).1,2 In contrast, the single electron reduction of CO2 to CO2˙− occurs at E0 = −1.90 V, E6, due to a large reorganizational energy between the linear molecule and bent radical anion.| CO2 + 2H+ + 2e−→ CO + H2O E0 = −0.53 V | (E1) |
| CO2 + 2H+ + 2e−→ HCO2H E0 = −0.61 V | (E2) |
| CO2 + 4H+ + 4e−→ HCHO + H2O E0 = −0.48 V | (E3) |
| CO2 + 6H+ + 6e−→ CH3OH + H2O E0 = −0.38 V | (E4) |
| CO2 + 8H+ + 8e−→ CH4 + 2H2O E0 = −0.24 V | (E5) |
| CO2 + e−→ CO2˙− E0 = −1.90 V | (E6) |
1.2 The challenge of CO2 reduction, kinetic considerations
One key problem in the conversion of CO2 to liquid fuels is the assembly of the nuclei and formation of chemical bonds to convert the relatively simple CO2 molecule into more complex and energetic molecules. Strategically, there are two primary ways that this can be accomplished. The first is to convert CO2 and H2O into CO and H2 (synthesis gas), and then to use well proven Fischer–Tröpsch technologies to convert the synthesis gas to liquid fuels, including gasoline. The advantage here is that it is considerably easier to convert CO2 to CO and H2O to H2 than it is to make even a simple liquid fuel such as methanol by electrocatalytic processes. The second primary option, then, is to attempt to “do it the hard way” by converting CO2 directly to liquid fuels by electrocatalytic processes. Here, the kinetic challenges are great. One possibility is to identify a single catalyst that can direct the complete sequence of steps necessary for converting CO2 to CO, then to H2CO, then to hydrocarbons or alcohols, all with low kinetic barriers. Catalysts that bring required functionalities into the proper position at the proper time will be required. A second possibility is to identify catalyst “panels,” where each panel contains optimal catalysts for each of the steps in the overall transformation of CO2 to a liquid fuel. An advantage of the parallel approach is that the catalysts for each step can be optimized independently using combinatorial or traditional ligand tuning methods, and then the catalyst panel can be assembled from the proven catalyst components.1.3 Scope of the review
This review article will begin with a brief general introduction to the field of electrocatalysis. This will be followed by the presentation of each of the best studied CO2 reduction catalysts reported to date, classified by general ligand type. We will then summarize the recent findings of the bioinorganic chemistry community on carbon monoxide dehydrogenases (CODHs). These enzymes have been optimized by nature for the equilibration of CO2 and CO. They offer lessons for the future design of artificial catalysts for CO2 reduction. We also offer our views on de novo synthetic catalyst design. Finally, we will attempt to draw conclusions from the prior art dealing with the conversion of CO2 to CO and liquid fuels, and suggest avenues of inquiry that investigators may wish to pursue in the future. For a more comprehensive review of homogeneous CO2 reduction catalysis the reader is referred to work by DuBois3 and Savéant.42. Tutorial on electrocatalysis
If the reduction of carbon dioxide to liquid fuels is to be accomplished through photovoltaic or other electrochemical means, the deployment of efficient electrocatalysts will be essential for the development of practical industrial processes. An electrocatalyst both participates in an electron transfer reaction (at an electrode) and facilitates acceleration of a chemical reaction. Both the electron transfer and chemical kinetics must be fast for an efficient electrocatalyst. Additionally, an optimal electrocatalyst must display a good thermodynamic match between the redox potential (E0) for the electron transfer reaction and the chemical reaction that is being catalyzed (e.g. reduction of CO2 to CO). These factors can be optimized by chemical tuning of the electrocatalyst metal centers via appropriate ligand design. Electrocatalysts are typically screened for their redox potentials, current efficiencies, electron transfer rate and chemical kinetics in order to determine the best overall catalysts.In the general sense, electrocatalysts are electron transfer agents that ideally operate near the thermodynamic potential of the reaction to be driven, E0(products/substrates). Direct electrochemical reduction of carbon dioxide on most electrode surfaces requires large overvoltages which consequently lowers the conversion efficiency. The overvoltage can be considered to be the difference between the applied electrode potential, Vapplied, and E0(products/substrates), at a given current density. Both thermodynamic and kinetic considerations are important here. Clearly, in order to minimize overvoltages, catalysts need to be developed that have formal potentials, E0(Catn+/0) well matched to E0(products/substrates), and appreciable rate constants, kcat, for the chemical reduction of substrates to products at this potential. In addition, the heterogeneous rate constant, kh, for reduction of the electrocatalyst at the electrode must be high for Vapplied near E0(Catn+/0). A general approach for an electrocatalytic system is given in Scheme 1.
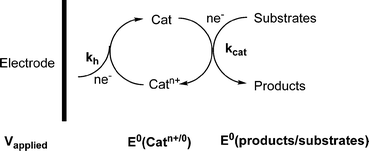 | ||
| Scheme 1 Electrocatalysis with electron source. | ||
Reaction rates for these processes can be estimated from the steady-state limiting current in cyclic voltammetry, or by rotating disk voltammetry studies of the heterogeneous electron transfer kinetics. Identification of electrocatalytic activity can be seen easily in cyclic voltammetry (CV) (Fig. 1). In a CV under a dry inert atmosphere, an electrocatalyst should show a reversible redox couple. Upon addition of CO2, the diffusion limited current should increase significantly, while the potential shifts anodically, and the reversibility in the return oxidation wave is lost due to the chemical reaction between CO2 and the electrocatalyst. Electrocatalysts offer critical solutions to lowering the overpotentials, improving selectivity, and increasing the reaction kinetics of carbon dioxide conversion.
