New approaches to hydrogen storage†
Jason
Graetz
*
Brookhaven National Laboratory, Upton, NY, USA. E-mail: graetz@bnl.gov; Fax: 1-631-344-7905; Tel: 1-631-344-3242
First published on 22nd October 2008
Abstract
The emergence of a Hydrogen Economy will require the development of new media capable of safely storing hydrogen in a compact and light weight package. Metal hydrides and complex hydrides, where hydrogen is chemically bonded to the metal atoms in the bulk, offer some hope of overcoming the challenges associated with hydrogen storage. The objective is to find a material with a high volumetric and gravimetric hydrogen density that can also meet the unique demands of a low temperature automotive fuel cell. Currently, there is considerable effort to develop new materials with tunable thermodynamic and kinetic properties. This tutorial review provides an overview of the different types of metal hydrides and complex hydrides being investigated for on-board (reversible) and off-board (non-reversible) hydrogen storage along with a few new approaches to improving the hydrogenation–dehydrogenation properties.
 Jason Graetz | Dr Jason Graetz received his PhD in Materials Science from the California Institute of Technology under Professor Brent Fultz in 2003. Later that year he joined the Hydrogen Storage Group at Brookhaven National Lab and is currently an Associate Scientist at Brookhaven. His research is focused on energy materials for lithium batteries and solid-state hydrogen storage. Dr Graetz is a US Expert of the International Energy Agency and was the recipient of the 2006 Ewald Wicke Award for his work on metal hydrides. |
1. Introduction
1.1 Why hydrogen?
It is estimated that the world energy consumption will double over the next half century.1 Meeting this demand will be challenging, but meeting this demand while maintaining or reducing our current environmental footprint will require significant changes in the way we produce, distribute, store and use energy. A crucial step in this process will be reducing our dependence on carbon-based fossil fuels.Hydrogen is a promising alternative energy carrier. It undergoes a perfectly clean combustion reaction‡ with oxygen (2H2g + O2g→ 2H2Og) to produce energy (ΔH = 120 kJ g−1) and water. Since hydrogen is a “carbon free” energy carrier, it has the highest possible energy density of any hydrogen-based fuel. In a combustion reaction (e.g. CH4 + 2O2→ CO2 + 2H2O), the energy contribution from hydrogen is 120 kJ g−1 while the contribution from carbon is only 33 kJ g−1. Therefore, the energy densities of hydrocarbon fuels scale directly with the hydrogen-to-carbon ratio. The carbon reaction also produces the undesirable greenhouse gas CO2. For a direct comparison, 3 kg of gasoline (or 1 gallon = 3.79 L) has the same energy as 1 kg of H2, but the gasoline also produces approximately 9 kg of CO2. In addition, gasoline is currently made from oil, a finite resource. The increase in demand for oil will likely out pace production within a few decades. Although gasoline can easily be produced from coal through a Fischer–Tropsch process, this method is costly and will likely result in greater greenhouse gas (CO2) emissions. Therefore, the technology for clean alternative energy carriers, like hydrogen, need to be developed today to facilitate the transition to a sustainable, carbon-neutral energy economy tomorrow.
