Cobalt–phosphate oxygen-evolving compound†
Matthew W.
Kanan
,
Yogesh
Surendranath
and
Daniel G.
Nocera
*
Department of Chemistry, 6-335, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA. E-mail: nocera@mit.edu
First published on 28th November 2008
Abstract
The utilization of solar energy on a large scale requires efficient storage. Solar-to-fuels has the capacity to meet large scale storage needs as demonstrated by natural photosynthesis. This process uses sunlight to rearrange the bonds of water to furnish O2 and an H2-equivalent. We present a tutorial review of our efforts to develop an amorphous cobalt–phosphate catalyst that oxidizes water to O2. The use of earth-abundant materials, operation in water at neutral pH, and the formation of the catalystin situ captures functional elements of the oxygen evolving complex of Photosystem II.
 Yogesh Surendranath, Daniel Nocera and Matthew Kanan | Matthew Kanan (right) received his BA in Chemistry from Rice University in 2000. He then moved on to Harvard where he completed his PhD in 2005 under the mentorship of Professor David Liu. Currently, he studies water oxidation at MIT as a NIH Ruth Kirchenstein Postdoctoral Fellow. |
Yogesh Surendranath (left) received his BS in Chemistry from the University of Virginia in 2006. As a graduate student at MIT, he examines water oxidation catalysis as a DoD NDSEG Fellow. |
Daniel Nocera (middle) is the Henry Dreyfus Professor of Energy at the Massachusetts Institute of Technology. He received his BS degree from Rutgers University in 1979 and his PhD degree from Caltech in 1984. He studies the mechanisms of biological and chemical energy conversion. |
Introduction
Modern day society relies on a continuous energy supply that must be available day and night. Although solar energy is of sufficient scale to meet future energy needs, it is diurnal.1 Consequently, solar energy will not be used as a large scale energy supply for society unless it can be stored. Unfortunately, most current methods of solar storage are characterized by low energy densities and therefore present formidable challenges for large scale solar implementation. For instance, consider the energy densities by mass of the following storage methods: compressed air (300 atm) ∼0.5 MJ kg−1, batteries ∼0.1–0.5 MJ kg−1, flywheels ∼0.5 MJ kg−1, supercapacitors ∼0.01 MJ kg−1, and water pumped uphill (100 m) ∼0.001 MJ kg−1. Conversely, the energy density of liquid fuels (∼50 MJ kg−1) is ×102 larger than the best of the foregoing storage methods and H2 (700 atm) possesses an even greater energy density at 140 MJ kg−1. Indeed, society has intuitively understood this disparity in energy density as it has developed over the last century as all large scale energy storage in our society is in the form of fuels. But these fuels are carbon-based. The imperative for the discipline of chemistry, and more generally science, is to develop fuel storage methods that are easily scalable, carbon-neutral and sustainable.A fuel-forming reaction that meets this imperative is:
| 2H2O + light = 2H2 + O2 | (1) |
A key design element of photosynthesis is the separation of the functions of light collection and conversion from catalysis. Light is collected and converted by Photosystem II (PSII) into a wireless current. The holes of this current are fed to the oxygen-evolving complex (OEC) where water is oxidized to O2 and the electrons are fed to Photosystem I where additional light capture occurs to provide sufficient reducing power for the reduction of NADP+ to NADPH by ferredoxin:NADP+ oxidoreductase. The separation of collection/conversion from catalysis is dictated by the thermodynamics of the water splitting reaction. To match the solar spectrum and at the same time deliver oxidizing and reducing equivalents of sufficient potential to split water, PSII is confined to generating an electron/hole pair one photon at a time. However, water splitting is a four-electron/hole process.4 Hence, multielectron catalysts at the terminus of the charge-separating network are compulsory so that the one photon-one electron/hole equivalency can be bridged to the four-electron/hole chemistry of water splitting. Additionally, the catalysts must couple protons to the multielectron transformation in order to avoid high-energy intermediates.