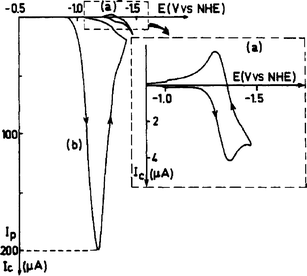 | ||
| Fig. 1 Example cyclic voltammogram (CV) under (a) N2 and (b) CO2. Under a CO2 environment is readily observed: (1) anodic potential shift, (2) large increase in current, (3) non-reversible waveform. Figure reproduced from ref. 11. Copyright 1986 J. Amer. Chem. Soc. USA. | ||
3. A review of CO2 reduction catalysts to date
The efficient electrochemical reduction of carbon dioxide to useful molecules such as carbon monoxide, formic acid, methanol, ethanol, and methane, presents an important challenge and a great opportunity for chemistry today. The field of transition metal catalyzed reduction of CO2 is relatively new, its origins tracing back to the 1970s, but the field has gained in breadth and intensity over the past 30 years.Before we review the important studies in the electrocatalytic reduction of CO2, we must consider several molecular chemistry studies of the reactivity of CO2 toward transition metal complexes that provide important insights concerning the activation and reduction of CO2. Aresta and Nobile first published the crystal structure of CO2 bound to a transition metal complex in 1975 and reported an η2-bidentate binding mode involving the carbon atom and one oxygen atom, with significant bending in the CO2 structure.5 Another important study came in 1981 when Darensbourg and co-workers reported that anionic group 6B metal hydrides would react readily with CO2 to form the metal formates.6 This reaction proceeds according to eqn (7).
| HM(CO)5− + CO2↔ HC(O)OM(CO)5− | (E7) |
In subsequent years, many reports of apparently homogeneous electrocatalysts for the reduction of CO2 appeared in the literature. We have divided the catalysts into three major categories that depend on ligand type: (1) metal catalysts with macrocyclic ligands, (2) metal catalysts with bipyridine ligands, and (3) metal catalysts with phosphine ligands. These seminal studies are summarized in the following subsections. It is important to note that this review is concerned with the electrocatalytic and homogeneous reduction of carbon dioxide and therefore may omit other types of carbon dioxide activation such as photocatalytic reduction, heterogeneous catalysis, and various others.
3.1 Metal complexes with macrocyclic ligands
Important early work in this area of electrocatalysis was done by Meshitsuka and Eisenberg. In 1974 Meshitsuka and co-workers reported the first electrocatalysis of CO2 using cobalt and nickel phthalocyanines.8 This communication did not report turnover numbers or current efficiencies and it was not clear what products were formed, but the research represented a first step in the understanding of the types of transition metal complexes that could display activity for the electrocatalytic reduction of CO2.The first reported transition metal catalysts with high current efficiencies and turnover numbers were demonstrated by Eisenberg and co-workers in 1980.9 In this work, tetraazomacrocyclic complexes of cobalt and nickel were employed as shown in Fig. 2. These complexes were able to reduce carbon dioxide to carbon monoxide or a combination of CO and molecular hydrogen at potentials ranging from −1.3 to −1.6 V vs. SCE. These catalysts were also able to provide high current efficiencies, up to 98% (complex 3), but displayed low turnover frequencies between 2 and 9 per hour at 23 °C.
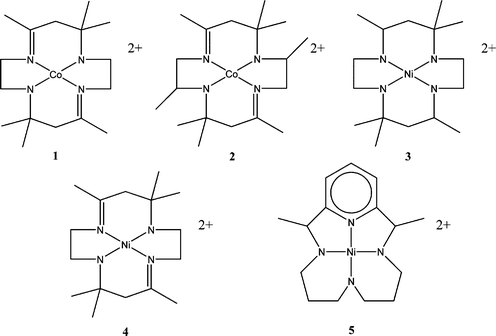 | ||
| Fig. 2 Eisenberg catalysts for the reduction of CO2 to CO.9 All complexes (1–5) were effective for the reduction with varying degrees of success. System suffered from a requirement of high overpotentials for the reduction as well as the coincidental and competing production of H2. | ||
Sauvage and co-workers have extensively studied the reaction of CO2 with NiII(cyclam) complexes.10–12 The Sauvage complexes are extremely stable, highly selective, and can show faradaic efficiencies up to 96% in the production of CO at −0.86 V vs. SCE, even under aqueous conditions. The ligand geometry allows for a highly accessible metal center. It was noted that unsaturated or open chain complexes of similar moieties showed poor catalytic activity. The nickel macrocycle complexes were shown to be very sensitive to the pH and required an Hg electrode surface to turnover. The anion in the supporting electrolyte was shown to affect the selectivity, with KNO3 and KClO4 showing the fastest observed rates. A subsequent study on a binuclear transition-metal centered nickel complex using bicyclam showed similar reactivity towards CO2 but no coupling products were observed. As shown in later studies by Balazs the high electrocatalytic activity originates with Ni(cyclam)+ strongly absorbed on a mercury electrode surface.13 It was also found that CO limits the long-term stability of the catalyst in an unstirred solution by the deposition of an insoluble precipitate believed to be Ni(cyclam)(CO).
Iron(0) porphyrins were reported by the Savéant group in 1991 to reduce CO2 to CO at −1.8 V vs. SCE in DMF.14 However the porphyrins were unstable and degraded after a few catalytic cycles. With the addition of a hard electrophile, such as Mg2+, the stability and reactivity of the catalyst improved noticeably. It is believed through mechanistic studies that the Mg2+ ion assists in the breaking of the CO2 bound to iron creating Fe(II)CO and MgCO3. This is an example where the electron rich iron center initiates the reduction while the Lewis acidic Mg2+ helps complete the process, by sequestering an oxygen atom in MgCO3.