1.2 Hydrogen storage background
The transition to a Hydrogen Economy will require a number of technological advancements. Hydrogen is an energy carrier (not an energy source) and therefore energy must be generated before hydrogen can be produced. Currently, hydrogen is primarily produced from natural gas reforming. This approach doesn’t eliminate carbon emissions, but simply transfers the source of the emissions from the user (e.g. vehicle) to the reforming plant. Improvements in carbon neutral hydrogen production (wind, hydro, solar, etc.), fuel cells and the development of hydrogen infrastructure are critical for the emergence of a Hydrogen Economy. However, one of the more challenging technological barriers to the advancement of hydrogen fuel cell technologies for mobile applications is storage. At the heart of the issue is that hydrogen is a gas at standard pressure and temperature (SPT) and therefore has a low volumetric density. As previously mentioned, one kilogram of H2 can replace about 3 kg or one gallon (3.79 L) of gasoline. However, on a volumetric scale more than 3500 gallons (1.3 × 104 L) of H2 gas are necessary (volume of a midsize car) to replace just one gallon (3.79 L) of gasoline at SPT. The low volumetric hydrogen densities of compressed H2 gas are illustrated in Fig. 1, which compares the gravimetric and volumetric energy densities of a number of different hydrogen storage media based on the lower heating value of H2 (120 kJ g−1).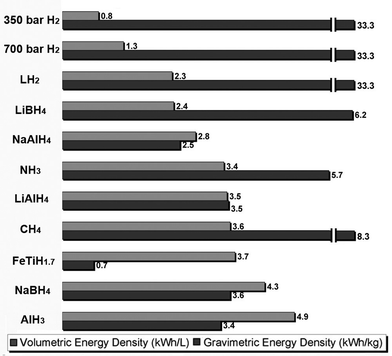 | ||
| Fig. 1 Volumetric (upper bar) and gravimetric (lower bar) energy densities of several metal hydrides and other hydrogen storage media. The energy was determined from the lower heating value of hydrogen (120 kJ g−1) and does not include any contribution from the system (container, valves, tubing, etc.). | ||
Compressed gas and liquid hydrogen storage are unlikely to meet the aggressive US Department of Energy (DOE) targets for on-board H2storage systems.2 Alternatives, such as solid-state hydrogen storage where a host material is used as a hydrogen carrier, offer improved volumetric energy densities. In fact, the energy density of a metal hydride fuel cell system is especially attractive when compared with conventional battery systems as demonstrated in Table 1, which assumes a penalty of 50% to account for system weight and an additional 50% penalty for the fuel cell (50 kW) conversion efficiency (system energy density is 25% of material density). Under this rather conservative scenario, a hydrogen fuel cell using a metal hydride (AlH3) as a H2 source yields gravimetric and volumetric energy densities over four times greater than even the best performing conventional batteries (Li polymer).
| Energy source | Specific energy/W h kg−1 | Energy density/W h L−1 |
|---|---|---|
| AlH3/fuel cell | 850 | 1200 |
| Li polymer | 200 | 300 |
| Ni–Metal hydride | 65 | 100 |
| Ni–Cd | 50 | 100 |
| Pb–acid | 30 | 40 |
This tutorial briefly reviews recent research efforts in solid state hydrogen storage. The reversible complex hydrides (alanates, borohydrides and amides) are briefly covered along with a few of the relevant materials challenges and possible solutions. In addition, a less well-explored class of materials, the non-reversible, or kinetically stabilized hydrides, are discussed as a possible alternative to the on-board reversible metal hydrides. Specific examples are given for aluminum hydride (AlH3) with a few details on controlling hydrogen rates and regeneration. A more thorough review of hydrogen and methods of hydrogen storage can be found elsewhere.3,4
The prospect of using metal hydrides as a hydrogen source for a fuel cell or internal combustion engine may appear promising due to their potential for high volumetric and gravimetric capacities. Ultimately, however, the true utility of these hydrides must be evaluated on a system basis (i.e. the complete tank design with valves, heat exchangers, etc.) rather than just a materials basis. Therefore, much more system and life cycle and safety analyses are necessary to understand the true strengths and weakness of different metal hydridehydrogen storage systems.