In addition to the separation of light collection/conversion from catalysis:
OEC is an all-inorganic metal oxide core . The recent X-ray diffraction and spectroscopic studies reveal PSII to be very complex5–7 However, most of this complexity is associated with the generation of a wireless current at high efficiency. OEC is postulated to be a distorted Mn3Ca cube with oxygen atoms at alternating corners of the cube and a dangling Mn atom.8,9
The OEC core self-assembles upon oxidation of incoming metal ions . Light drives the oxidation of Mn2+ to higher oxidation states, which then leads to OEC self-assembly.10
The OEC – protein complex is not structurally stable and hence a repair mechanism is required . Water oxidation at OEC produces reactive oxygen species, which damage the associated proteins of the PSII complex. Oxygenic photosynthetic organisms have evolved to replace the D1 protein in which the OEC resides with a newly synthesized copy every ∼30 minutes.11 Thus functional stability is maintained despite structural instability.
Proton management is required for water oxidation catalysis in aqueous solutions at pH = 7 . The protons released upon water oxidation cannot be captured and transported by H2O because it is a very weak base. Moreover, the [OH−] = 10−7 M at pH = 7. Proton transport from OEC is accomplished along water channels lined by Lewis basic amino acid side chains.12,13
Within the foregoing framework, an OECmpd is presented. The catalyst self-assembles from aqueous solution upon oxidation of Co2+ to Co3+. Phosphate anion manages the protons released from water oxidation and also provides a mechanism for repair. The catalyst is extremely versatile and it can form on diverse conducting surfaces of varying geometry. Thus the catalyst can be easily interfaced with a variety of light absorbing and charge separating materials.
Catalyst formation and characterization
Controlled potential electrolysis of Co2+ salts in pH 7 phosphate (Pi) buffer at 1.3 V (vs. NHE) results in observed currents that asymptotically approach 1.5 mA cm−2 over several hours. During this time, a dark green-black film forms on the surface of the electrode surface. Similar behavior is observed if methyl phosphonate (MePi) is used as the supporting electrolyte instead of phosphate.14Fig. 1 shows SEMs of the electrode surface at various times during the electrolysis. The ITO substrate can be seen through cracks in the film that form upon drying, as evidenced by particles that are split into complementary pieces. To date, indium-tin-oxide (ITO) and fluorine-tin-oxide (FTO) have been the electrodes of choice because these materials exhibit high overpotentials for O2 production from water and thus ensure minimal background activity. Nevertheless, the thin film forms on many conducting surfaces including glassy carbon, carbon felt, Ni and other metals. The thickness of the electrodeposited catalyst is determined by the length of the electrodeposition and the concentration of Co2+ in the deposition solution. Prolonged electrolysis (passage of 40 C cm−2) produces films with limiting thicknesses of ∼3 μm with the concomitant formation of spherical nodules of 1–5 μm in diameter. The nodules are of similar composition to the film. Prepared films do not need to be used immediately. They can be stored under ambient conditions and subsequently used as an anode in Co-free solutions.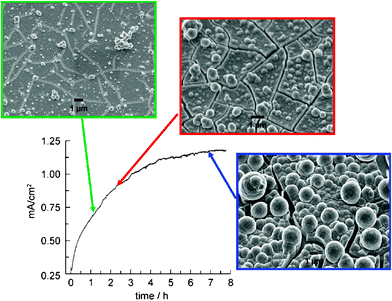 | ||
| Fig. 1 Current density profile for bulk electrolysis at 1.29 V (vs.NHE) in 0.1 M KPi electrolyte at pH 7.0 containing 0.5 mM Co2+. SEM images of the electrode surface taken at indicated time points during the electrodeposition of the catalyst film. The ITO substrate can be seen through cracks in the dried film. | ||
Cyclic voltammetry (CV) of solutions of 1 mM Co2+ in Pi electrolyte suggests that a catalytically active film forms immediately following oxidation of Co2+. A quasi-reversible wave in the CV is observed at 1.14 V vs.NHE (pH 7.0) which is more similar to the Co3+/2+ couple for cobalt ion with hydroxo ligands (E[Co(OH)2+/0] = 1.1 V vs.NHE).15 A large catalytic wave is observed immediately beyond the Co2+/3+ couple and catalyst appears to deposit immediately following oxidation of Co2+ to Co3+.
