Small molecule mimics of hydrogenases: hydrides and redox†
Frédéric Gloaguen*a and Thomas B. Rauchfuss*b
aUMR CNRS 6521, Université de Bretagne Occidentale, CS 93837, 29238 Brest cedex 3, France. E-mail: fgloaguen@gmail.com
bSchool of Chemical Sciences, University of Illinois, Urbana, IL 61801, USA. E-mail: rauchfuz@uiuc.edu
First published on 31st October 2008
Abstract
This tutorial review is aimed at chemical scientists interested in understanding and exploiting the remarkable catalytic behavior of the hydrogenases. The key structural features are analyzed for the active sites of the two most important hydrogenases. Reactivity is emphasized, focusing on mechanism and catalysis. Through this analysis, gaps are identified in the synthesis of functional replicas of these fascinating and potentially useful enzymes.
 Frédéric Gloaguen | Frédéric Gloaguen received his undergraduate degree in Brest in 1990 and his PhD in Grenoble in 1994. He worked at the University of Poitiers and was hired by CNRS in 1996. After a sabbatical leave in 2001 at the University of Illinois, he joined the University of Brest in 2002. He is interested in all aspects of the catalysis of electrochemical reactions involved in energy storage and utilization. |
 Thomas Rauchfuss | Thomas Rauchfuss received his PhD degree at Washington State University in 1975. After a postdoc in Australian National University, he joined the faculty at the University of Illinois where he conducts research in synthetic inorganic and organometallic chemistry. In recent years he has focused on models for the active sites of the hydrogenases. In collaboration with Gloaguen, his group discovered the catalytic hydrogen evolution by diiron dithiolates. |
1. Introduction
Since the 1930’s, biologists have known that some microorganisms produce molecular hydrogen (dihydrogen to organometallic chemists) in the course of their normal metabolism. The underlying biochemistry of the associated enzymes, called hydrogenases (H2ases), has been actively studied since that time. Beginning with the crystallographic characterization of the [NiFe]-H2ase from D. gigas in 1995, the level of activity increased, in part because of the startling structure of the active site, which features an iron carbonyl cyanide. Almost five years later, the phylogenetically unrelated [FeFe]-H2ase was also characterized crystallographically, again revealing yet another kind of iron carbonyl cyanide center.1In an effort to understand the molecular mechanisms by which H2ases operate, much research has been aimed at mimicking the structures of their active sites. In recent years, these models have begun to yield biochemically significant insights, although gaps remain. Perhaps most perplexing are the high rates achieved by these enzymes, especially in view of the fact that they utilize first row metals that typically display diminished affinities for dihydrogen.2 Furthermore and still more challenging, the H2ases effect their reactions via apparent 1e− changes, which require odd-electron intermediates. The one-electron chemistry of metal hydrides and metal–dihydrogen complexes is lightly studied; thus, the biochemical mechanisms present opportunities for learning new organometallic chemistry relevant to dihydrogen.
The literature on the production of hydrogen in solution, homogeneous hydrogenogenesis, is not extensive,3,4 but the coordination chemistry of dihydrogen has been active for decades2 and is obviously relevant to biological processes. Although H2 itself exhibits neither redox nor any acid–base reactivity, its metal complexes exhibit both, i.e. dihydrogen complexes can be highly acidic and, the derived metal hydrides can be oxidized. It therefore makes sense that the active sites of both the [NiFe]- and the [FeFe]-H2ases feature metals. This generalization extends to the recently discovered hydrogen-transfer enzyme Hmd.5 Interestingly, the active sites of all three classes of “hydrogen-processing” enzymes feature thiolato iron carbonyl entities.
This review brings a particular focus on the reactivity of models for the H2ases. Reactivities of interest include protonation, binding of H2 and CO (a common inhibitor of H2ases), and redox.
2. Recent progress in functional modeling of the active site of the [FeFe]-H2ases‡
Overview of the H-cluster
Crystal structures of the [FeFe]-H2ases have been solved for proteins obtained from C. pasteurianum and D. desulfuricans. The active sites are very similar and are viewed by inorganic chemists as derivatives of the reference compound Fe2(pdt)(CO)6 (Fig. 1). The active site features trans dibasal cyanide ligands with one apical position bound to the cysteinate of a 4Fe-4S cluster, and the other apical position being either vacant or bound to water. The two Fe centers are bridged or semi-bridged by a CO ligand. Substrate turnover appears to occur at the iron that is distal from the 4Fe-4S cluster. Carbon monoxide, a potent inhibitor of H2ases, binds stereospecifically at the apical site on the distal Fe. The central atom of the dithiolate cofactor is probably the nitrogen atom of an amine (see below). Some enzymes feature channels that relay reagents (H+, H2) into and out of the active site, as well as a chain of electron-transporting Fe-S clusters. Enzymes isolated from some algae are smaller and simpler in design, which promises to be of biotechnological value as they are more easily overexpressed.6 Spectroscopic study of such algal proteins also provide simplified, more interpretable spectra. The cyanide ligands are hydrogen-bonded to the protein backbone; otherwise the active site is anchored in the protein via the single cysteinyl ligand. Lightly anchored active sites are characteristic of all three H2ases, a fact that encourages the idea that their functionality might be replicated outside of the protein.5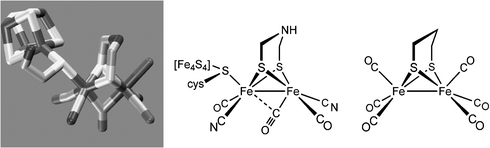 | ||
| Fig. 1 Structure of H-cluster from C. pasteurianum, a line drawing of the same, omitting the water situated near the distal (rightmost) Fe center, and the structure of the prototypical Fe2(pdt)(CO)6. | ||
Cyanides of the diiron dithiolates
