Oxidative addition of water to transition metal complexes†
Oleg V.
Ozerov
*
Department of Chemistry, Brandeis University, 415 South Street, Waltham, MA 02454, USA. E-mail: ozerov@brandeis.edu; Fax: +1 (781) 736-2516; Tel: +1 (781) 736-2508
First published on 23rd October 2008
Abstract
Possible modes of reactivity of water with transition metal complexes are analysed and examples of oxidative addition of water are discussed in this tutorial review.
 Oleg Ozerov | Oleg Ozerov is a native of Russia and a graduate (1998) of the Higher Chemical College of the Russian Academy of Sciences. He received his PhD degree in 2000 from the University of Kentucky with Prof. Folami Ladipo. Following two years at Indiana University as a postdoctoral fellow with Prof. Kenneth Caulton, he joined the department of chemistry at Brandeis University in 2002. In early 2009, he moves to take up a position at Texas A&M University. His interests span a wide range of topics in transition metal and main group chemistry. |
Introduction
Oxidative addition (OA) is one of the fundamental elementary reactions in organometallic chemistry and is essential in homogeneous catalysis.1 OA of water to a transition metal complex leads to the cleavage of the O–H bond and the formation of the metal–hydride and metal–hydroxide entities (Scheme 1). OA is most commonly contemplated in a mononuclear sense, where the resultant hydride and hydroxide ligands are bound to a single metal center. However, binuclear OA is also possible,2 where the involvement of two metals leads to the hydride and hydroxide being bound to two different metal centers. | ||
| Scheme 1 | ||
Oxidative addition of water is of potential relevance to solar energy conversion routes that rely on catalysis by transition metal complexes of the photo- or electric current-driven conversion of water to H2 and O2.3 OA of water and the ensuing hydride/hydroxide complexes may be involved in other catalytic organometallic processes such as hydration of unsaturated organics and the water gas shift reaction.4,5 In addition to the consideration of water as a reagent, understanding of the reactivity towards water is also important because of the appeal of water as a “green” solvent and because of water’s ubiquitous presence as an impurity in the majority of chemical reactions.
By analogy with C–H activation, the term “O–H activation” is often employed to describe O–H OA. In the case of C–H, the word “activation” carries the connotation of the metal activating an otherwise unreactive moiety. For O–H, the term “activation” ought to be considered a misnomer. Unlike most C–H bonds, O–H bonds in water and other RO–H compounds are easily broken heterolytically (via deprotonation) and can hardly be considered unreactive.
Thermodynamic considerations
The bond dissociation energy (BDE) of the O–H bond in water is 499 kJ mol−1.6 This is a high value; indeed, among H–X bonds, only H–F has a higher BDE. However, the magnitude of this parameter cannot be taken to mean that water OA is universally more challenging thermodynamically than OA of other, weaker H–X bonds. It has been observed that the increase in C–M BDE follows the trend in the increase of C–H BDE, but the slope in the C–M trend is steeper (consistent with a thermodynamic preference for activation of stronger C–H bonds).7 Naturally, for water, the question is in whether the M–O and the M–H bonds formed as a result of OA are strong enough to compensate for the cleavage of the O–H bond. The BDE data for M–H bonds8 and especially for M–O bonds9 are in relatively short supply. Furthermore, these BDE values tend to vary considerably in different complexes, making simple BDE-based predictions difficult.In considering the viability of OA of water, it is useful to examine the plausible elementary modes of addition of a water molecule to a transition metal complex (Scheme 2). A saturated (i.e., 18-electron) metal complex cannot accept water as a two-electron ligand; thus, the potential interaction is limited to protonation or hydrogen bonding in the secondary coordination sphere. For an unsaturated transition metal complex other modes of addition are also available. Direct abstraction of a hydrogen atom from water can be removed from consideration because the strengths of M–H bonds cannot come close to compensating for cleavage of the O–H bond in water. Abstraction of a hydroxyl radical may be energetically possible for early transition metals in low oxidation states, however, other pathways typically dominate (vide infra). Coordination of water through the oxygen is a very common mode. Formation of an η2-O,H sigma complex can probably be ruled out as a viable ground-state option as it is hard to imagine it being thermodynamically preferred to the coordination through O or separated from the latter by a meaningful kinetic barrier. A sufficiently Lewis-acidic unsaturated metal complex may abstract hydroxide from water; this, of course, gives the conjugate base of a O-bound water complex. Lastly, OA requires both the unsaturation at the metal center and the availability of the N + 2 oxidation state (for a mononuclear case).10
 | ||
| Scheme 2 | ||
The favorability of OA must thus be considered not only vs. the separated reactants, but also vs. the other modes of addition. In other words, in order for OA of water to occur, the newly formed M–O and M–H bonds must be strong enough to compensate not only for the cleavage of the O–H bond, but also for any potential thermodynamic gain from another mode of interaction (e.g., water coordination).