Powder X-ray diffraction patterns of the electrodeposited catalysts exhibit broad amorphous features; no peaks indicative of crystalline phases are observed other than the peaks associated with the ITO sublayer. Films prepared from Pi exhibit a 2 : 1 Co : P ratio as determined by energy-dispersive X-ray (EDX) and elemental analyses. Elemental analysis of films prepared from MePi permits the carbon content of the film to be ascertained. A P : C ratio of ∼2 : 1 indicates partial decomposition of methylphosphonate to phosphate within the film; phosphate signals in the {1H}31P NMR spectrum of films isolated from the electrode support this observation. In contrast, MePi and Pi buffer solutions remain intact under prolonged electrolysis. No other major signals excepting those from the buffer are observed in the 31P NMR spectra of solutions taken from the working or auxiliary compartments.
Together, the XRD and analytical results indicate that electrolysis of a Co2+ solution in aqueous phosphate buffer results in the electrodeposition of an amorphous Co oxide or hydroxide incorporating a substantial amount of phosphate anion. X-Ray absorption spectroscopy (XAS) studies are underway to provide additional structural information.
Oxygen catalysis
Electrodeposition of the film is accompanied by vigorous effervescence of O2, as confirmed by mass spectrometric analysis. Mass spectrometric detection of O2 in real-time from 18OH2 enriched Pi and MePi indicate an isotopic ratio of 16,16O2, 18,16O2 and 18,18O2 in agreement with the predicted statistical ratio, indicating that water is the source of the O-atoms in the evolved O2. The data shown in Fig. 2a for films prepared in Co2+/Pi solutions is exemplary. Signals for all three isotopes of O2 rise from their baseline levels minutes after the onset of electrolysis and then they slowly decay after electrolysis is terminated and O2 is purged from the head space. The O2 isotopic ratios are invariant over hours (Fig. 2b). The Faradaic efficiency of the catalyst is most conveniently measured by a fluorescence-based O2 sensor. Fig. 2c shows the current passed during an electrolysis performed at 1.3 V (blue line) is completely accounted for by the quantity of O2 produced (red line). Moreover, the amount of O2 produced (95 μmoles, 3.0 mg) greatly exceeds the amount of catalyst (∼0.2 mg), which shows no perceptible decomposition over the course of the experiment. Thus, all current passed through the catalyst is used for O2 production. | ||
Fig. 2 (a) Mass spectrometric detection of isotopically-labeled 16,16O2 ( ), 16,18O2 ( ), 16,18O2 ( ) and 18,18O2 ( ) and 18,18O2 ( ) during electrolysis of a catalyst film on ITO in KPi electrolyte containing 14.6% 18OH2. Green and red arrows indicate initiation and termination of electrolysis at 1.29 V (NHE). Inset: expansion of the 18,18O2 signal. (b) Percent abundance of each isotope over the course of the experiment. Average observed abundance ±2σ indicated above each line and calculated statistical abundances are indicated in the parenthesis. (c) O2 production measured by fluorescent sensor ( ) during electrolysis of a catalyst film on ITO in KPi electrolyte containing 14.6% 18OH2. Green and red arrows indicate initiation and termination of electrolysis at 1.29 V (NHE). Inset: expansion of the 18,18O2 signal. (b) Percent abundance of each isotope over the course of the