The presence of two cyanide ligands is one of the most distinctive features of the [FeFe]-H2ases, and closely related dicyano diiron carbonyls are easily prepared. Treatment of most derivatives of the type Fe2(SR)2(CO)6 with cyanide salts gives the dicyanides [Fe2(SR)2(CN)2(CO)4]2−. Under normal preparative conditions, the monocyanides form only in trace amounts and the tricyanide never forms.7 Salts of these dicyanides have been isolated as several isomers (rotamers) that differ with respect to the basal vs. apical location of the cyanide ligands. DNMR studies indicate that these species are stereochemically nonrigid due to rapid turnstile rotation, as is expected for derivatives of the type M2(μ-X)2L6 (but not those of the type M2(μ-X)3L6). Replacement of one further CO ligand in [Fe2(SR)2(CN)2(CO)4]2− with a [4Fe-4S] cluster would give a near-perfect protein-free model for the H-cluster. The anion [Fe2(SR)2(CN)2(CO)4]2−, however, resists substitution reactions.Redox is a key attribute of the H2ases, and thus early studies attempted, fruitlessly, to oxidize the dicyanides to mixed-valence derivatives. Useful insights have however been obtained from the low-temperature oxidation of [Fe2[(SCH2)2C(Me)CH2SR](CN)2(CO)4]2−, wherein the dithiolato ligand bears a pendant, uncoordinated thioether group. At −40 °C, this species undergoes 1e− oxidation to give a mixed-valence intermediate, which is proposed to be stabilized by coordination of the thioether. Characterization by EPR and IR spectroscopies confirm the similarity of this mixed valence species to the CO-inhibited (HCOox) state of the enzyme.8
Oxidation of the diiron dicyanides typically affords intractable solids that appear to be polymers containing μ-CN linkages. Some details of the redox-induced aggregation of the diiron cyanides were elucidated in the case of [Fe2(edt)(CN)(CO)3(dppv)]−. This species undergoes complete consumption upon treatment with only one equivalent of Fc+ to give a diamagnetic Fe4 derivative [FeI2(S2C2H4)(CO)3(dppv)](μ-CN)[FeII2(S2C2H4)(μ-CO)(CN)(CO)2(dppv)]. The result is consistent with the disproportionation of a mixed-valence diiron intermediate into a diamagnetic mixed valence tetrairon species, i.e., 2FeIFeII→ FeIFeIFeIIFeII (eqn (1)).9
 | (1) |
Hydride derivatives
The [FeFe]-H2ases characteristically convert protons to dihydrogen, thus there is great interest in the protonation of diiron dithiolato carbonyls. Protonation of [Fe2(edt)(CN)2(CO)4]2− gives three isomers of the hydride [Fe2(SR)2(μ-H)(CN)2(CO)4]−.10 The observation of isomeric hydrides is unusual because protonations of many other diiron(I) dithiolates affords single isomers (see below). The presence of isomers indicates either high barriers to isomerization or that the three isomers are of comparable energy, possibly reflecting the low steric profile of CO and CN−. Solutions of [Fe2(SR)2(μ-H)(CN)2(CO)4]− are unstable and decompose in the presence of acids.11Particularly significant advances in biomimetic modeling have resulted from the study of diiron compounds where the cyanides are replaced all or in part by tertiary phosphine ligands, as well as carbenes and isocyanides. Phosphine complexes of the type Fe2(SR)2(CO)6−x(PR3)x, especially for x≥ 2, protonate readily to give characterizable hydrides. Whereas the isomeric hydrides of [Fe2(SR)2(μ-H)(CN)2(CO)4]− are observable by NMR spectroscopy, hydrides containing monodentate phosphine ligands typically are isolated as single isomers.12 Mixed phosphine-cyanide complexes represent a compromise between the uncomplicated properties of phosphines and the biologically relevant, highly basic cyanide. In contrast, the parent hexacarbonyls protonate only with the strongest acids, e.g. triflic acid in CH2Cl2 solution.
A representative and well-studied “μ-hydride” is [Fe2(edt)(μ-H)(CO)4(PMe3)2]+, wherein CO ligands are trans to the hydride and the phosphines occupy trans basal sites. Such μ-hydrides are invoked as intermediates in some pathways for electrocatalytic hydrogenogenesis (see section 4). Protonation strongly affects the diiron center: in contrast to its unprotonated precursor, the hydrides are susceptible to substitution reactions involving replacement of CO ligands with donor ligands, such as cyanide and phosphines. Further indicating the lability of the CO ligands, upon illumination the hydrido complexes also catalyze isotopic exchange between D2 and H2O. H2ases characteristically catalyze such exchange, although photoactivation is not required.13
Since the time of the original crystallographic characterization, the enzymatic reactions have been assumed to proceed via the intermediacy of a hydride located on the apical site of the distal iron center. A model for such a terminal hydrido species has been characterized crystallographically in the form of [HFe2(edt)(CO)2(PMe3)4]BF4. The hydride is located at one of the two apical sites and one CO group semibridges the two Fe centers. Unlike the isomeric μ-hydrido complex [Fe2(edt)(μ-H)(CO)2(PMe3)4]BF4, the terminal hydride reacts with strong acids to release H2 (eqn (2)).14
 | (2) |
In situ analyses show that diiron dithiolato carbonyls initially protonate a single iron center.15,16 In most cases, the resulting terminal hydride complexes rapidly rearrange to give the bridged hydride isomer. Unsymmetrical complexes such as Fe2(pdt)(CO)4(dppe) protonate mainly at the Fe(CO)3 site, not the ostensibly more basic Fe(CO)(dppe) site. The terminal hydrides are more stable when the diiron dithiolato center is both electron-rich and has bulky ligands that hinder turnstile rotation. Such species can even be observed near room temperature and interrogated spectroscopically in detail (Scheme 1). Thus, protonation of Fe2(pdt)(CO)2(dppv)2 with HBF4 gives the terminal hydride [HFe2(pdt)(CO)2(dppv)2]+, which unimolecularly isomerizes to a series of isomeric μ-hydrides. The terminal hydride reduces at potentials ca. 200 mV more positive than the isomeric bridging hydride [Fe2(pdt)(μ-H)(CO)2(dppv)2]+. This lowered reduction potential points to a thermodynamic advantage for terminal hydrides as precursors to H2.17 Reduction of this diferrous hydride generates a mixed-valence hydride that is highly reactive toward protons to release H2. Overall, these studies indicate several important aspects: terminal hydride ligands form readily by protonation, they more easily undergo protonolysis to H2, and they reduce more easily than the isomeric bridging hydrides.