The entropic factors must play a role as well. At first glance, as any addition reaction that decreases the number of free particles, oxidative addition of water should bear an entropic penalty.11 However, the contributions arising from changes in solvent ordering (e.g., loss of strong hydrogen bonding with free water) will likely often render such simple analysis moot.
From a practical point of view, identification of a reaction with water as OA, even when that is thermodynamically favorable, can be hampered by the subsequent transformations of the hydride/hydroxide products. Hydrolysis of M–H with excess water, dehydration and formation of bridging oxo ligands, reactions of M–H or M–OH with other ligands represent some potential culprits.
Mononuclear oxidative addition with late metals
The first report detailing OA of water appeared in 1970.12 The overall number of examples of OA of water is relatively small. That in itself may owe more to the lack of sustained interest in specifically exploring this reaction (this looks bound to change with the rise in importance of solar energy conversion problems). Nonetheless, some generalizations can be made. The known examples involve OA of water to Ru0, Os0, RhI, IrI, and Pt0 complexes (vide infra). These are all electron-rich configurations for which OA in general is common. Late metals generally form weaker M–OR bonds than early metals.9,13 This can be attributed to the general soft-acid/hard-base mismatch and to dπ–pπ repulsion between the filled dπ orbitals of the (saturated) metal center and the lone pairs on the oxygen. The weaker M–OH bonds that would be formed upon OA of water are a challenge to the thermodynamic feasibility of the process. On the other hand, (a) a late metal (soft) Lewis acid is a poorer match for water as an oxygenous Lewis base (hard) and (b) the above mentioned side reactions common for early metals (hydrolysis of ancillary ligands and of the product M–H) are not typical of late metal complexes. Consequently, while the thermodynamics of OA of water to a late metal complex versus the reactants is handicapped compared to early metals, the favorability of competing reactions is diminished. Still, compared to various other common OA reactions (H–H, C–H, Si–H, C–Cl, etc.), OA of water to late metals appears to be among the most thermodynamically difficult. In a few cases it is possible to establish that the thermodynamics of the OA of water is comparable to the OA of C(sp3)–H bonds (vide infra).Examples
Bergman reported OA of water to the (Me3P)4Ru0 fragment arising from 1 (Scheme 3).143 was thermally stable and while 2 was not, its decomposition probably involved pathways other than reductive elimination of water. 2 was also obtained through reaction of water with 4; the same reaction was also reported for the Os analog 6.15 It is not clear whether here the reaction of water with 4 or 5 (or even with 1) proceeds via OA to the unobserved (Me3P)4M or, alternatively, by hydrolysis of the M–C bond in the three-membered metallacycle in 4 or 5. | ||
| Scheme 3 | ||
Gillard et al. described the reduction of trans-[(en)2RhCl2]+ (7) in water leading to the formation of trans-[(en)2RhH(OH)]+ (9).12 The authors proposed a sequence of reactions depicted in Scheme 4 and noted that solvolysis of the Rh–OH bond in 9 also takes place. 9 was stable enough to be isolated in an analytically pure form as a tetraphenylborate salt. However, in highly alkaline solutions, reductive elimination of water generating trans-[(en)2Rh]+ (8) was reported. In a related study of RhI/water reactivity, the chemistry of water gas shift reaction (H2O + CO → H2 + CO2) catalysis by HRhI(PPri3)3 was described by Otsuka.16
 | ||
| Scheme 4 | ||
Several examples are known for OA of water to IrI (Scheme 5). Jensen reported OA of water to the pincer-supported (PCP)Ir fragment 12; unlike all other Ir examples, this resulted in a 16-electron hydrido/hydroxide product (13).1713 functioned as a pre-catalyst for dehydrogenation of alkanes, which implies that reductive elimination of water to generate 12 is kinetically accessible. Reactions with both cationic [IrL4]+ (14)18 and neutral L3IrCl (19 and 21)5,19 or [L2IrCl]2 (16 and 22)19–21 complexes have been described. Reactions of water with 16 and 22 involve a bimetallic reactant and a bimetallic product, but they are not examples of binuclear oxidative addition. There is no metal–metal bond in either the reactant or the product, and the stoichiometry of addition is one molecule of water per Ir atom, not per dimer.