Later Savéant found that iron(0) porphyrins (Fig. 3) were able to catalyze the reduction of CO2 to CO in the presence of weak Brønsted acids.15 For the system where weak Brønsted acids such as 1-propanol, 2-pyrrolidine, and CF3CH2OH were added, Savéant found that catalysis was significantly improved in terms of both the efficiency and lifetime, without significant formation of H2. They were also able to reach turnover numbers as high as 350 h−1 at a catalyst decay rate of 1% per catalytic cycle. This system does, however, require reduction potentials that are still too negative for practical use (approximately −1.5 V vs. SCE) and the necessity of a mercury electrode, which we have noted above can display significant activity in the reduction of CO2sans catalyst.
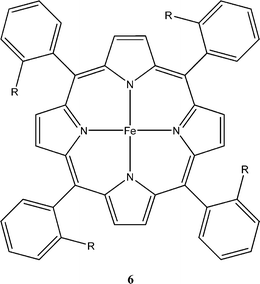 | ||
| Fig. 3 Savéant iron porphyrin catalyst structure shown to reduce CO2 to CO in the presence of weak Brønsted acids at a potential of −1.5 V vs. SCE.15 Porphyrins of this nature reduce CO2 to CO with high turnover frequency and low catalyst degradation, but require potentials too high for practical applications. | ||
In 2002 Fujita and co-workers published their findings on cobalt and iron corroles and their ability to catalyze the reduction of CO2 to CO.16 These complexes of the structure shown in Fig. 4 displayed a catalytic current with onset at approximately −1.7 V vs. SCE in the presence of CO2. The majority of analytical studies of the products for this reaction were done during photochemical reduction, but it was found that the major product of reduction was CO. Photochemical reduction of the complexes was achieved in deoxygenated solvents containing “sacrificial” triethylamine (Et3N) reducing agent and p-terphenyl (TP) as a sensitizer. The catalysts would produce varying amounts of CO and H2 (depending on catalyst) for up to 10 hours. Problems with this catalyst system include high overpotentials for reduction of CO2 and catalyst breakdown after long periods of irradiation.

3.2 Metal complexes with bipyridine ligands
In 1984 the Lehn group reported the electrocatalytic reduction of CO2 by the use of a Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine) complex.17 Using this rhenium bipyridine complex they were able to show the selective reduction of CO2 to CO at a potential of −1.49 V vs. SCE using a 9 : 1 DMF–H2O solution. It was also noted that as the percentage of water was increased the selectivity for CO was diminished, and when the reduction was run under an atmosphere of argon, only molecular hydrogen was produced. While this system had high current efficiencies (98%), and excellent selectivity for carbon monoxide over hydrogen production, the limiting factor was the low TOF of 21.4 h−1.It was later reported by Tanaka and co-workers that bipyridine complexes of ruthenium could catalyze the reduction of CO2.18 Ru(bipy)(CO)22+ and Ru(bipy)(CO)Cl+ were found to electrocatalytically reduce CO2 to CO, H2, and HCOO−. Both complexes were shown to reduce CO2 at −1.40 V vs. SCE. Following a two electron reduction of the complex, CO is lost to form a five coordinate neutral complex. In the presence of CO2 the complex forms an η1-CO2 adduct of Ru(0). This species can also be formed by addition of two equivalents of OH− to Ru(bipy)(CO)22+. Addition of a proton forms the LRu(CO)(COOH) species which under acidic conditions (pH 6.0) gains another proton to lose water and regenerate the catalyst as shown in Scheme 2. Under basic conditions (pH 9.5) the catalyst may undergo a two electron reduction with the participation of a proton to create HCOO− and regenerate the five coordinate Ru(0) complex. Even with the limitations of this system, such as low turnover numbers and low selectivity, it helped to elucidate several of the key intermediates in the reduction of CO2.
 | ||
| Scheme 2 Proposed mechanism for the Tanaka catalyst. | ||
In a similar system studied by the Meyer group, it was found that 2,2′-bipyridine complexes of rhodium and iridium act as electrocatalysts for the reduction of CO2.19 They found that cis-[Rh(bpy)2X2]+ (X is Cl or OTf) reduces CO2 at −1.55 V vs. SCE to predominantly form formate. It is interesting to note that no CO was detected in any of the electrolysis experiments; however, H2 was formed presumably by the degradation of the supporting electrolyte. This system was found to have both low turnovers (6.8 to 12.3) and poor current efficiencies (64% for formate, and 12% for H2). Meyer and co-workers also found that [M(bpy)2(CO)H]+ (M = Os, Ru) were electrocatalysts for the reduction of CO2.20 Under anhydrous conditions the major product was CO, and with the addition of H2O up to 25% formate was observed.
3.3 CO2 reduction by transition metal phosphine complexes
The first reported transition metal electrocatalyst containing phosphine ligands was the Rh(dppe)2Cl (dppe = 1,2-bis(diphenylphosphino)ethane) complex reported in 1984 by the Wagenknecht group.21 In this system the products upon reduction of CO2 were found to be the formate anion with small percentages of cyanoacetate. Current efficiencies for the generation of the formate anion were approximately 42% for short electrolysis runs and down to 22% for longer runs. While mechanistic studies were not undertaken, it was hypothesized that the reduction of the complex resulted in either hydrogen abstraction from acetonitrile to form the transition metal hydride followed by insertion of CO2 to form the formate complex, or through the formation of a CO2 adduct followed by abstraction of the proton from acetonitrile. While the reduction of CO2 will occur at −2.21 V vs. SCE in neat DMF, the reported reduction of CO2 was nearly 700 mV lower at −1.55 V vs. SCE using the Rh(dppe)2Cl complexes.Palladium complexes using polydentate phosphine ligands represent some of the most extensively studied CO2 reduction catalysts to date. First reported in 1991 by the Dubois group22 these systems have shown significant development over the past 15 years.23,24 The typical catalyst system is based on a tridentate phosphine ligand that was initially coordinated to Co, Fe, or Ni. While the iron system showed catalysis of CO2 reduction, the overpotentials were high and the rates were slow. However, the Ni system displayed two one-electron reductions in the area of interest for CO2 reduction. This observation ultimately led this group to study the palladium triphos complexes as catalysts. The first reported complex, [Pd(triphos)(PR3)](BF4)2 (Fig. 5), would catalyze the reduction of CO2 to CO in acidic acetonitrile solutions. The active species was later found to be the phosphine-dissociated solvent complex [Pd(triphos)(solvent)](BF4)2.