Dithiolate cofactors
Protein crystallographic studies indicate that the two iron centers in the diiron dithiolato subsite are linked by a dithiolate of the type SCH2YCH2S, where, based on its X-ray-scattering power, Y is CH2, NH, or O. None of these dithiolates were previously known to nature. In 2001, Fontecilla-Camps proposed that the dithiolate is an amine, which we referred to as an azadithiolate.18 Such an amine (pKa≈ 10 in water) is well suited to participate in the heterolytic reactivity of dihydrogen. In fact, it is conceivable that the amine is protonated in at least one of the resting states of the enzyme. The ammonium center is poised to convey protons to and away from the apical site on the distal iron center. Furthermore, because the amine is tightly constrained by its attachment to the two bridging thiolato ligands, it lacks the flexibility to coordinate directly to Fe, which would quench the coordinative unsaturation at that metal. This constrained base motif (CBM) is generalizable and has been replicated in the design of mono-Fe catalysts for the processing of hydrogen.19 Recent DFT calculations point to the plausibility of oxadithiolate, O(CH2S)22−, as the cofactor and “strongly disfavor” a role for the azadithiolate.20 Models promise to play a key role in resolving the assignment of O vs. N problem, since X-ray crystallography cannot distinguish these atoms and the free cofactors would be unstable, thus precluding their isolation.N-Protonation is favored thermodynamically for Fe2(adt)(CO)6 (with pKa’s ≈ 8 in MeCN solution). Replacement of the carbonyl ligands with alkyl phosphines (and presumably cyanides) enhances the basicity of the iron centers. Even in Fe2(adt)(CO)4(PMe3)2, where the FeFe bond is 1000× more basic than the amine, N-protonation is kinetically favored.21 In cases where the terminal hydride is more stabilized, equilibration between the hydride and ammonium derivative can be observed (Scheme 1). The emerging mechanistic picture is that N-protonation precedes and facilitates the formation and deprotonation of the terminal hydride.
 | ||
| Scheme 1 N- and Fe-protonation of a diiron azadithiolato complex and its 1e−-reduction. | ||
Even propanedithiolate noticeably affects the coordinating tendency of the Fe centers since the central methylene group projects over this apical site. This steric shielding is evidenced by the effect of the pdt vs. edt on the stereochemistry of Fe2(edt/pdt)(CO)4(diphosphine). For edt, the diphosphine exclusively spans apical and basal sites, whereas in the pdt derivative, one observes significant amounts of the dibasal isomer. The rates of carbonylation of the unsaturated Hox models containing pdt are also slower for the edt derivatives (see below).
Redox auxiliary
Hydrogen redox is assumed to be a 2e− process, both for the reductive coupling of two protons or the oxidative cleavage of dihydrogen. Redox by one electron otherwise yields hydrogen radicals, high energy species that are rarely observed near ambient temperatures. For the [FeFe]-H2ases, the 2Fe subunit provides one redox equivalent, with the second equivalent being supplied by the appended 4Fe-4S cluster. Thus, even though most modeling studies focus on diiron dithiolato complexes, the entire H-cluster is required for turnover. Model systems rely on the ability of electrodes to supply the additional redox equivalents.A model for the entire H-cluster has been prepared with a 4Fe-4S subunit linked via thiolato bridges to one or more Fe2(SR)2(CO)5 centers (Fig. 2). In MeCN solution, the synthetic 6Fe ensemble is an electrocatalyst for hydrogen evolution using 3,5-dimethylpyridinium as a proton source. In addition to representing a significant synthetic achievement, this 6Fe cluster provides insights into the nature of the linkage between the 2Fe and the 4Fe subunits, insights potentially relevant to engineering biomimetic catalysts.22 First, attachment of the Fe2(SR)2(CO)5 fragment to one thiolate of Fe4S4(SR)42− shifts the reduction potential of the 4Fe cluster in the positive direction by 150 mV. It remains unclear how the redox properties of the 4Fe-4S cluster will be affected by more realistic diiron fragments such as [Fe2(SR)2(CN)2(CO)3]x−. Second, IR measurements in the νCO region suggest that the 4Fe-4S cluster-ligand is thioether-like in its Lewis basicity. This μ-SR linkage between the [4Fe-4S] and the Fe2(SR)2(CO)5 subunits breaks easily upon reduction of the dianionic 6Fe, whereas in the enzyme this bond is enforced by the protein.
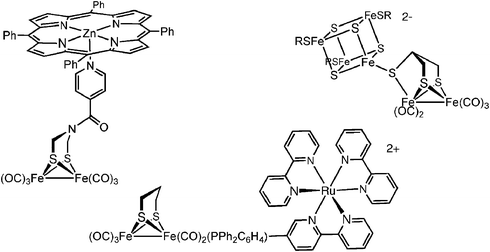
Fe2(SR)2(CO)6 centers have also been modified with abiological redox auxiliaries.23,24 These assemblies are designed to couple the hydrogen producing properties of the diiron dithiolato carbonyls with sources of photogenerated reducing equivalents. To this end, diiron dithiolato carbonyls have been covalently linked to light-harvesting chromophores, such as [Ru(bipy)3]2+ and Zn(TPP), which serve as antennae. Excitation of the sensitizer produces a reducing equivalent that, when transferred to the diiron assembly, could potentially reduce protons to H2. Initial designs have focused on matching the properties of the photoreductant—lifetimes, quantum yields, and reducing properties—with the redox properties of the diiron center. A long-range goal of such work is the development of a catalyst for water splitting.