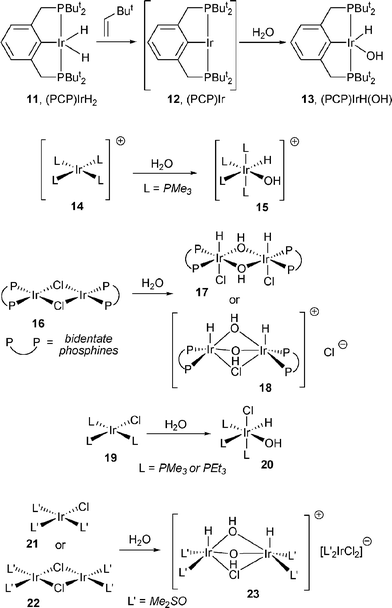 | ||
| Scheme 5 | ||
OA of water to Pt0 was studied by Otsuka and co-workers (Scheme 6).22 When (iPr3P)2Pt or (iPr3P)3Pt (24) was treated with a large excess (>300 : 1) of water in THF, the formation of trans-(iPr3P)2PtH(OH) (25) was observed. The latter was isolated in a low yield and decomposed readily in solution in the absence of excess water. In contrast, the reaction of water with (Et3P)3Pt only yielded [(Et3P)3PtH]OH (26). Ostensibly, the difference in the outcome is due the thermodynamics of the equilibrium between species like 25 and 26. With the smaller Et3P, displacement of hydroxide is favored.
 | ||
| Scheme 6 | ||
Oxidative addition of water to Pd(II) and Pt(II) has not been experimentally recorded, but in two studies OA of water may well have been part of the overall transformation. Dedieu and Canty used computational studies on a simplified model to propose a mechanism for the reduction of water to H2 by 27 (Scheme 7).23 Their proposal involves stepwise, pyrazolyl-assisted addition of a proton and hydroxide to the Pd(II) center to give the Pd(IV) hydride/hydroxide 29 (Scheme 7). The dangling pyrazolyl ligand also participates in the H2 formation (30→31) upon protonation of 29. Pudephatt reported oxidation of the Pt(II) complex 32 (Scheme 7) by water and alcohols to give Pt(IV) hydro- or alkoxides 33 and H2 (similarly to the preceding Pd example).24 While other mechanisms could not be ruled out, the authors suggested that OA of water is a plausible initial step.
 | ||
| Scheme 7 | ||
Mechanistic and thermodynamic considerations
Experimental mechanistic information for the OA of water is limited. Scheme 8 presents the simplified plausible options. Addition of water may require prior dissociation of a ligand (e.g., 34→35). The addition of water may proceed either in the concerted fashion (via a three-center transition state 37) or stepwise. The stepwise addition would involve either initial protonation (intermediate 36) or initial abstraction of hydroxide (intermediate 38).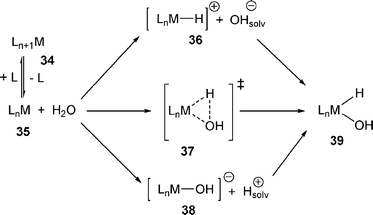 | ||
| Scheme 8 | ||
Evidence was presented to support the notion that 21 and 22 react with watervia prior dissociation of a ligand, via a 14-electron IrI intermediate.19 By analogy with other OA reactions occurring in the (PCP)Ir system, OA of water to give 13 likely also proceeds via a 14-electron (PCP)Ir (12).17 On the other hand, in the study of the closely related reaction of MeOH with 19 by Blum and Milstein it was concluded that the O–H OA occurred to the 4-coordinate 16-electron Ir complex.5 The experimental evidence was in support of concerted addition characterized as a nucleophilic attack on the H of the hydroxy group.