experiment. Average observed abundance ±2σ indicated above each line and calculated statistical abundances are indicated in the parenthesis. (c) O2 production measured by fluorescent sensor ( ) and the theoretical amount of O2 produced ( ) and the theoretical amount of O2 produced ( ) assuming a Faradaic efficiency of 100%. Green arrow indicates initiation of electrolysis at 1.29 V and red arrow indicates termination of electrolysis. Reproduced with permission from Science 2008, 321, 1072. Copyright 2008 American Association for the Advancement of Science. ) assuming a Faradaic efficiency of 100%. Green arrow indicates initiation of electrolysis at 1.29 V and red arrow indicates termination of electrolysis. Reproduced with permission from Science 2008, 321, 1072. Copyright 2008 American Association for the Advancement of Science. | ||
Fig. 3 shows the Tafel plot for a CoPi catalyst run in Pi; a similar Tafel plot is obtained for the catalyst run in MePi. Appreciable current densities are obtained beginning at 0.28 V overpotential. The red circles on Fig. 3 show the current density measured at a constant applied potential (Vappl = 1.24 V) at various pH values. The overpotential (η) was determined by correcting Vappl for iR drop and assuming Nernstian behavior for the potential at various pH values according to the following:
| η = (Vappl + 0.059ΔpH − iR) −E(pH 7) | (2) |
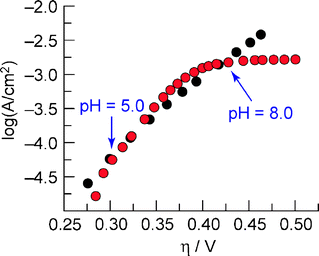 | ||
Fig. 3 Tafel plot (●), η = (Vappl− iR) −E(pH 7), of a catalyst film on ITO in 0.1 M KPi electrolyte pH = 7.0, corrected for the iR drop of the solution. pH data converted into a Tafel plot ( ) using eqn (2). The pH = 5 and pH = 8 data points are indicated by arrows. Reproduced with permission from Science 2008, 321, 1072. Copyright 2008 American Association for the Advancement of Science. ) using eqn (2). The pH = 5 and pH = 8 data points are indicated by arrows. Reproduced with permission from Science 2008, 321, 1072. Copyright 2008 American Association for the Advancement of Science. | ||
 | ||
| Fig. 4 (top) The PCET activation of tyrosine upon excitation of a Re(I) complex. The pH dependence of the PCET rate constant arises from the H2PO4−/HPO42− equilibrium. The increase in rate constant with increasing pH establishes HPO42− as the proton acceptor. (bottom) Oxidation to the active catalysts from which O2 generation occurs may proceed by the PCET reaction of a Co3+–hydroxide intermediate in which HPO42− is the proton acceptor. | ||
These results are reminiscent of the oxidation of tyrosine in model PCET systems shown in Fig. 4 (top). A metal-to-ligand charge transfer excited state of a Re polypyridyl is capable of oxidizing an appended tyrosine, but only if HPO42− is present.20 The reaction kinetics are pH dependent and consistent with a PCET mechanism in which ET from Y to the oxidant is accompanied by PT from Y to HPO42−. Accompanying theoretical work on the model system supports such a concerted PCET mechanism with HPO42− acting as the proton acceptor.21
Catalyst function does not require pure water. Catalyst prepared from Pi or MePi solutions retain high activity for oxygen production from Co-free buffer solutions containing high salt concentration.14 Although the catalyst operates (1.30 V, pH 7.0) near the formal HOCl/Cl− potential (1.28 V at pH 7), it is oxygen that is produced at high Faradaic efficiency from 0.5 M NaCl solutions.