Highlighting the versatility and robustness of the Fe2(SR)2(CO)6 species, photosensitizers have been attached via several means, including linking to the azadithiolate and through phosphine ligands (Fig. 2). The diiron(I) dithiolato framework is compatible with numerous conjugation and assembly methods which will enable matching of the reducing potential of the photogenerated reducing equivalents with that of the underlying diiron center (ca.−1.2 V vs. SCE).23
Mixed valency
Mixed valency is a defining feature of the oxidized state (Hox) of the enzyme. Protein crystallography and DFT show that this S = 1/2 state adopts an unusual structure whereby the coordination environment of the distal Fe(CO)2CN subunit is “rotated” (see Fig. 1).25 In the rotated structure, one CO ligand shifts from an apical site to a semi-bridging position, exposing a vacant coordination site (Scheme 2).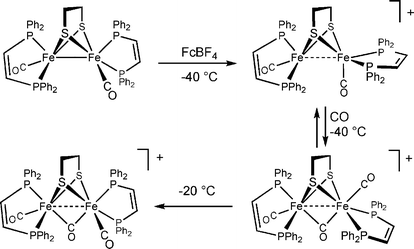 | ||
| Scheme 2 Generation of models for the Hox and HoxCO states. | ||
Modeling studies have yielded related coordinatively unsaturated diiron species exhibiting the expected magnetism, structure, and tendency to carbonylate.26 The mixed phosphine-carbene complex Fe2(pdt)(CO)4(PMe3)(IMes) undergoes oxidation at very mild potentials (−450 mV vs. Fc0/+) to give [Fe2(pdt)(CO)4(PMe3)(IMes)]+, isolated as its PF6− salt. The tetra- and trisubstituted derivatives of the type Fe2(edt/pdt)(CO)2(PR3)4 and Fe2(edt/pdt)(CO)3(PR3)3 can also be oxidized by ferrocenium to yield the corresponding monocations. The cations are somewhat thermally sensitive, typically decomposing within minutes near room temperature, but they are amenable to EPR, IR, and crystallographic analyses that show a strong similarity to the active site in the Hox state.
For these mixed valence species, EPR studies indicate that the rotated Fe center is Fe(I), thus oxidation causes “rotation without redox” at distal Fe and “redox without rotation” at the proximal Fe. In [Fe2(pdt)(CO)4(PMe3)(IMes)]+, the rotated (Fe(I)) site carries the bulky IMes ligand. DFT calculations suggest that the rotated Fe(I) site is more electrophilic, despite its lower oxidation state. Diamagnetic analogues of Hox, prepared using NO in place of CO, indicate that rotation of the distal Fe is caused by the electronic asymmetry that induces the shift of one CO ligand to a semi-bridging site.27
The unsaturated [Fe2(SR)2(CO)6−x(PR3)x]+ species reversibly bind CO, as observed also for the Hox state of the enzyme.28 IR studies of the 13CO-labeled adduct, a model for the HoxCO state, indicate that the exogenous CO binds to the vacant apical site. EPR results, in particular the 31P hyperfine values, indicate that the resulting 35e− adducts are valence-delocalized, in contrast to the Hox models. At 2.5 Å, the Fe–Fe distance in the Hox models is virtually unchanged relative to the 34e− precursor complexes,29 but upon binding CO, the Fe–Fe distance elongates by 0.1 Å. Electrochemical studies show that these HoxCO models are susceptible to oxidation to diamagnetic diferrous species, which are akin to the diamagnetic Hoxair state. CO is virtually unique in forming stable adducts with the Hox models, stronger donors result in disproportionation (eqn (3)).
| 2[Fe2(SR)2(μ-CO)L6]+⇌ [Fe2(SR)2(μ-CO)L6]2+ + Fe2(SR)2L6 + CO | (3) |
3. Biomimetics for the active site of the [NiFe]-H2ases
Overview of the active site
High resolution crystal structures of the [NiFe]-H2ases have been solved for proteins obtained from D. gigas, D. vulgaris, and D. desulfuricans.30 The active site resembles that for the [FeFe]-H2ase in key respects: a bimetallic center is bridged by two thiolates (cysteines, not a cofactor). The iron center has one CO and two cyanide ligands, and the coordination sphere about nickel is completed with two terminal cysteinyl thiolates (Fig. 3), although in some proteins one terminal cysteine is replaced by selenocysteine. In contrast to the [FeFe]-H2ases, substrate turnover involves hydride ligands that bridge the two metals. When the bridging site is occupied, the Fe center becomes octahedral and the Ni distorted trigonal pyramidal. Oxygen-inhibited enzymes feature oxide or hydroperoxide ligands in place of the hydride, and the removal of these poisons determines the “readiness” of enzyme preparations.30![Structure of the active site of the [NiFe]-H2ase and line drawing of the same.](/image/article/2009/CS/b801796b/b801796b-f3.gif) | ||
| Fig. 3 Structure of the active site of the [NiFe]-H2ase and line drawing of the same. | ||
Although the [NiFe]-H2ases are pervasive and the structure announced several years ago, functional models have lagged structural models.31 The barriers to functional modeling result from several factors: (i) the heterometallic nature of the [NiFe] site is inherently more challenging synthetically than homodimetallic species, (ii) the Ni center is structurally unusual, (iii) the ambidentate character of both the cyanide (on Fe) and terminal thiolato (on Ni) ligands complicates the assembly of discrete species, and (iv) easily accessed models are cationic, whereas it is likely that the active site is anionic or charge-neutral. Underscoring the synthetic challenge is the corresponding complexity of the biological assembly pathway.30
The Ni(SR)4 subunit
The Ni(cysteinate)4 site in the [NiFe]-H2ase adopts a distorted tetrahedral geometry (not unlike SF4), which is unusual for Ni(II). Mononuclear nickel tetrathiolates are rare, the prototype being [Ni(SPh)4]2−, which is high spin and kinetically labile. The extensive inventory of bisdithiolenes, e.g. Ni(S2C2Ph2)2, presumably are not reasonable building blocks because of their weak basicity. For these reasons, modeling has emphasized nickel complexes of diaminodithiolates and electronically related dianionic ligands, the idea being that amines and phosphines are acceptable surrogates of terminal thiolates.The [NiFe]-H2ases are characteristically redox active. Mononuclear iron(II) carbonyls resist oxidation, thus it is logical that redox is centered on Ni, as implicated in several biophysical studies. Nickel shuttles between S = 0 Ni(II) and the otherwise uncommon S = 1/2 Ni(III) near −250 mV (vs. NHE). Model studies confirm that thiolates stabilize Ni(III).32 Well defined reactivity of these Ni(III) thiolates toward H2 has not been demonstrated.