2. Metal hydride fundamentals
There are many different ways of categorizing hydrogen storage materials which seem to change frequently as new materials are developed that fall outside the previous classification system. This tutorial review is focused on compounds that decompose endothermically (with the exception of Ca(AlH4)2, which is slightly exothermic in the first decomposition step), therefore, all enthalpy values reported here correspond to the heat input required to release the hydrogen. In general hydrogen storage materials can be divided into two categories, those that bind molecular hydrogen to surfaces via weak van der Waals interactions (typically <10 kJ/mol H2 (ref. 5, and references therein) in a process known as physisorption (e.g. activated carbons) and those that trap atomic hydrogenvia a strong (typically >10 kJ/mol H2) chemisorption process (e.g.metal hydrides, complex hydrides and chemical hydrides). Storage media that trap hydrogen in a chemical process come in two flavors, reversible and non-reversible. The label “non-reversible” implies that the material does not readily hydrogenate under reasonable pressure and temperature conditions. The barrier to hydrogenation can be thermodynamic, kinetic or both. Therefore, the non-reversible hydrides are found with a range of decomposition enthalpies from the weak (e.g. AlH3ΔH = 7 kJ/mol H2) to the strong (e.g. LiBH4ΔH = 67 kJ/mol H2 for partial decomposition). Reversible hydrogen carriers typically have moderate decomposition enthalpies 20–55 kJ/mol H2.Much of the current activity on metal hydrides for hydrogen storage is focused on developing new materials that can meet the demands of a low temperature automotive fuel cell system. For most automotive applications the hydride must have a high gravimetric (>6 wt% H2 or 2 kW h−1 kg−1) and volumetric (>0.045 kg H2 L−1 or 1.5 kW h L−1) hydrogen density and must also satisfy the kinetic and thermodynamic requirements for hydrogen uptake and release. In a typical reversible metal hydride the hydrogen atoms are chemically bonded to the metal and the hydrogen is released by a change in the thermodynamic conditions (decreasing pressure or increasing temperature). The hydrogenation reaction is exothermic, and therefore the release of H2 requires the addition of energy, generally in the form of heat. Idealy this energy can be supplied by the waste heat from the power source (e.g. engine or fuel cell). However, proton exchange membrane (PEM) fuel cells operate at low temperature and can supply heat at a temperature not much greater than 80 °C. This low temperature limit makes it challenging to meet the fuel flow needs for a 100 kW PEM fuel cell, which requires a H2 pressure greater than 1 bar (thermodynamic requirement) and a flow rate of around 2 g H2 s−1 (kinetic requirement). It is important to note that additional heat can be added by combusting some of the stored hydrogen. However, this process is undesirable since it consumes a useful form of energy (as opposed to waste heat) and reduces the overall storage capacity of the system. There are no known materials that have the desired high capacity, thermodynamics and kinetics for this application and therefore much of the current research is focused on fundamentals of metal hydrides and materials discovery.
Metal hydride thermodynamics are typically measured by calorimetric methods or by performing pressure–composition–temperature (PCT) measurements. In the PCT procedure aliquots of H2 are added (or removed) from a reactor with a calibrated volume. After the system has equilibrated the equilibrium pressure is recorded and the hydrogen uptake (or release) from the sample is determined from the pressure change. This process is repeated until the material is fully hydrogenated (or dehydrogenated). Fig. 2(a) shows an example of three hypothetical PCT isotherms that are overlayed onto a metal hydride phase diagram. At the early stage of hydrogenation (e.g.H/M ≤ 0.1) the hydrogen is initially incorporated into the metal (M) in a solid solution. After a critical concentration is reached the hydride phase nucleates forming a two-phase system (M + MH) and the isotherm plateaus (a consequence of the Gibbs phase rule). As additional hydrogen is added, the hydride phase (MH) grows at the expense of the M phase until the M phase is completely exhausted. The thermodynamics of the hydrogenation reaction can be determined by plotting the plateau pressures and temperatures in a van’t Hoff plot (usually plotted as ln P vs. 1/T) as shown in Fig. 2(b). At equilibrium, the relationship between H2 pressure (P) and temperature (T) is given by the van’t Hoff equation:
R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(P298 K) = ΔG298 K/T = ΔH298 K /T−ΔS298 K ln(P298 K) = ΔG298 K/T = ΔH298 K /T−ΔS298 K | (1) |
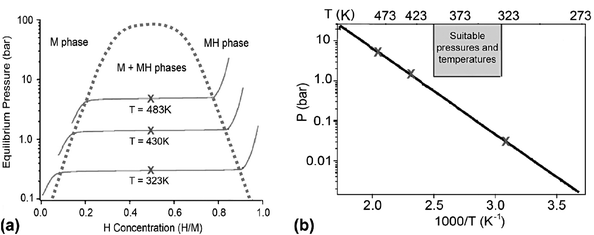 | ||
| Fig. 2 (a) Pressure-composition phase diagram for a hypothetical metal hydride showing two-phase region and PCT isotherms. (b) van’t Hoff plot showing a linear fit to the equilibrium pressure/temperature data (X) and a region of suitable pressures and temperatures for on-board hydrogen storage (box). | ||
3. Reversible hydrides: complex hydrides
Conventional metal hydrides that can readily supply hydrogen at room temperature typically have storage capacities <2 wt%6 and cannot meet the demands of an automotive PEM fuel cell. In the interest of high gravimetric capacities, much of the current metal hydride research is focused on hydrides composed primarily of light elements (Z≤ 13). Recently much attention has been devoted to the complex hydrides, many of which have appreciable gravimetric hydrogen storage capacities as shown in Fig. 1. In a complex hydride the hydrogen atoms are covalently bonded to a central atom in an anion complex (e.g. [AlH6]3−, [AlH4]−, [BH4]−, [NH2]−) and stabilized by a cation, typically an alkali (e.g.Li) or alkaline earth metal (e.g.Mg) or a transition metal (e.g. Zn). In general, the complex hydrides of interest for hydrogen storage applications can be distilled down to three categories: tetra and hexahydroaluminates (alanates), tetrahydroboartes (borohydrides) and amides. A brief introduction to the complex hydrides is presented here, but a more thorough review of these materials can be found elsewhere.73.1 Alanates
The group of complex hydrides that have received the most attention over the past decade are the alanates, which can be found as hexahydroaluminates (Al octahedrally coordinated by H) or tetrahydroaluminates (Al tetrahedrally coordinated by H). A comprehensive summary of the known structures of all aluminum-based hydrides (including alanes) was recently prepared by Hauback.8 One of the best performing and most well studied reversible complex hydride is catalyzed sodium alanate (NaAlH4 + 2% Ti):9| 3NaAlH4↔ Na3AlH6 + 2Al + 3H2↔ 3NaH + 3Al + 9/2H2 | (2) |
In addition to NaAlH4 and Na3AlH6 there are a number of other alanates capable of rehydrogenation under moderate pressures and temperatures including Na2LiAlH6, KAlH4, K3AlH6, K2LiAlH6 and K2NaAlH6. However, all of these compounds have lower gravimetric hydrogen densities and are more stable than sodium alanate. On the other hand, there are quite a few alanates that are known to be less stable than sodium alanate, which also exhibit high gravimetric hydrogen densities such as LiAlH4, Li3AlH6, Mg(AlH4)2, Ca(AlH4)2, Ti(AlH4)4 among others. Despite their promising capacities, these materials are not known to be reversible under moderate pressure conditions (see section on non-reversible hydrides below).
3.2 Borohydrides
Recently, the quest to find a high capacity reversible hydride has shifted more to the amides and the borohydrides. The crystal structures of the various borohydrides can be found in a review by Orimo et al.7 and references therein. In general, the alkali borohydrides have a large decomposition enthalpy and require prohibitively high temperatures for H2 release. One of the more popular borohydrides, LiBH4, decomposes at temperatures >380 °C to LiH and B and releases 13.5 wt% hydrogen in the following reaction:| 2LiBH4→ 2LiH + 2B + 3H2 | (3) |
There has also been considerable recent interest in the alkaline earth and transition metal borohydrides (Mg(BH4)2,12 Ca(BH4)2,13 Zr(BH4)4,14 Zn(BH4)214), which are much less stable that the Group 1 borohydrides. These materials have demonstrated promising capacities at reasonable decomposition temperatures, but similar to the less stable alanates, there remains a problem of reversibility. Some evidence for reversibility in these less stable borohydrides was recently demonstrated by Ronnebro et al. who prepared Ca(BH4)2 directly from CaB6, CaH2 and H2 gas at pressures of 700 bar and 400–440 °C.13
3.3 Amides
Another class of materials that has received much attention in recent years is the amides. The crystal structures of the various amides and imides of interest can also be found in the review by Orimo et al.7 and references therein. The reversible reaction that initially ignited a flurry of activity on the amides is a two-step reaction between lithium amide and lithium hydride to yield a theoretical capacity of 10.3 wt% H2:15| LiNH2 + 2LiH ↔ Li2NH + LiH + H2↔ Li3N + 2H2 | (4) |