Binuclear oxidative addition with late metals
The examples of binuclear OA of water that have thus far been reported can be classified into two categories (Scheme 9). The Type I binuclear OA of water proceeds with loss of ligands, retention of the metal–metal bond, and formation of bridging hydride and hydroxide ligands in 41. Type II is encouraged by tightly bound, non-dissociable chelating ligands and results in the formation of monometallic, terminal, hydride and hydroxide complexes (43 and 44). | ||
| Scheme 9 | ||
Type I
Lewis and co-workers showed that Ru and Os clusters can react with water. Thermolysis of Ru3(CO)12 did not produce any identified hydrido/hydroxide complexes, instead, only Ru4(CO)12H4 was isolated.25Thermolysis (230 °C) of Os3(CO)12 led primarily to a mixture of numerous Os carbonyl and carbonyl/hydrido clusters, but traces of Os3(CO)10H(OH) were also observed.25 The complex Os3(CO)10(η-cyclohexadiene) proved to be a more convenient reagent (presumably because of easier loss of the diene ligand) and its reaction with water at 80 °C led to a modest (20%) yield of Os3(CO)10H(OH).26 A phosphine-ligated Ru dimer, (Me3P)6Ru2(μ-CH2)3 was shown to react with water to give (Me3P)6Ru2(μ-H)(μ-OH),27 but it is not clear whether this reaction should be classified as water OA.Type II
The Wayland group developed rich small molecule activation chemistry of rhodium porphyrins. Porphyrins with sulfonated appendages confer aqueous solubility on the Rh complexes. Dimeric RhII complexes were shown to react with water with formation of hydride (47) and hydroxide (48) RhIII products (Scheme 10).4,28 The reactions proceed via the intermediacy of monomeric RhII complexes (46); with a bulkier porphyrin ligand only the monomeric 46 was observed. The mechanism of the reaction with water is not known in detail. For a closely related O–H OA of methanol a mechanism was proposed involving disproportionation of the RhII methanol complex into RhIII and RhI, with subsequent proton transfer.29 Wayland et al. determined the bond dissociation free energy (BDFE) of both Rh–H and Rh–OH bonds to be ca. 250 kJ mol−1 and the free energy for OA of water to be on the order of −10 kJ mol−1. Interestingly, the free energy for O–H OA of methanol was ca. 25 kJ mol−1 less favorable than for the isomeric C–H OA of methanol. | ||
| Scheme 10 | ||
Fafard et al. reported a binuclear OA of water to the dimeric complex 49 under thermo- or photolytic conditions.30 The resultant 50 and 51 were formed in a strictly 1 : 1 ratio. No intermediates were observed. The mechanism remains unknown, however, isolobal similarity with Rh porphyrins might be suggestive of mechanistic similarities. As well, in both cases, the metal is supported by a potentially redox-active ligand, which may play a role in electron transfer and/or stabilization of various oxidation states. It should be noted that, in either case, abstraction of H atom (or hydroxyl radical) from free water by a monomeric metal complex is not viable as part of the mechanism. It would correspond to a reaction barrier of at least 250 kJ mol−1 in the Rh case (and likely similarly high for Pd), which is incompatible with the experimental observation of the reaction. On the other hand, it is conceivable that abstraction of H atom from water coordinated to an odd-electron metal center (e.g., Rh(II) or Pd(I)) might be a more energetically viable path.
Binuclear OA of water of type II circumvents some of the inherent challenges (vide supra). Coordination of water to an odd-electron monomer (or its dimer) does not seem a likely thermodynamic sink. The presence of only rigid, multidentate auxiliary ligands excludes undesirable ligand-based reactions.