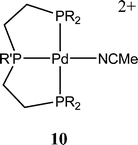 | ||
| Fig. 5 DuBois tridentate phosphine catalyst. | ||
In later mechanistic studies it was determined that as the Pd(II) complex gained an electron to form a Pd(I) intermediate, a reaction with CO2 occurred to form a five-coordinate CO2 adduct. Upon transfer of the second electron, the Pd(0) intermediate dissociated the solvent. Protonation of one of the oxygen atoms of coordinated CO2 affords a metallocarboxylic acid intermediate, Pd–COOH. It is believed that the metallocarboxylic acid is protonated again to form a “dihydroxy carbene”, and that CO is then formed by dehydration of the dihydroxy carbene. CO dissociation and solvent association regenerates the initial complex. This proposed catalytic cycle is shown in Scheme 3. In solutions of high acid concentration the rate determining step was found to be the reaction of the Pd(I) intermediate with CO2. However, in solutions of low acid concentrations the cleavage of the C–O bond to form carbon monoxide and water limits the rate of the catalytic cycle.
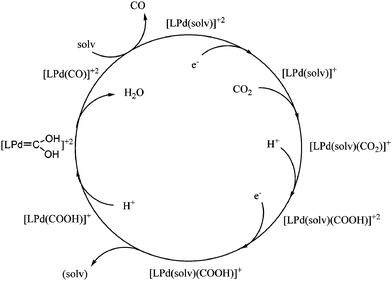
The triphosphine ligand system allows for variation of both electronic and steric effects. Changing the substituent on the central phosphorus to a mesityl group effectively blocked one of the open coordination sites cutting the rate of reaction in half, excluding the possibility of a six-coordinate intermediate that has been proposed for some Ni(I) macrocyclic complexes.13,25 The central donating atom of the tridentate ligand was varied from P to C, N, S, and As as shown in Fig. 6. None were as effective as the original triphosphine ligand, however mechanistic insight into the production of molecular hydrogen was gained. With the [Pd(PCP)(CH3CN)](BF4)2 (11) complex, CO2 was found to be a cofactor in the production of hydrogen suggesting that H2 production goes through the same intermediate that produces CO. From these studies DuBois hypothesized that the preference for forming hydrogen or carbon monoxide depends on the basicity/redox potential of the catalyst. The more negative redox potential favors the protonation of the Pd to form a hydride, the less negative redox potential favors protonation of the coordinated CO2 oxygen to form CO and H2O.
 | ||
| Fig. 6 Varied tridentate phosphine ligands. | ||
Recent studies have focused on complexes incorporating two or more independent Pd triphosphine units.23 Early studies with dendrimers of the Pd catalyst showed decreased activity and selectivity, however with the methylene bridged monomers of the catalysts rates were found to increase by three orders of magnitude, suggesting a cooperative binding of CO2. Along with this increase in catalytic activity came an increase in the formation of Pd(I)–Pd(I) bonds, thus decreasing catalyst lifetimes.
These classes of Pd phosphine complexes have shown catalytic rates in the range of 10 to 300 M−1 s−1 and with >90% current efficiencies for CO production. Overpotentials were in the range of 100–300 mV, yet turnover numbers were low (ca. 10–100) and the decomposition to Pd(I) dimers and hydrides eventually causes cessation of catalytic activity.
Catalytic CO2 reduction has been reported by our group as early as 1987 with a binuclear Ni(0) “cradle” complex, [Ni2(CNMe)3(dppm)2][PF6]2 operating around −0.87 V vs. SCE.26 However over extended time periods the complete carbonylation of the complex occurs to give Ni2(CO)3(dppm)2. More recently, we have returned to a binuclear [Ni2(μ-dppa)2(μ-CNR)(CNR)2] system.27 This system also suffers from the CO produced being trapped by the catalyst.
A key finding was made in 1992 when a new trinuclear nickel cluster [Ni3(μ3-I)(μ3-CNMe)(μ2-dppm)3]+ (dppm = bis(diphenylphosphino)methane) (15) was found to be an electrocatalyst for the reduction of CO2. We have extended this class of nickel cluster electrocatalysts significantly over the past several years to include other isocyanide capped clusters (16–20),28,29 the CO capped cluster 21 (Table 1),28 and chalcogenide capped clusters.30 Results of studies of the electrochemical kinetics of the reduction of CO2 by clusters 15–21 have been reported.29 These results are summarized here briefly. Under an atmosphere of CO2 in dry acetonitrile, the reduction of 15 affords CO and CO32− as the only products observed by GC, HPLC, and IR spectroscopy. The use of 13CO2 results in 13CO and 13CO32−, and no oxalate was observed. The relative rates of reaction of the alkyl or aryl substituted isocyanide or carbonyl capped clusters with CO2 follow the order: CNCH3 (15) ∼ CN(i-C3H7) (16) > CNC6H11 (17) > CNCH2C6H5 (18) > CO (21) > CN(t-C4H9) (19) ∼ CN(2,6-Me2C6H3) (20) (Table 2). It is important to note that although the values of E1/2(+/0) fall into a relatively narrow range of −1.18 (15) to −1.08 V (20) vs. SCE, small differences in E1/2(+/0) dramatically affect the rates of the reaction with CO2. The data indicate that the reaction rates of the isocyanide capped nickel clusters 16–20 with CO2 are primarily influenced by the reduction potentials of the clusters and that the size and geometry of the substituents of the capping ligand play a secondary role. The interaction of CO2 with the reduced forms of 16–20 can occur either at the isocyanide ligand or at the nickel core. In these clusters, we have favored a metal-based mechanism as molecular orbital studies have shown that the LUMO which becomes singly occupied in the reduction of this class of clusters is almost entirely metal-centered.28 The electrochemical studies also suggest that the energy of this orbital correlates with reactivity toward CO2. The secondary steric effect observed in the electrochemical kinetics study further suggests that CO2 activation is occurring on the isocyanide capped face of the clusters.