| Mg(NH2)2 + 2LiH ↔ Li2Mg(NH)2 + 2H2 | (5) |
4. Reversible hydrides: new approaches
None of the known reversible complex hydride storage systems come close to meeting the three primary targets (capacity, thermodynamics and kinetics) necessary for a practical on-board storage system. Since there are established methods to improve kinetics (e.g. reduce particle size, catalysts, etc.), current efforts are primarily focused on developing high capacity reversible hydrides that can meet the thermodynamic target. As a result a number of promising new approaches are being explored to tune the (de)hydrogenation reaction thermodynamics of the complex hydrides.Since the purpose of the hydride is to supply H2 to a fuel cell, the ideal thermodynamics are generally determined from the decomposition reaction. As previously mentioned, hydrogen must be released at low temperature (T≈ 80 °C) and useful pressures (P80 °C≈ 1–10 bar). Based on these values the decomposition enthalpy for a hypothetical material with ideal thermodynamics can easily be calculated using eqn (1). For direct hydrogenation with H2 gas, the entropy term is typically dominated by the entropy of the gas (SH2 = 131 J mol−1K−1). Using this value as an approximation for the entropy change, the decomposition enthalpy of the ideal reversible material is around 39–46 kJ/mol H2. The thermodynamics of most hydrides fall outside this range and are either too stable (evolve H2 at high temperature) or too unstable (requiring extremely high pressures to rehydrogenate) for a low temperature PEM fuel cell.
4.1 Thermodynamic tuning
One simple approach to meeting both the capacity and the thermodynamic requirements is to develop general methods to tune the thermodynamics of the high capacity complex hydrides. This is a relatively straightforward process with interstitial metal hydrides where the initial metal can be alloyed with a second metal to alter the hydride stability. This process is a little trickier with the complex hydrides, which are composed of a metal hydride anion complex (e.g. [MH6]3− or [MH4]− where M = B, Al) and a metal cation (e.g.Li, Mg) that acts to stabilize the structure. Altering the decomposition thermodynamics therefore involves making changes to either the cation or the anion complex.17,18,19An example of thermodynamic tuning through cation substitution is found in the hexahydroaluminates. These compounds favor a structure with two different cation sites such as the fcc structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m).17 It was recently demonstrated that two different alkali cations (M and M′) could be accommodated on different sites to yield a bialkali alanate of the form M2M′AlH6 as shown in Fig. 3. Changing the cation composition significantly alters the decomposition thermodynamics. As an example, the hexahydroaluminate, Na3AlH6, has Na ions located in two different cation sites and has an equilibrium H2 pressure of 63 bar at 245 °C. When the Na cation is replaced with a Li cation on the octahedra site the bialkali alanate Na2LiAlH6 is formed. This new compound has an equilibrium pressure of 32 bar at 245 °C, which is a pressure reduction by almost fifty percent. Although some control over thermodynamics is clearly possible through cation substitution, only a few specific combinations have been shown to form stable compounds.
m).17 It was recently demonstrated that two different alkali cations (M and M′) could be accommodated on different sites to yield a bialkali alanate of the form M2M′AlH6 as shown in Fig. 3. Changing the cation composition significantly alters the decomposition thermodynamics. As an example, the hexahydroaluminate, Na3AlH6, has Na ions located in two different cation sites and has an equilibrium H2 pressure of 63 bar at 245 °C. When the Na cation is replaced with a Li cation on the octahedra site the bialkali alanate Na2LiAlH6 is formed. This new compound has an equilibrium pressure of 32 bar at 245 °C, which is a pressure reduction by almost fifty percent. Although some control over thermodynamics is clearly possible through cation substitution, only a few specific combinations have been shown to form stable compounds.
 | ||
| Fig. 3 Structure of bialkali alanate M2M′AlH6 showing large (M) and small (M′) alkali metal cations along with AlH6 octahedra. | ||
More recently a number of researchers have been exploring the alkali and alkaline earth metal mixed cation alanates (e.g. LiMg(AlH4)3, LiMgAlH6).20,21 Although some of these materials exhibit favorable decomposition temperatures at the moment they have not been demonstrated as reversible hydrides.