Early metals
Early transition metals in high oxidation states form very strong bonds to anionic oxygen-based donors.13 It may thus be expected that OA of water to give an early metal hydride/hydroxide complex should be thermodynamically sound. Indeed, laser-ablated group 4 metal atoms were shown to react with water spontaneously in a frozen argon matrix to give HMOH and H2M(OH)2 (M = Ti, Zr, Hf).31 In conventional experimental practice, however, there are virtually no examples of OA of water to early transition metals. Two reasons can be identified for such scarcity.Oxidative addition requires a transition metal precursor in a low oxidation state; for early metals this would most commonly correspond to the d2 configuration. Complexes of metals of groups 4 and 5 with an unambiguous d2 configuration are rather rare (and unknown for group 3). More commonly, the d2 metal center is stabilized by coordination to an unsaturated organic π-acceptor (alkene, alkyne, diene, arene, etc.). In such complexes, the high degree of back-donation to the π-acceptor leads to an oxidation state dichotomy, as illustrated in Scheme 11 for an alkyne complex (d2), alternatively described as metallacyclopropene (d0). The d0 description is typically reflected in the reactivity with electrophiles, including protic electrophiles. For example (Scheme 11), the zirconacyclopropene (alkyne complex of zirconocene) 56 reacts with CO2 to give 57 and with water to give 58.32 In these reactions, the α-C of the metallacycle acts as a nucleophile. All in all, hydrolysis of auxiliary ligands in early metal complexes would often be the dominant reaction pathway with water.
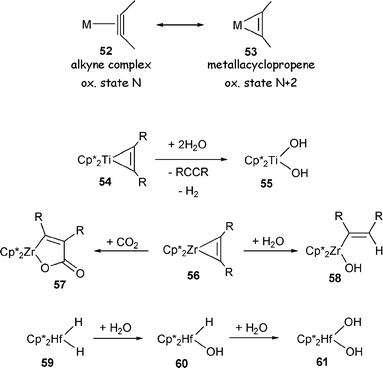 | ||
| Scheme 11 | ||
The other obstacle to observing well-defined OA of water to early transition metals is the likelihood of subsequent transformations of the hydride/hydroxide products. The newly formed metal hydride may react with water or the metal hydroxide moiety further with evolution of H2 and formation of a bis(hydroxide) or a bridging oxo. Scheme 11 provides examples of such reactivity from the work or Rosenthal (Ti)32 and Bercaw (Hf).33 In Rosenthal’s case, it is quite possible that OA of water takes place in a reaction with 54 to give the unobserved hydride/hydroxide that undergoes further reaction with water to give 55; this is in curious contrast to the reactivity of the Zr analog 56 (Scheme 11). In Bercaw’s study, the hydride/hydroxide (60) was observed, but the deuterium labeling experiments led the authors to conclude that “the mechanism of hydrogen elimination [in the reaction of water with 59] is best described as a protonolysis of the hydride instead of base (H2O) induced reductive elimination of H2 followed by (or concomitant with) oxidative addition of an O–H bond of water”.
The “earliest” metal shown to participate in an OA reaction with water is tungsten. A study by Tyler described the formation of Cp*2W(H)(OH) in low yield upon photolysis of Cp*2WH2 (known to generate Cp*2W) in the presence of water.34 Notably, Cp*2W(H)(OH) was not stable under the conditions of synthesis, gradually evolving into Cp*2WO.
Conclusions
Oxidative addition of water is a reaction with multiple potential uses. Conversion of water into metal–hydride and metal–hydroxide functionalities is an attractive step towards incorporation of the elements of water into organic molecules or in photo- and electrocatalytic conversion of water into fuel and O2. Virtually all known examples of OA of water come from late transition metal chemistry. With late metals, OA of water is thermodynamically rather difficult and many of the known examples display reversibility. The lack of water OA reactions with early transition metal complexes is in part attributable to the preponderance of non-OA reaction pathways with water.References
- P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer Academic Publishers, Dordrecht, Boston, London, 2004, pp. 271–298, 387–402 Search PubMed.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley-Interscience, New York, 2005, 4th edn, pp. 160–161 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS.
- X. Fu, S. Li and B. B. Wayland, Inorg. Chem., 2006, 45, 9884 CrossRef CAS and references within.