| L | E1/2(+/0)a | FT-IRb | 31P NMRc | UV-Visd | |
|---|---|---|---|---|---|
| (V vs. SCE) | ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) N) | δ (ppm) | λmax (ε) | ||
| a Cyclic voltammograms recorded in CH3CN.b Recorded as KBr pellets.c Recorded at 121.6 MHz in CD3CN.d Recorded in CH3CN, λmax in nm, ε (M−1 cm−1× 103) given in parentheses.e Broad.f ν(CO). | |||||
| 15 | CNCH3 | −1.18 | 1927, 1871 | 0.4 s | 527.0 (3.8) |
| 16 | CN(i-C3H7) | −1.18 | 1876, 1815 | −0.2 s | 534.4 (4.5) |
| 17 | CNC6H11 | −1.17 | 1885, 1832 | 0.3 s | 536.5 (5.0) |
| 18 | CNCH2C6H5 | −1.11 | 1887, 1801 | 0.4 s | 529.3 (4.3) |
| 19 | CN(t-C4H9) | −1.12 | 1780e | −1.4 s | 539.2 (3.8) |
| 20 | CN(2,6-Me2C6H3) | −1.08 | 1849, 1822 | −1.9 s | 542.2 (3.4) |
| 21 | CO | −1.12 | 1726f | 3.1 s | 520.0 (6.3) |
| L | E1/2(+/0)a | kCO2b | |
|---|---|---|---|
| (V vs. SCE) | (M−1 s−1) | ||
| a Cyclic voltammograms recorded in CH3CN.b Rates determined by rotating disk voltammetry. | |||
| 15 | CNCH3 | −1.18 | 1.6 ± 0.3 |
| 16 | CN(i-C3H7) | −1.18 | 1.4 ± 0.3 |
| 17 | CNC6H11 | −1.17 | 0.5 ± 0.1 |
| 18 | CNCH2C6H5 | −1.11 | 0.2 ± 0.05 |
| 19 | CN(t-C4H9) | −1.12 | 0.0 ± 0.05 |
| 20 | CN(2,6-Me2C6H3) | −1.08 | 0.0 ± 0.05 |
| 21 | CO | −1.12 | 0.1 ± 0.1 |
The binuclear copper complex, [Cu2(μ-PPh2bipy)2(MeCN)2][PF6]2, 22, (PPh2bipy = 6-diphenylphosphino-2,2′-bipyridyl), and its pyridine analog, [Cu2(μ-PPh2bipy)2(py)2][PF6]2, 23 (Fig. 7), were also found to be efficient electrocatalysts for the reduction of CO2.31 Two sequential single electron transfers to 22 are observed at E1/2(2+/+) = −1.35 V and E1/2(+/0) = −1.53 V vs. SCE in MeCN. Both are required to effect CO2 reduction. Again, the CO2 derived products correspond mainly to the reductive disproportionation to CO and CO32−. The only gaseous product was found to be CO. A turn-over frequency of >2 h−1 was maintained over the course of a 24 hour experiment. The catalyst was still active at the end of this experiment, and 22 was recovered quantitatively in its original form. The homogeneous electron transfer kinetics for the reduction of CO2 by complex 22 were studied by chronoamperometry. The limiting rate constant, kCO2, for the reaction of the doubly reduced state of 22 with CO2, was determined to be 0.7 M−1 s−1 in acetonitrile. The rate constant, kCO2, for 22 in methylene chloride solvent is comparable, 0.6 M−1 s−1. However, the rate constant, kCO2, for the pyridine adduct, 23, in methylene chloride solvent is significantly less, 0.1 M−1 s−1. These data suggest that substitution of the labile acetonitrile or pyridine ligands of 22 and 23 respectively is required for CO2 reduction. Significantly, 22 is a 2e− electrocatalyst for the reduction of carbon dioxide. The 2e− redox cycle of 22 appears to lead to at least an order of magnitude increase in the steady state catalytic currents for CO2 reduction compared to our nickel cluster electrocatalysts described above. The difference in overall rates is significant since the limiting [CO2] dependent derived rate constants are comparable. This suggests that the nickel cluster catalysts are slow because they operate via a single electron redox cycle while the overall reduction of CO2 is a 2e− process. On the other hand, the potentials required by the copper catalysts to reduce CO2 are between 350 and 450 mV, more negative than those of the trinuclear nickel catalysts.