The Ni(SR)2Fe core
Synthesis of heterobimetallic cores have mainly relied on the binding of nickel(II) dithiolates to electrophilic iron(II) species.33 These routes exploit the well-known ability of thiolato ligands to form strong bridges between metals. The incorporation of even one CO ligand ensures that the Fe center is low-spin (Scheme 3), which is required for H2ase-like reactivity. Of the several Ni(SR)2Fe-containing models, none exhibit any reactivity toward H2, as well as “easier substrates” such as silanes. The apparent problem is that structural models feature coordinatively saturated ferrous centers that lack labile ligands.![Selected models for the [NiFe]-H2ases active site featuring the Ni(SR)2 core and at least one biomimetic ligand.5,29,34,35](/image/article/2009/CS/b801796b/b801796b-s3.gif) | ||
| Scheme 3 Selected models for the [NiFe]-H2ases active site featuring the Ni(SR)2 core and at least one biomimetic ligand.5,29,34,35 | ||
Hydrides
Dihydrogen complexes are unknown for nickel, in sharp contrast to iron(II), and fewer nickel hydrides are known as well. Work by DuBois et al. shows that nickel(II) can be an excellent hydride acceptor, but a pendant base, not unlike the azadithiolate (section 2) is required for reasonable rates of H2 activation.36 Thus, complexes of the type NiIIL42+ form the corresponding hydrides HNiIIL4+ both by protonation of Ni0L437 and by oxidation of H2, as is typically required of the enzyme. In the [NiFe]-H2ases, a terminal thiolato ligand may serve as an internal base. The hydride acceptor ability of NiIIL42+ complexes correlates with their distortion toward tetrahedral geometry,38 consistent with the distorted coordination sphere seen in the active site. Some square-planar charge-neutral nickel dithiolato complexes slowly catalyze H–D exchange between D2 and proton-bearing ligands.39The Fe cyanide subsite
The facial Fe(CN)2(CO) subunit in the active site was unprecedented in coordination chemistry but subsequent model compounds with fac-L3Fe(CN)2(CO) centers exhibit spectra in the νCN and νCO regions that closely resemble vibrational spectra obtained for the enzyme. The CO ligand is situated trans to the hydride, a pattern also seen in μ-hydride derivatives of the [FeFe]-H2ases.Prior to the structural characterization of the [NiFe]-H2ases, hydride and carbonyl derivatives of ferrocyanides were essentially unknown. The analogy between the coordination properties of FeII(CN)5 and RuII(NH3)5, noted by Taube,40 suggested the possibility that the cyanoferrous centers would bind H2, as has been seen for the Ru(II) ammines.2 Although the iron hexacyanides have long been known, the systematic chemistry of mixed ligand [Fe(CN)6−x(CO)x](4−x)− complexes has only recently been developed and four such species are now known. These studies show that the presence of even one CO ligand strongly stabilizes the ferrous state, arguing against a redox role for the iron.41 The emerging picture is that the FeII(CN)2(CO) center is well suited to serve as a Lewis acidic site for stabilizing the bridging hydride.
The chemistry of iron hydrido and dihydrogen complexes is well developed, largely due to the pioneering studies of Morris et al. on the [Fe(H2)L(diphosphine)2]2+ systems.42 The hydrido carbonyl cyanide [HFe(CN)2(CO)3]− has been prepared as a mixture of two major isomers. This anion not only features several of the ligands observed in active site, it releases H2 upon protonation.43 The unanswered question is whether CO ligands in this complex could be further substituted to accommodate thiolate ligands as seen in the active site.
4. Catalysis of H2 production by models
In principle, catalysis of H2 evolution can be achieved through the protonation of a suitable Brønsted base followed or preceded by one or several reduction steps. To this end, chemists have favored low oxidation state transition metal complexes, because the basicity and the redox potential can be both varied by an appropriate choice of the metal and the ligands. Early H2 evolution catalysts included organometallic complexes such as cyclopentadienylcobalt phosphines, as well as macrocyclic systems such as cobaloxime, nickel cyclam, and various metalloporphyrins. With the exception of the cobaloximes, most homogeneous catalysts are either unstable or require high overpotentials.3 In 2001 diiron dithiolates were shown to be good electrocatalysts for proton reduction,44 and this finding led to intense study of related complexes.Proton reduction catalysis is generally indicated by changes in cyclic voltammetry upon addition of acid HA. The main criterion is that the height of the reduction peak of the homogeneous catalyst, or its protonated form, increases with [HA] (Fig. 4). The increase in peak current is due to a catalytic cycle that produces H2 after electron transfers to the protonated and oxidized forms of the catalysts giving more current than in the absence of acid. At high [HA]/[catalyst] ratios, the catalytic current reaches a plateau value indicating that the current is limited by the rate of the catalytic cycle, not by proton diffusion. With respect to their catalytic properties, [FeFe]-H2ase models can be classified into two categories, depending on the sequence of protonation and reduction.