- O. Blum and D. Milstein, J. Am. Chem. Soc., 2002, 124, 11456 CrossRef CAS and references within.
- A. K. Chandra and T. Uchimaru, J. Phys. Chem. A, 2000, 104, 9244 CrossRef CAS.
- E. Clot, C. Megret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2006, 128, 8350 CrossRef CAS.
- M. Tilset and V. D. Parker, J. Am. Chem. Soc., 1989, 111, 6711 CrossRef CAS.
- J. R. Fulton, A. W. Holland, D. J. Fox and R. G. Bergman, Acc. Chem. Res., 2002, 35, 44 CrossRef CAS.
- Protonation is also a process formally resulting in a +2 oxidation state change at the metal, but it is not OA because only a part of the water molecule (a proton) is added to the metal.
- L. A. Watson and O. Eisenstein, J. Chem. Educ., 2001, 79, 1269.
- R. D. Gillard, B. T. Heaton and D. H. Vaughan, J. Chem. Soc. A, 1970, 3126 RSC.
- M. F. Lappert, D. S. Patil and J. B. Pedley, J. Chem. Soc., Chem. Commun., 1975, 830 RSC.
- M. J. Burn, M. G. Fickes, J. F. Hartwig, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 5875 CrossRef CAS.
- J. Gotzig, R. Werner and H. Werner, J. Organomet. Chem., 1985, 285, 99 CrossRef.
- T. Yoshida, T. Okano, Y. Ueda and S. Otsuka, J. Am. Chem. Soc., 1981, 103, 3411 CrossRef CAS.
- D. Morales-Morales, D. W. Lee, Z. Wang and C. M. Jensen, Organometallics, 2001, 20, 1144 CrossRef CAS.
- D. Milstein, J. C. Calabrese and I. D. Williams, J. Am. Chem. Soc., 1986, 108, 6387 CrossRef CAS.
- R. Dorta, H. Rozenberg, L. J. W. Shimon and D. Milstein, Chem.–Eur. J., 2003, 9, 5237 CrossRef CAS.
- R. Dorta and A. Togni, Organometallics, 1998, 17, 3423 CrossRef CAS.
- K. Tani, A. Iseki and T. Yamagata, Angew. Chem., Int. Ed., 1998, 37, 3381 CrossRef CAS.
- T. Yoshida, T. Matsuda, T. Okano, T. Kitani and S. Otsuka, J. Am. Chem. Soc., 1979, 101, 2027 CrossRef CAS.
- A. Milet, A. Dedieu and A. J. Canty, Organometallics, 1997, 16, 5331 CrossRef CAS.
- P. K. Monaghan and R. J. Puddephatt, Organometallics, 1984, 3, 444 CrossRef CAS.
- C. R. Eady, B. F. G. Johnson and J. Lewis, J. Chem. Soc., Dalton Trans., 1976, 838 RSC.
- E. G. Bryan, B. F. G. Johnson and J. Lewis, J. Chem. Soc., Dalton Trans., 1977, 1328 RSC.
- R. A. Jones, G. Wilkinson, I. J. Colquohoun, W. McFarlane, A. M. R. Galas and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1980, 2480 RSC.
- X. Fu and B. B. Wayland, J. Am. Chem. Soc., 2004, 126, 2623 CrossRef CAS.
- S. Li, W. Cui and B. B. Wayland, Chem. Commun., 2007, 4024 RSC.
- C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318 CrossRef CAS.
- M. Zhou, L. Zhang, J. Dong and Q. Qin, J. Am. Chem. Soc., 2000, 122, 10680 CrossRef CAS.
- P.-M. Pellny, V. V. Burlakov, W. Baumann, A. Spannenberg and U. Rosenthal, Z. Anorg. Allg. Chem., 1999, 625, 910 CrossRef CAS.
- G. L. Hillhouse and J. E. Bercaw, J. Am. Chem. Soc., 1984, 106, 5472 CrossRef CAS.
- M. Yoon and D. R. Tyler, Chem. Commun., 1997, 639 RSC.
Footnote |
| † Part of the renewable energy theme issue. |
| This journal is © The Royal Society of Chemistry 2009 |