The size and electronegativities of the cation and anion complexes determine the structure and bonding of the compound. A direct correlation exists between cation electronegativity and bond strength, which can be seen as a function of decomposition temperature12 or reaction enthalpy, as shown in Fig. 4. When the average cation electronegativities are used for the bialkali alanates the plot reveals a linear relationship between ΔH and Pauling electronegativity for the hexahydroaluminates. A similar trend in reaction enthalpy is observed in the metal-borohydrides.22 Currently there is much interest in destabilizing the metal borohydrides by cation substitution to produce mixed-metal borohydrides (e.g. LiK(BH4)2).23
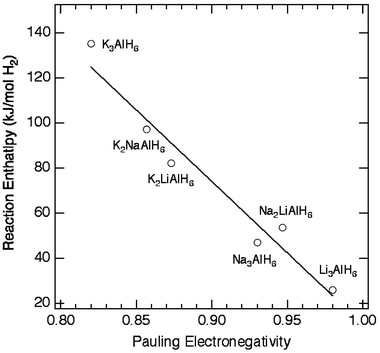 | ||
| Fig. 4 Correlation between reaction enthalpy (ΔH) and Pauling electronegativity of the cation for a few hexahydroaluminates. For the bialkali alanates (M2M′AlH6) the average cation electronegativity was used. | ||
Thermodynamic tuning can also be achieved by altering the anion complex. Brinks et al. recently demonstrated a significant destabilization associated with fluorine substitutions in the hydrogen sublattice of Na3AlH6.19 The mixed hydride fluoride Na3AlH6−xFx (with x≈ 3) exhibits a much greater equilibrium pressure (25 bar) than the pure hydride (1.5 bar) at 120 °C.19,24 An alternative route to changing the anion complex is to partially replace Al with a different metal (e.g.B). There has been considerable interest recently in the mixed B–N systems with the preparation of multiple new quaternary phases (Li4(NH2)3BH4).25 Preliminary data suggests these materials are destabilized and release hydrogen at lower temperatures than pure LiBH4, but have limited reversibility.
4.2 Destabilization
Another approach being explored to improve the dehydrogenation thermodynamics (i.e. reducing decomposition enthalpy) of the complex hydrides involves incorporating a second species into the reaction to stabilize the reaction product (Fig. 5).26 It is important to note that in this approach the individual compounds are not destabilized. Rather, the overall reaction is destabilized by decomposing to a more stable alloy (AHx + yB → ABy + x/2H2) rather than the elements (AHx→ A + x/2H2). Since the reaction now stops at a lower energy state the total enthalpy of the reaction is reduced and less energy (heat) is required for decomposition. The amide/LiH reaction (reaction (4)) is an example of a destabilized reaction since it forms a stable Li3N product as opposed to the elements Li and N. Vajo et al. have demonstrated this concept with a number of different hydride systems (LiH/Si, MgH2/Si, LiBH4/MgH2).27 Recent computational studies have identified a number of new destabilized systems with high hydrogen capacity and favorable thermodynamics.28,29 Although this approach has proven to be effective, the addition of a second species that is not a hydride ultimately reduces the total gravimetric capacity of the system. Many of the recently proposed destabilized schemes utilize two hydrides (e.g.reactions (4) and (5)) and thereby maintain a high theoretical hydrogen capacity.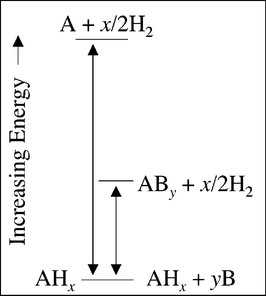 | ||
| Fig. 5 Energy diagram of the destabilization scheme showing the original decomposition reaction (left) and the destabilized reaction (right). The reaction enthalpy is reduced by stabilizing the final state as an alloy. | ||
The primary challenge with this approach is obtaining a decomposition reaction that occurs along the desired reaction pathway. In the simplest case the decomposition of the hydride (AHx) and the alloy formation (ABy) must occur simultaneously in order to achieve the desired thermodynamic effect. This becomes more complicated when two hydrides are involved and multiple reaction pathways are possible. One of the keys to this process is engineering a system with extremely fast kinetics to allow decomposition and alloy formation to occur simultaneously and avoid local thermodynamic minima. Much of the ongoing effort in this area involves improving the reaction kinetics by using nanocrystalline material incorporated into nanoporous frameworks to increase surface area and avoid agglomeration.27