![Cyclic voltammograms of a solution 2.2 mM Fe2(bdt)(CO)6 in MeCN–Bu4NPF6 upon addition of an increasing amount of toluenesulfonic acid (HOTs) up to 33 mM. Inset: dependence of the catalytic peak current vs. [HOTs] (conditions: glassy carbon electrode 0.071 cm2 in surface area; scan rate 0.1 V s−1).](/image/article/2009/CS/b801796b/b801796b-f4.gif) | ||
| Fig. 4 Cyclic voltammograms of a solution 2.2 mM Fe2(bdt)(CO)6 in MeCN–Bu4NPF6 upon addition of an increasing amount of toluenesulfonic acid (HOTs) up to 33 mM. Inset: dependence of the catalytic peak current vs. [HOTs] (conditions: glassy carbon electrode 0.071 cm2 in surface area; scan rate 0.1 V s−1). | ||
Proton reduction by diiron dithiolato hexacarbonyls
In the case of hexacarbonyl diiron dithiolates, the first step in catalytic hydrogenesis is an electron-transfer. Fe2(bdt)(CO)6 reduces at E1/2,red = −1.27 V (vs. Fc+/0) to its dianion in a chemically reversible two-electron process.45 The two-electron character of the reduction arises from an inversion of the two individual electron transfers, i.e. the second reduction occurs at a potential less negative than the first. Such an inversion is indicative of a large structural change induced by the first reduction, which was confirmed by DFT calculations showing scission of one of the Fe–S bonds and a shifting of a CO to a bridging position (eqn 4).46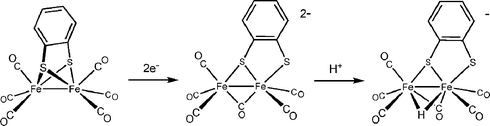 | (4) |
 | ||
| Scheme 4 | ||
The primary reduction of the propanedithiolate Fe2(pdt)(CO)6 occurs at E1/2,red = −1.63 V, 360 mV more negative than the bdt derivatives, indicating the superior donor ability of the alkyldithiolate.48 The chemical reversibility of the [Fe2(pdt)(CO)6]0/− couple depends on the solvent and is improved in the presence of CO. The charge passed upon electrolysis under CO atmosphere is consistent with an overall two-electron reduction. However the one-electron reduced species [Fe2(pdt)(CO)6]−, which is proposed to be structurally similar to its neutral precursor, is involved in several subsequent chemical reactions to give eventually an Fe4 product that is also catalytically active. Voltammetry of Fe2(pdt)(CO)6 in the presence of HOTs shows two different proton reduction waves. The first one (process I, Scheme 5) is associated with the reduction of the pdt complex. The second one (process II), which occurs at a potential more negative, appears in the presence of excess acid. Simulation of the cyclic voltammograms suggests that process I corresponds to H2 production through an ECEC mechanism with slow liberation of H2 from the doubly reduced intermediate, nominally H2Fe2(pdt)(CO)6. As a result, an additional electron transfer occurs (process II) leading to a 3-electron-2-proton intermediate that quickly releases H2.
 | ||
| Scheme 5 | ||
The preceding examples show that the nature of the dithiolate bridge strongly influences the reduction potential of the Fe2(SR)2(CO)6 complexes, and thus the potential at which H+/H2 catalysis occurs. In addition, at least two mechanisms must be considered depending on thermodynamic (pKa) and/or kinetic factors (H2 liberation rate). When the reduction process is not chemically reversible, as for the pdt derivative, products (dimers) formed in the absence of acid may also be involved in the electrocatalytic proton reduction process.49
Proton reduction by substituted diiron dithiolates
The substituted diiron dithiolates are clearly more relevant to the H-cluster where the cyanide ligands substantially enhance the basicity of the diiron center. The anion [Fe2(pdt)(CO)4(PMe3)(CN)]− (Ep,red = −2.56 V) reacts with HOTs to form a derivative that reduces at Ep,red = −1.45 V.50 Spectroscopy and crystallography establish the thermodynamic preference for protonation of the Fe–Fe bond to form a bridging hydride, the cyanide ligand being less basic. In situ studies however suggest that protonation at cyanide is kinetically favored.11 Reduction of [Fe2(pdt)(μ-H)(CO)4(PMe3)(CNH)]+ in the presence of excess acid leads to catalytic H2 production. Similarly, the bisphosphine Fe2(pdt)(CO)4(PMe3)2 (Ep,red = −2.29 V) protonates at the Fe–Fe bond to form a μ-hydride species (Ep,red = −1.41 V), which also catalyzes proton reduction. Qualitative studies suggest that the rate determining step of the H+/H2 catalytic cycle is protonation of the diiron site. This result again highlights the kinetic barriers to protonation of metal centers51 and indicates the advantages of cofactors that would facilitate protonation. Although similar behavior may be anticipated for other complexes of the type Fe2(pdt)(CO)4L2 (L is a good donor ligand), the bis(PMe3) derivative appears to be a slower catalyst than the phosphine-cyanide derivative (L = PMe3/CN−).50 Since the bis(PMe3) and PMe3/CN− have similar pKa values, the differing rates may point to a possible role of the cyanide ligand in facilitating proton transfer to the diiron site. The doubly protonated complex [Fe2(pdt)(μ-H)(CO)4(PMe3)(CNH)]+ is reduced, and thus catalyzes proton reduction, at a potential about 0.15 V milder than Fe2(pdt)(CO)6.Although monosubstituted derivatives Fe2(SR)2(CO)5L do not readily protonate, solutions of both Fe2(pdt)(CO)5(P(OMe)3) (Ep,red = −1.98 V) and Fe2(bdt)(CO)5(P(OMe)3) (Ep,red = −1.53 V) exhibit acid-responsive peaks at Ep,red = −1.65 and −1.18 V, respectively.16,52 A CE mechanism implicates the facile reduction of [(H)Fe2(SR)2(CO)5L]+. Simulations of voltammetric responses and electrolysis experiments suggest that hydrogen evolution catalysis may occur by a bimolecular reaction, i.e., 2HFe2(SR)2(CO)5L → 2Fe2(SR)2(CO)5L + H2, even in the presence of excess acid. Similarly, proton reduction by Fe2(bdt)(CO)5(P(OMe)3), which occurs at mild potentials, is also slow, due also to a bimolecular reaction.
Comments on overpotential
The “best” catalysts for hydrogen evolution exhibit large catalytic currents at mildly negative potentials. An important parameter is the potential Ecat at which catalysis occurs relative to the standard potential E0HA for the HA/A−, H2 half-reaction. This overpotential (η) is the difference Ecat−E0HA. In the case of weaker acids (HA), E0HA can be estimated from , the potential of the proton/hydrogen couple and pKa,HA, the acid dissociation constant (eqn (5)).