 | ||
| Fig. 7 Kubiak dinuclear copper complexes that reduce CO2 to CO and CO32− at −1.53 V vs. SCE. Catalyst shows turnover numbers of greater than 2 h−1 over 24 hour periods and is still intact after catalytic period. Catalysts are effective, but suffer from low turnover and high overpotentials. | ||
4. Lessons from nature
Of all of the synthetic systems reported for the electrochemical reduction of carbon dioxide5–31 none are as efficient and selective as the systems found in nature. The classes of enzymes that catalyze the oxidation of carbon monoxide are designated as carbon monoxide dehydrogenases (CODHs). They are the only catalysts kinetically and thermodynamically optimized to equilibrate CO2 and CO at room temperature. CODHs reversibly catalyze the reaction of carbon monoxide with water to form carbon dioxide, protons and electrons (E8).| CO + H2O ↔ CO2 + 2H+ + 2e− | (E8) |
4.1 Ni-Fe-S
The crystal structure of CO dehydrogenase from the anaerobic bacteria Carboxydothermus hydrogenoformans reveals an active site containing a complex NiFe4S4 center.32 The structure was recently solved in three different reduced states, one held at a potential of −320 mV, another at −600 mV, and a third at −600 mV in combination with CO2. In both of its reduced states the nickel is coordinated by three sulfur ligands forming a distorted T-shaped geometry.Upon addition of CO2 to the reduced state, CO2 binds to both the Ni and Fe. This binding causes minimal geometry changes and occupies the fourth position around Ni completing the square planar geometry. In the coordination of CO2, nickel acts as the Lewis base, while the iron acts as the Lewis acid, and the partial negative charge on the oxygen is stabilized through hydrogen bonding provided by the protein surroundings. The positions of the Ni and Fe are held in place by the Fe3S4 framework and are essentially unchanged by the presence or absence of CO2. The cluster also serves to act as an electronic buffer stabilizing the electronic charges on Fe and Ni during the catalytic cycle (Scheme 4). It is this low reorganization energy that allows for a catalyst with a turnover rate of 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1.32
000 s−1.32
 | ||
| Scheme 4 Proposed catalytic cycle for anaerobic CO dehydrogenases. | ||
4.2 Mo-S-Cu
The first crystal structure of the Mo-S-Cu active site isolated from the aerobic bacteria Oligotropha carboxidovorans was reported in 1999 by Dobbek and co-workers33. The structure was reported to contain an active site consisting of molybdenum with three oxo ligands, molybdopterin-cytosine and S-selanylcysteine. Upon further review the active site was reported to be a Mo(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)SCu active site, while the presence of selenium could not be confirmed (Fig. 8, Scheme 5).34
O)SCu active site, while the presence of selenium could not be confirmed (Fig. 8, Scheme 5).34 | ||
| Fig. 8 Active site of aerobic CO dehydrogenase. | ||
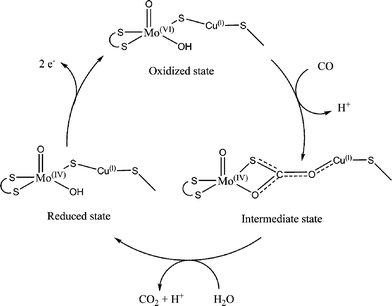 | ||
| Scheme 5 Proposed catalytic pathway for aerobic CO dehydrogenase. | ||
The active site consists of distorted square pyramidal molybdenum with an apical oxo ligand, a hydroxyl group, bridging sulfur to the copper and bound to the protein through molybdopterin cytosine dinucleotide cofactor. The copper is attached to the protein through the Sγ atom of Cys-388. Within the second coordination sphere of the active site there are several groups within hydrogen bonding distance, allowing for stabilization of charges. In the CO reduced, air oxidized, cyanide inactivated, and n-butyl isocyanide bound states the hydrogen bonding distances remain essentially the same. The active site lies 17 Å deep within the protein with a hydrophobic channel that averages 7 Å in diameter.
In the refined structures of the reduced state, the active site remains structurally similar to the air oxidized state. Several of the bond lengths increase (Mo–Cu 3.74 Å to 3.93 Å and Mo–OH 1.87 Å to 2.03 Å) yet the overall geometry remains the same. The enzyme has a catalytic rate of 107 s−1. The active site of O. Carboxidovorans can be inhibited by the use of isocyanides. When inhibited by n-butyl isocyanide, an “intermediate” in the reduction of CO2 can be observed.
From the crystal structures reported to date the requirements for efficient reduction of carbon dioxide are becoming evident. Each of these structures exhibit late first row transition metals in low oxidation states, Cu(I) and Ni(0), and a redox reservoir (Mo(IV) and Fe3S4) to supply electrons for the reduction of carbon dioxide. A key feature of both enzymes is the minimal change in energy between the reduced and oxidized states.
Although most of the work mimicking active sites of proteins has focused on the hydrogenases,35 some research has been done on “bio-inspired” reduction of CO2. Tezuka and co-workers first reported the reduction of CO2 by using a Fe4S4(SR)42− cluster in DMF.36 With this cluster they showed that they were able to reduce CO2 to formate at a potential of −1.7 V vs. SCE. They also report considerable amounts of C3 hydrocarbons in the gas analyses after controlled potential electrolysis, however this may be due to the reduction of the electrolyte.
More recently, Tatsumi and co-workers reported the synthesis of sulfide bridged molybdenum-copper complexes related to the active site of O. carboxidovorans.37 Several compounds were synthesized mimicking the active site, however none exhibited reactivity towards either carbon monoxide or tert-butyl isocyanide. This emphasizes the difficulty in creating a working mimic of biological systems.