5. Reversible hydrides: the heat problem
One of the often-overlooked issues concerning the thermodynamics of metal hydride reactions is the vast quantity of heat generated during hydrogenation (refueling). Current research efforts are primarily focused on optimizing the decomposition thermodynamics so that the H2 is released at reasonable pressures and temperatures. As previously mentioned, the ideal hydride has a decomposition enthalpy of around 39–46 kJ/mol H2 (H2 pressure of 1–10 bar at 80 °C). Therefore, in a system that accommodates 5 kg of H2 the total energy released during hydrogenation is around 100 MJ. Although this is a lot of wasted energy, the real problem arises when we consider the rapid refueling rates necessary to complete the process in approximately 3 minutes (1.67 kg H2/min). The 100 MJ of heat generated during refueling will need to be dissipated at a rate of 0.5 MW, which is an extremely challenging engineering problem that is not likely to be solved by moderate improvements in heat exchangers.30 This problem may be alievated somewhat by using hybrid (metal hydride/compressed gas) tanks or by increasing the time for refueling (probably not going to be a popular option). A third option is to exchange the spent material with fresh hydrogenated material and allow the spent fuel to recharge slowly off-board. Off-board hydrogenation would also allow for the recovery of some of the exothermic heat released during the reaction. The importance of this point is that the hydrides that have traditionally been considered “on-board reversible” may need to be taken off-board for hydrogenation to satisfy the short refueling time. Therefore the search for the ideal material should not be restricted to the reversible hydrides, but should also include many non-reversible hydrides, which have traditionally been overlooked because they are too unstable and not easily hydrogenated.6. Non-reversible hydrides: kinetically stabilized hydrides
A number of high capacity hydrides are not easily rehydrogenated due to limitations of thermodynamics, kinetics or both. The non-reversible hydrides have received less attention in recent years due to the cost of regeneration and the off-board refueling requirement. However, a number of recent studies have demonstrated that some of these compounds may offer higher capacities and lower decomposition temperatures than the reversible hydrides. Currently, much interest is focused on the ammonia–boranes and progress in this area can be found in a few recent reviews31–33 and references therein. Here we will limit the scope of discussion to metal hydrides with very small decomposition enthalpies.The kinetically stabilized hydrides are characterized by a low decomposition enthalpy (<30 kJ mol−1) and although the decomposition reaction is thermodynamically favorable at room temperature, H2 evolution is limited by the kinetics. Depending upon the composition, these trapped metastable phases have shelf lives from only a few minutes to many years. In fact, it was recently demonstrated that a sample of metastable α-AlH3 retained 96% of its original hydrogen content after thirty years of storage in air.30 Although the mechanism(s) responsible for stabilizing these materials is not well understood, a few likely possibilities include slow hydrogen/metal diffusion and surface barriers that inhibit the formation of molecular hydrogen. A few examples of kinetically stabilized hydrides (AlH3, Li3AlH6, Mg(AlH4)2, Ca(AlH4)2, etc.) are shown in the van’t Hoff plot in Fig. 6 along with a few well known reversible hydrides.20,24,34–36
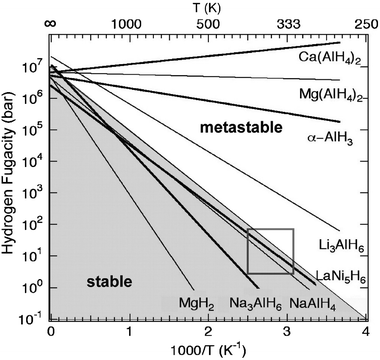 | ||