, the potential of the proton/hydrogen couple and pKa,HA, the acid dissociation constant (eqn (5)).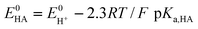 | (5) |
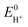 and pKa,HA have been measured in a large number of solvents (Table 1).53
and pKa,HA have been measured in a large number of solvents (Table 1).53
| Catalyst | Ecat/V | Acid | E0HA/Va | −η/V | Ref. |
|---|---|---|---|---|---|
a E0HA =  − 0.059 pKa,HA, with − 0.059 pKa,HA, with 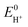 = −0.14 and −0.77 V in MeCN and DMF, respectively. = −0.14 and −0.77 V in MeCN and DMF, respectively. | |||||
| Fe2(bdt)(CO)6 | −1.31 | HOTs | −0.65 | 0.66 | 47 |
| Fe2(bdt)(CO)6 | −2.10 | HOAc | −1.46 | 0.64 | 46 |
| Fe2(pdt)(CO)6 (process I) | −1.59 | HOTs | −0.65 | 0.94 | 54 |
| Fe2(pdt)(CO)6 (process II) | −1.81 | HOTs | −0.65 | 1.16 | 48 |
| [Fe2(pdt)(μ-H)(CO)4(PMe3)(CNH)]+ | −1.43 | HOTs | −0.65 | 0.78 | 50 |
| Fe2(bdt)(CO)5P(OMe)3 | −1.18 | HOTs | −0.65 | 0.53 | 52 |
| Co(dmgBF2)2(MeCN)2 | −0.93 | CF3CO2H | −0.89 | 0.04 | 4 |
The possible role of the adt cofactor raises the possibility that an internal base in the diiron models would enhance the rate of H+/H2 catalysis, thereby lowering the overpotential. The rate-determining step for proton reduction catalysis by Fe2(adt)(CO)6 is, however, the slow H2 release from the 2-electron-2-proton intermediate,55 as seen for Fe2(pdt)(CO)6. Moreover, protonation of Fe2(adt)(CO)4(PMe3)2 gives the μ-hydride, which is too distant to be affected by the adt.12 Intramolecular proton–hydride coupling will be favored by minimizing the protonation-induced reorganization of the iron coordination sphere. A second condition for low overpotential is a matching of the pKa values of the H2-binding site with the protonated pendant base. These requirements have not been fulfilled in the models described to date. Progress is however imminent as protonation of Fe2(adt)(CO)2(dppv)2 gives terminal hydride [HFe2(adt)(CO)2(dppv)2]+ that is reactive towards protons upon reduction.17
5. Future prospects
The considerable success with modeling the different aspects of the [FeFe]-H2ases also highlights several gaps. First, the current models for the [FeFe]-H2ases fail to exhibit any reactivity toward H2. Second although bio-inspired hydrogenogenesis has been achieved with [FeFe]-models, synthetic catalysts operating via the intermediacy of terminal hydrides are just emerging.17 The catalytic properties exhibited by [Fe2(μ-H)(μ-SR)2(CO)6−x(PR3)x]+ may in fact be better described as models for the [NiFe]-H2ases, although it is striking to note the absence of reactivity toward H2, which is the main biological substrate for the [NiFe]-H2ases.By deviating from the Fe2(SR)2 and NiFe(SR)2 stoichiometries of the active sites, it is possible to generate new families of hydrogen-processing catalysts, those that are more bio-inspired than they are biomimetic. It appears that many combinations of nickel, iron carbonyls, and sulfide will activate H2 or reduce protons catalytically. Recent examples include catalysts with Fe4(SR)6,48 NiFe2(SR)2,33 and RuNi(SR)2 cores.35
Acknowledgements
We thank the CNRS-UIUC cooperation program. Research at UIUC was supported by NIH. We thank Bryan Barton, Matt Olsen, and Matt Whaley for comments on the review.References
- R. Cammack, M. Frey and R. Robson, ‘Hydrogen as a Fuel: Learning from Nature’, Taylor & Francis, 2001 Search PubMed.
- G. J. Kubas, ‘Metal Dihydrogen and σ-Bond Complexes’, Kluwer Academic/Plenum, 2001 Search PubMed.
- U. Koelle, New J. Chem., 1992, 16, 157 Search PubMed.
- X. L. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988 CrossRef CAS.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572 CrossRef CAS.
- C. Kamp, A. Silakov, M. Winkler, E. J. Reijerse, W. Lubitz and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 410 CrossRef CAS.
- F. Gloaguen, J. D. Lawrence, M. Schmidt, S. R. Wilson and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 12518 CrossRef CAS.
- M. Razavet, S. J. Borg, S. J. George, S. P. Best, S. A. Fairhurst and C. J. Pickett, Chem. Commun., 2002, 700 RSC.
- A. K. Justice, L. De Gioia, M. J. Nilges, T. B. Rauchfuss, S. R. Wilson and G. Zampella, Inorg. Chem., 2008, 47, 7405 CrossRef CAS.
- X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 9710 CrossRef CAS.
- C. A. Boyke, PhD Thesis, University of Illinois, Urbana-Champaign, 2005.
- G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Chem.–Eur. J., 2007, 13, 7075 CrossRef CAS.
- X. Zhao, C.-Y. Chiang, M. L. Miller, M. V. Rampersad and M. Y. Darensbourg, J. Am. Chem. Soc., 2003, 125, 518 CrossRef CAS.
- J. I. van der Vlugt, T. B. Rauchfuss, C. M. Whaley and S. R. Wilson, J. Am. Chem. Soc., 2005, 127, 16012 CrossRef.
- S. Ezzaher, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer, J. Talarmin, R. Pichon and N. Kervarec, Inorg. Chem., 2007, 46, 3426 CrossRef CAS; P.-Y. Orain, J.-F. Capon, N. Kervarec, F. Gloaguen, F. Pétillon, R. Pichon, P. Schollhammer and J. Talarmin, Dalton Trans., 2007, 3754 RSC.
- D. Morvan, J.-F. Capon, F. Gloaguen, A. Le Goff, M. Marchivie, F. Michaud, P. Schollhammer, J. Talarmin and J.-J. Yaouanc, Organometallics, 2007, 26, 2042 CrossRef CAS.
- B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2008, 47, 2261 CrossRef CAS.
- Y. Nicolet, A. L. de Lacey, X. Vernede, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596 CrossRef CAS.
- R. M. Henry, R. K. Shoemaker, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2006, 128, 3002 CrossRef CAS.
- A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, J. Am. Chem. Soc., 2008, 130, 4533 CrossRef CAS.
- J. L. Stanley, Z. M. Heiden, T. B. Rauchfuss, S. R. Wilson, L. De Gioia and G. Zampella, Organometallics, 2008, 27, 119 CrossRef CAS.
- C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610 CrossRef CAS.
- L. Sun, B. Åkermark and S. Ott, Coord. Chem. Rev., 2005, 249, 1653 CrossRef CAS.