5. De novo synthetic catalysts
In the previous section the operating principles of the biological and biologically inspired catalysts were reviewed as they provide important leads for the future of CO2 reduction at low potentials. At this stage of development, however, it is not clear whether the best catalysts will be developed based on lessons learned from nature, or whether de novo synthetic catalysts will be superior for the production of synthetic fuels from carbon dioxide. Synthetic catalysts for the reduction of CO2 reported to date do not possess the efficiencies or stabilities needed for useful large-scale technology. The major obstacle preventing efficient conversion of carbon dioxide into energy-bearing products is the high potential at which carbon dioxide is reduced coupled with the lack of a catalyst that can use an abundant renewable energy source to perform the reaction (e.g., electricity from solar, wind, or geothermal sources). The difficulties in synthesizing a legitimate electrocatalyst for the reduction of CO2 arise from a variety of sources, including, but not limited to, gaps in understanding about what types of complexes will make the best catalysts, the fact that carbon dioxide is a relatively inert molecule, and the problem of performing multi-electron reductions to produce a usable end product. Currently, a literature search of the best catalysts reported to date yields some that have good current efficiencies, some that are robust, and some that give reasonable turnover numbers, but none that accomplish all of these goals. There are also many catalysts with high efficiencies and good activity towards CO2 reduction, but they require the use of sacrificial reducing agents. Another of the notable absences from the current literature is a catalyst that can fix and transform CO2 at low overpotentials, specifically at less than 0.1 V.In the case of CO2, slow kinetics and high overpotentials for electrochemical reduction result from a large nuclear reorganization energy. The winning strategy for efficient reduction of CO2 must involve simultaneous multi-electron transfers and catalytic sites that direct nuclear configurations of reactants favorably for product formation. The development of catalysts to enable two-electron transfer from the same molecule will allow for kinetically more efficient reduction of CO2.
Past examples of carbon dioxide reduction catalysts have cleared some paths for understanding what remains to be done in order to solve the problems summarized above. In most of these past studies the major products have been carbon monoxide and formic acid, and on relatively small scales. In only a few studies has methane or methanol been the primary product of the reductions.
In order to do a multi-electron reduction of CO2 to a usable liquid fuel we will have to develop new methods for activating the carbon dioxide molecule and it will probably be necessary to develop a catalyst that can perform several different kinds of reduction or a series of catalysts that can each work together to perform the required steps.
As an example, consider the first logical step in a CO2 reduction scheme: the hydrogenation of CO2 to formic acid, HCOOH. CO2 is an “amphoteric” molecule (possessing both acidic and basic properties). The carbon atom is susceptible to attack by nucleophiles and the oxygen atoms are susceptible to attack by electrophiles. In the hydrogenation of CO2, therefore, one can think of activation of CO2 being initiated by attack of a nucleophilic metal hydride (H−) at the CO2 carbon atom. The transfer of charge from the hydride to the carbon atom in turn causes negative charge to develop on the oxygen atoms. This charge can be stabilized by a Brønsted acid (H+). In the limit of these interactions, one can consider the hydrogenation of CO2 to result from a heterolytic process that adds H− to the carbon and H+ to the oxygen as shown in Fig. 9. The next logical step in the reduction of CO2 is the deoxygenation of formic acid (HCOOH) to formaldehyde (H2CO). In photosynthesis, for example, CO2 is reduced to H2CO equivalents that are combined to form saccharides (H2CO)6. The reduction of formic acid to a formaldehyde equivalent can again be thought to proceed by H− addition to the carbon atom and H+ addition to the OH group. However, it is important to note that the details of this next step in the reduction (strength of hydride and proton equivalents, geometry of addition, etc.) can be expected to be quite different from those required in the production of formic acid in the first place. Similarly, the third step, namely reduction of formaldehyde to methanol (CH3OH), may proceed by a completely different mechanism such as the direct addition of H2 across the CO double bond of formaldehyde. This illustrates the need for detailed mechanistic and theoretical knowledge in the development of catalysts for the conversion of CO2 to liquid fuels.
 | ||
| Fig. 9 One way to envision proton-couple electron transfer to CO2 is from a metal hydride and a neighboring acid. | ||
6. Conclusions and future directions
In this discussion, we have attempted to review the prior art in the field of electrocatalytic and homogeneous approaches to the reduction of CO2. We also covered its conversion to molecules that are precursors to or are directly usable as fuels. At this stage, it is useful to draw conclusions from past work, and try to identify concepts that will provide a framework for research leading to the next major advances in the field.One important lesson from previous work is that the fundamental reorganization energies of both the CO2 molecule and the catalysts that reduce CO2 are extremely important considerations. An important distinction between the biological CODHs and the synthetic transition metal complex catalysts for CO2 reduction is that the CODHs can equilibrate CO2 and CO. In other words; they can catalyze the reduction of CO2 in both directions. This implies that the CODHs function at the thermodynamic potential for CO2 reduction. It further implies that the CODHs operate at low barriers that result from active sites that direct the linear CO2 molecular substrate toward a necessarily bent CO2 configuration in the reduction intermediates.
A second feature which prior studies also suggest to be important in guiding future work is a multi-electron transfer capacity of the electrocatalysts. For example, of all of the known synthetic electrocatalysts for CO2 reduction, only the [Ni3(μ3-I)(μ3-CNR)(μ2-dppm)3]+ can equilibrate CO2 with CO and CO32−. The rates, however, are quite slow and this has been attributed to the fact that the [Ni3(μ3-I)(μ3-CNR)(μ2-dppm)3]+ catalysts function only by a single-electron redox cycle, while the reduction of CO2 to CO and CO32− is a two-electron process. The development of CO2 reduction catalysts that have highly reversible electrochemistry, indicative of low catalyst reorganization energies, but that also possess multiple electron redox capacities appears to be a very promising area of research.