A robust catalyst
The design of a cobalt-based catalyst presents significant challenges to the inorganic chemist. Co2+ in an oxygen-atom ligand field is a high spin, d7, ion. Hence, the eg(M–Lσ*) orbital is populated and cobalt ion in the 2+ state is substitutionally labile. Conversely, Co3+ and higher oxidation states are low spin and substitutionally inert in an oxygen-atom ligand field. This long known reactivity22 of Co2+ and Co3+ (and higher oxidation states) explains the 109 difference in the substitution rates of aqua ligands on cobalt ions in solution. Substitution rates on metal ions in solution have been correlated with metal oxide dissolution rates.23 The Co2+, Co3+ and likely Co4+ oxidation states will be accessed in any 4e−water–oxygen redox cycle involving cobalt. Thus a quandary is presented. How can a “stable” catalyst be prepared? The typical approach of an inorganic chemist is to prepare a ligand that enforces, usually by chelation, a stable ligand environment. But in such a case, a ligand field that is appropriate for Co2+ will not be so for Co3+ and excess potential will be required to oxidize Co2+ to Co3+. Thus by pursuing the strategy of a stable ligand environment about the cobalt ion, excess overpotential will be introduced into the redox cycle.Though solubility products (Ksp) for cobalt ions and HPO42− are not easily found, the Ksp for Ca(HPO4) is 10−7.24 This value offers an “electrostatic” baseline for the Ksp of HPO42− with a 2+ ion. Overlaying ligand field considerations on this Ksp, an HPO42−salt of high spin Co2+ would be expected to be more soluble than Ca(HPO4) whereas one of low spin Co3+ would be expected to be less soluble than Ca(HPO4). The window provided by the surmised differences in the Ksp of HPO42− with Co2+ and Co3+ explains the electrodeposition process since the film only forms upon oxidation of Co2+ to Co3+. In light of in situ formation, a mechanism for reformation of the catalyst during cycling is viable. If Co2+ is produced during the cycle and released from the film prior to re-oxidation, a dynamic equilibrium between Co2+–HPO42− in solution and Co3+–HPO42− on the electrode may be established. Isotope labeling studies are underway to observe the exchange of ions directly between the electrodeposited film and solution. In addition, if Co2+ is part of a cluster of higher oxidation state cobalt centers, the 2+ oxidation state may be shared over the entire cluster core thus retarding the release of cobalt ions from the cluster prior to re-oxidization.
A working model for the cycle
Several useful concepts of catalysis are embodied by the CoPi OECmpd. A working model for operation of the catalyst is shown in Fig. 5. The Co2+ is oxidized to Co3+ and then is deposited on the electrode in the presence of HPO42−. The pH dependence of the current density is consistent with the oxidation of Co2+–OH2 to Co3+–OH and/or the oxidation of Co3+–OH to Co4+–oxo; in Fig. 5 we emphasize the latter PCET process to produce a Co4+–oxo from which O2 is produced and cobalt is returned to the 2+ oxidation state. In situ EXAFS experiments on an active electrode are currently underway to establish the nature of the catalytically active state of cobalt ion. The overall cycle of catalysis need not proceed strictly in the heterogeneous or homogeneous phase. The catalyst may cycle through both phases, which communicate with each other via equilibria processes. Importantly, we believe that catalysis is occurring at molecular centers with discrete electronic structure. Unlike most solid state catalysts, an electronic structure of the bulk or a nanodomain does not prevail. We suspect that this is one reason why the catalyst works well in salt solution at modest overpotentials. Presumably, outer sphere electron transfer mechanism involving chloride does not effectively compete with the inner sphere redox processes involving oxygen. In light of the foregoing considerations, Table 1 proposes several parallels between OEC and the CoPi OECmpd. | ||
| Fig. 5 A working model for the Co–phosphate OECmpd. | ||
| Photosystem II OEC | CoPi OECmpd | |
|---|---|---|
| Self-assembly | Earth-abundant metal (Mn) | Earth-abundant metal (Co) |
| All oxo core | All oxo framework | |
| Self-assembled from water upon metal oxidation | Self-assembled from water upon metal oxidation | |
| Repair | D1 protein | HPO42−/Co3+ equilibrium |
| O2 generation | From neutral water | From neutral water |
| At 1 atm and RT | At 1 atm and RT | |
| At low overpotential | At low overpotential | |
| Proton carrier (amino acid) | Proton carrier (HPO42−) | |
Future prospects
The CoPi OECmpd advances the viability of water-splitting as a solar storage mechanism by enabling the solar-to-fuels conversion to be performed in neutral water under benign conditions. Mechanistic studies of CoPi OECmpd may provide insight into the requirements for O2 evolution under the conditions of natural photosynthesis. Additionally, CoPi OECmpd offers inroads to artificial systems aimed at mimicking photosynthesis. Although many ingenious synthetic charge separating networks comprising protein, inorganic and organic dyads and triads have been developed, the realization of artificial photosynthesis using these constructs has been limited by the difficulty of connecting them to water-splitting catalysts. Because most of these constructs are not stable in highly acidic or basic environments, CoPi OECmpd is a good candidate as a catalyst interface for these charge-separating networks. The catalyst can also be deposited on conductive or semi-conductive substrates with complicated geometries and large surface areas. As in the charge-separating networks, one-photon, one-electron charge separation of a photoanode can be accumulated by the catalyst to attain the four equivalents needed for water splitting. The ease of implementation of the CoPi OECmpd with such a diverse array of photoactive materials suggests that the catalyst will be of interest to many in their endeavors to store solar energy by water splitting.Acknowledgements
This research was supported by a Center for Chemical Innovation of the National Science Foundation (Grant CHE-0533150) and a grant from the Chesonis Family Foundation. Grants from the NSF also provided instrument support to the DCIF at MIT (CHE-9808061, DBI-9729592). M.W.K. is a Ruth Kirchenstein NIH Postdoctoral Fellow. Y.S. gratefully acknowledges the Department of Defense for a pre-doctoral fellowship.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS.