- L. C. Song, M. Y. Tang, S. Z. Mei, J. H. Huang and Q. M. Hu, Organometallics, 2007, 26, 1575 CrossRef CAS; Y. Na, M. Wang, J. Pan, P. Zhang, B. Åkermark and L. Sun, Inorg. Chem., 2008, 47, 2805 CrossRef CAS; J. Ekström, M. Abrahamsson, C. Olson, J. Bergquist, F. B. Kaynak, L. Eriksson, L. Sun, H.-C. Becker, B. Åkermark, L. Hammarström and S. Ott, Dalton Trans., 2006, 4599 RSC.
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414 CrossRef CAS.
- T. Liu and M. Y. Darensbourg, J. Am. Chem. Soc., 2007, 129, 7008 CrossRef CAS; A. K. Justice, T. B. Rauchfuss and S. R. Wilson, Angew. Chem., Int. Ed., 2007, 46, 6152 CrossRef CAS.
- M. T. Olsen, M. Bruschi, L. De Gioia, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2008, 130, 12021 CrossRef CAS.
- A. K. Justice, M. Nilges, T. B. Rauchfuss, S. R. Wilson, L. De Gioia and G. Zampella, J. Am. Chem. Soc., 2008, 130, 5293 CrossRef CAS.
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268 RSC.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273 CrossRef CAS.
- A. C. Marr, D. J. E. Spencer and M. Schröder, Coord. Chem. Rev., 2001, 219–221, 1055 CrossRef CAS.
- S. Fox, Y. Wang, A. Silver and M. Millar, J. Am. Chem. Soc., 1990, 112, 3218 CrossRef CAS; T. Yamamura, H. Arai, H. Kurihara and R. Kuroda, Chem. Lett., 1990, 19, 1975 CrossRef; H.-J. Krüger, G. Peng and R. H. Holm, Inorg. Chem., 1991, 30, 734 CrossRef CAS; J. Hanss and H.-J. Krüger, Angew. Chem., Int. Ed., 1998, 37, 361; C.-H. Chen, G.-H. Lee and W.-F. Liaw, Inorg. Chem., 2006, 45, 2307 CrossRef CAS.
- A. Perra, E. S. Davies, J. R. Hyde, Q. Wang, J. McMaster and M. Schröder, Chem. Commun., 2006, 1103 RSC.
- Y. Ohki, K. Yasumura, K. Kuge, S. Tanino, M. Ando, Z. Li and K. Tatsumi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7652 CrossRef CAS.
- S. Ogo, T. Kabe, K. Uehara, B. Kure, T. Nishimura, S. C. Menon, R. Harada and S. Fukuzumi, Science, 2007, 316, 585 CrossRef CAS.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358 CrossRef CAS.
- H.-F. Klein, A. Dal, T. Jung, U. Flörke and H.-J. Haupt, Eur. J. Inorg. Chem., 1998, 2027 CrossRef CAS.
- M. R. Nimlos, C. H. Chang, C. J. Curtis, A. Miedaner, H. M. Pilath and D. L. DuBois, Organometallics, 2008, 27, 2715 CrossRef CAS.
- D. Sellmann, P. Prakash and F. W. Heinemann, Eur. J. Inorg. Chem., 2004, 1847 CrossRef CAS.
- H. Taube, Pure Appl. Chem., 1979, 51, 901 CAS.
- S. M. Contakes, S. C. N. Hsu, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 2002, 41, 1670 CrossRef CAS; J. Jiang and S. A. Koch, Angew. Chem., Int. Ed., 2001, 40, 2629 CrossRef CAS.
- E. Rocchini, P. Rigo, A. Mezzetti, T. Stephan, R. H. Morris, A. J. Lough, C. E. Forde, T. P. Fong and S. D. Drouin, J. Chem. Soc., Dalton Trans., 2000, 3591 RSC.
- A. Kayal and T. B. Rauchfuss, Inorg. Chem., 2003, 42, 5046 CrossRef CAS.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476 CrossRef CAS.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2004, 566, 241 CrossRef CAS.
- G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Am. Chem. Soc., 2007, 129, 12521 CrossRef CAS.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2006, 595, 47 CrossRef CAS.
- S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. Liu and C. J. Pickett, J. Am. Chem. Soc., 2004, 126, 16988 CrossRef CAS; M. H. Cheah, C. Tard, S. J. Borg, X. Liu, S. K. Ibrahim, C. J. Pickett and S. P. Best, J. Am. Chem. Soc., 2007, 129, 11085 CrossRef CAS.
- I. Aguirre de Carcer, A. DiPasquale, A. L. Rheingold and D. M. Heinekey, Inorg. Chem., 2006, 45, 8000 CrossRef CAS; S. P. Best, S. J. Borg, J. M. White, M. Razavet and C. J. Pickett, Chem. Commun., 2007, 4348 RSC.
- F. Gloaguen, J. D. Lawrence, T. B. Rauchfuss, M. Bénard and M.-M. Rohmer, Inorg. Chem., 2002, 41, 6573 CrossRef CAS.
- K. W. Kramarz and J. R. Norton, Prog. Inorg. Chem., 1994, 42, 1 CAS.
- F. Gloaguen, D. Morvan, J.-F. Capon, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2007, 603, 15 CrossRef CAS.
- A. M. Appel, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2005, 127, 12717 CrossRef CAS; G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181 CrossRef CAS.
- M. H. Cheah, S. J. Borg, M. I. Bondin and S. P. Best, Inorg. Chem., 2004, 43, 5635 CrossRef CAS.
- J.-F. Capon, S. Ezzaher, F. Gloaguen, F. Y. Petillon, P. Schollhammer and J. Talarmin, Chem.–Eur. J., 2008, 14, 1954 CrossRef.
Footnotes |
| † Part of the renewable energy theme issue. |
| ‡ Abbreviations: Fc+ = ferrocenium, pdt2− = 1,3-propanedithiolate, edt2− = 1,2-ethanedithiolate, adt2− = 2-aza-1,3-propanedithiolate, bdt2− = 1,2-benzenedithiolate, Imes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene, dppv = 1,2-bis(diphenylphosphino)ethylene, dppe = 1,2-bis(diphenylphosphino)ethane, TPP2− = tetraphenylporphyrinate, bipy = 2,2′-bipyridine. |
| This journal is © The Royal Society of Chemistry 2009 |