A third point which has been largely overlooked in previous work, but should figure importantly in future work is proton coupled electron transfer. Proton coupled electron transfer (PCET) is frequently discussed in mechanisms for water splitting. PCET may well be even more important in CO2 reduction catalysis. The redox potentials summarized in eqn (1)–(5) show that the coupling of C–H bond forming reactions with CO2 reduction can lead to very reasonable overall thermodynamics. The key challenge here then is not just one proton coupled electron transfer, but multiple proton coupled electron transfers to produce molecules such as methanol or methane. One observation that we make from this is that it is difficult to imagine one catalyst that can do it all. As we noted in the previous section, in the hydrogenation of CO2, it appears that the most effective initial hydrogenation of CO2 to formic acid would occur by the heterolytic hydride (H−)/proton (H+) addition of H2. The second step in the reduction of CO2 is the deoxygenation of formic acid (HCOOH) to formaldehyde (H2CO). The reduction of formic acid to a formaldehyde equivalent may again be thought to proceed by H− addition to the carbon atom and H+ addition to the OH group, but by a mechanism that could be quite different from that which led to the production of formic acid in the first place.
Finally, the fact that the reduction of formaldehyde to methanol (CH3OH) may proceed by a completely different mechanism still illustrates that either a multi-functional single catalyst or panel of catalysts would be needed for efficient conversion of CO2 to methanol. Presently, we favor the second approach as shown in Scheme 6. This approach includes a panel of three catalysts that each contributes to the overall reduction of CO2 to methanol in optimized single steps. There are various platforms for such catalysts to be developed. These include immobilization on beads, anchoring to separate or single supports or surfaces, etc.
 | ||
| Scheme 6 Proposed path for the tandem catalytic reduction of CO2 to methanol. A series of three catalysts that each contributes to the overall reduction of CO2 to methanol in optimized single steps. | ||
Acknowledgements
We gratefully acknowledge support from DOE and DARPA. CPK wishes to acknowledge the generous hospitality and support or Prof. Dr Karsten Meyer and the Institute of Inorganic Chemistry at the University of Erlangen – Nürnberg extended during a sabbatical leave, May 2008, when most of this article was written. AJS would like to acknowledge a Jacobs Fellowship for support.References
- M. M. Halmann and M. Steinberg, Greenhouse Gas Carbon Dioxide Mitigation Science and Technology, Lewis Publishers, 1999 Search PubMed
.
- N. Sutin, C. Creutz and E. Fujita, Comments Inorg. Chem., 1997, 19, 67 CrossRef CAS
- D. L. DuBois, Encyclopedia of Electrochemistry, 2006, 7a, 202 Search PubMed
- J. M. Saveant, Chem. Rev., 2008, 108, 2348 CrossRef CAS
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636 RSC
- D. J. Darensbourg, A. Rokicki and M. Y. Darensbourg, J. Am. Chem. Soc., 1981, 103, 3223 CrossRef CAS
- W. Paik, T. N. Andersen and H. Eyring, Electrochim. Acta, 1969, 14, 1217 CrossRef CAS
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158 RSC
- B. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361 CrossRef CAS
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315 RSC
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461 CrossRef CAS
- J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986 CrossRef CAS
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1993, 361, 149 CrossRef CAS
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455 CrossRef CAS
- I. Bhugun, D. Lexa and J. M. Saveant, J. Am. Chem. Soc., 1996, 118, 1769 CrossRef CAS
- J. Grodkowski, P. Neta, E. Fujita, A. Mahammed, L. Simkhovich and Z. Gross, J. Phys. Chem. A, 2002, 106, 4772 CrossRef CAS
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328 RSC
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181 CrossRef CAS
- C. M. Bolinger, N. Story, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582 CrossRef CAS
- M. R. M. Bruce, E. Megehee, B. P. Sullivan, H. Thorp, T. R. O’Toole, A. Downard and T. J. Meyer, Organometallics, 1988, 7, 238 CrossRef CAS
- S. Slater and J. H. Wagenknecht, J. Am. Chem. Soc., 1984, 106, 5367 CrossRef CAS
- D. L. DuBois, A. Miedaner and R. C. Haltiwanger, J. Am. Chem. Soc., 1991, 113, 8753 CrossRef CAS
- J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Organometallics, 2006, 25, 3345 CrossRef CAS
- D. L. Dubois, Comments Inorg. Chem., 1997, 19, 307 CAS
- S. Sakaki, J. Am. Chem. Soc., 1992, 114, 2055 CrossRef CAS
- D. L. Delaet, R. Delrosario, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1987, 109, 754 CrossRef CAS
- E. Simon-Manso and C. P. Kubiak, Organometallics, 2005, 24, 96 CrossRef CAS
- D. A. Morgenstern, G. M. Ferrence, J. Washington, J. I. Henderson, L. Rosenhein, J. D. Heise, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1996, 118, 2198 CrossRef CAS
- R. E. Wittrig, J. Washington, G. M. Ferrence and C. P. Kubiak, Comments Inorg. Chem., 1998, 270, 111 CAS
- G. M. Ferrence, P. E. Fanwick and C. P. Kubiak, Chem. Commun., 1996, 1575 RSC
- R. J. Haines, R. E. Wittrig and C. P. Kubiak, Inorg. Chem., 1994, 33, 4723 CrossRef CAS
- J. H. Jeoung and H. Dobbek, Science, 2007, 318, 1461 CrossRef CAS
- H. Dobbek, L. Gremer, O. Meyer and R. Huber, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8884 CrossRef CAS
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15971 CrossRef CAS
- C. J. Pickett and S. P. Best, Coord. Chem. Rev., 2005, 249, 1517–1690 CrossRef CAS
- M. Tezuka, T. Yajima, A. Tsuchiya, Y. Matsumoto, Y. Uchida and M. Hidai, J. Am. Chem. Soc., 1982, 104, 6834 CrossRef CAS
- M. Takuma, Y. Ohki and K. Tatsumi, Inorg. Chem., 2005, 44, 6034 CrossRef CAS
Footnote |
| † Part of the renewable energy theme issue. |
| This journal is © The Royal Society of Chemistry 2009 |