- J. Barber, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 1007 CrossRef CAS.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS.
- T. A. Betley, Q. Wu, T. Van Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS.
- B. Loll, J. Kern, W. Saenger, A. Zoun and J. Biesiadka, Nature, 2005, 438, 1040 CrossRef CAS.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821 CrossRef CAS.
- J. Barber, Inorg. Chem., 2008, 47, 1700 CrossRef CAS.
- J. M. Peloquin, K. A. Campbell, D. W. Randall, M. A. Evanchik, V. L. Pecoraro, W. H. Armstrong and R. D. Britt, J. Am. Chem. Soc., 2000, 122, 10926 CrossRef CAS.
- R. L. Burnap, Phys. Chem. Chem. Phys., 2004, 6, 4803 RSC.
- E.-M. Aro, M. Suorsa, A. Rokka, Y. Allahverdiyeva, V. Paakkarinen, A. Saleem, N. Battchikova and E. Rintamaki, J. Exp. Bot. E, 2005, 56, 347 Search PubMed.
- J. W. Murray and J. Barber, J. Struct. Biol., 2007, 159, 228 CrossRef CAS.
- H. Ishikita, W. Saenger, B. Loll, J. Biesiadka and E.-W. Knapp, Biochemistry, 2006, 45, 2063 CrossRef CAS.
- Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., submitted for publication Search PubMed.
- B. S. Brunschwig, M. H. Chou, C. Creutz, P. Ghosh and N. Sutin, J. Am. Chem. Soc., 1983, 105, 4832 CrossRef CAS.
- C. J. Chang, Z.-H. Loh, C. Shi, F. C. Anson and D. G. Nocera, J. Am. Chem. Soc., 2004, 126, 10013 CrossRef CAS.
- S. Y. Reece, J. M. Hodgkiss, J. Stubbe and D. G. Nocera, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 1351 CrossRef CAS.
- J. Rosenthal and D. G. Nocera, Acc. Chem. Res., 2007, 40, 543 CrossRef CAS.
- J. D. Soper, S. V. Kryatov, E. V. Rybak-Akimova and D. G. Nocera, J. Am. Chem. Soc., 2007, 129, 5069 CrossRef CAS.
- T. Irebo, S. Y. Reece, M. Sjödin, D. G. Nocera and L. Hammarström, J. Am. Chem. Soc., 2007, 129, 154622.
- H. Ishikita, A. V. Soudackov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2007, 129, 11146 CrossRef CAS.
- F. Basolo and R. G. Pearson, Mechanisms of Inorganic Reactions, John Wiley & Sons, London, 1958 Search PubMed.
- W. H. Casey, J. Colloid Interface Sci., 1991, 146, 586 CrossRef CAS.
- A. C. Bennett and F. Adams, Soil Sci. Soc. Am. J., 1976, 40, 39 CAS.
Footnote |
| † Part of the renewable energy theme issue. |
| This journal is © The Royal Society of Chemistry 2009 |