| Fig. 6 Approximate van’t Hoff plots for several reversible and non-reversible hydrides.20,24,34–36 The compounds in the lower left (shaded) region are stable while the compounds in the upper right are unstable or metastable at ambient pressure and temperature. The box represents the thermodynamic target for “on-board reversible” hydrides. | ||
6.1 Advantages
This new class of kinetically stabilized hydrides offers a number of advantages over the traditional reversible hydrides. First, the decomposition enthalpy is small (5–15 kJ/mol H2), which reduces the heat required to release the hydrogen at practical pressures. Therefore, there is less demand on the waste heat from the PEM fuel cell. Second, the large driving force for decomposition ensures that many of the kinetically stabilized hydrides will exhibit rapid decomposition rates at low temperatures (80–100 °C).6.2 Challenges
The metastable hydrides also present a new set of challenges. The first issue is developing a material with a simple temperature dependent decomposition so that the H2 evolution can be easily controlled. The material must be able to meet the rapid rate requirements, but also avoid unwanted H2 evolution at low temperatures (<40 °C). Hydrogen storage systems based on the reversible metal hydrides control the rate of decomposition through the tank pressure, which is kept near equilibrium (see eqn (1)). When the H2 evolution rate exceeds the fuel cell demand, the pressure in the tank rises above the plateau pressure and the reaction stops. On the other side, as the demand increases and the gas is consumed, the pressure is reduced below the plateau value and the H2 evolution resumes. For a metastable hydride the equilibrium pressure is much too high to limit decomposition and the release of H2 must be controlled kinetically. The desired H2 evolution rates range from 0–0.02 g H2 s−1kW−1 and must be achieved within the available temperature window of 40–100 °C. More research is needed to develop new materials and tools for a thermally controlled hydrogen storage system.The second and possibly more challenging problem is regeneration. The metastable hydrides cannot easily be formed by direct hydrogenation at low pressure and temperature. Therefore, these materials are generally prepared by an organometallic reaction typically involving a complex hydride (LiAlH4) or binary hydride (MgH2) with a metal chloride (AlCl3). These expensive one-way reactions yield stable byproducts that are energetically costly to recycle. New efficient methods to regenerate the kinetically stabilized hydrides directly from the spent material are needed to make these materials a viable candidate for automotive fuel cell applications.
7. Non-reversible hydrides: aluminum hydride
One of the more promising kinetically stabilized hydrides is aluminum hydride (AlH3), which has a volumetric hydrogen capacity (148 g L−1) greater than twice that of liquid hydrogen and a gravimetric hydrogen capacity exceeding 10 wt%. Aluminum hydride forms a number of different crystallographic phases,8 all of which are thermodynamically unstable under ambient conditions. Military research efforts on aluminum hydride date back to the 1960s when independent groups in the United States and the Former Soviet Union first synthesized the crystalline form of α-AlH3. The general interest in this highly energetic compound was primarily for solid rocket propellants and explosives. More recently, a renewed interest in AlH3 has developed since it exhibits rapid hydrogen evolution rates at low temperature (<100 °C).37 The decomposition of AlH3 occurs by the following single step reaction:| AlH3→ Al + 3/2H2. | (6) |
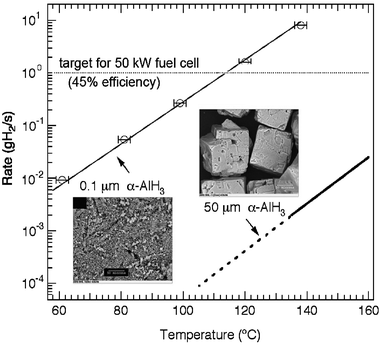 | ||
| Fig. 7 Hydrogen evolution rates and corresponding scanning electron microscopy images for α-AlH3 with crystallite sizes of 0.10 μm and 50 μm (dashed line represents extrapolated region). The rates are based upon 10 kg H2 (∼100 kg AlH3). The dotted line represents the fuel flow target 1.0 g H2 s−1 for a 50 kW fuel cell with 45% efficiency. | ||
7.1 Controlling decomposition
Kinetic studies of AlH3 have also demonstrated that the stability and decomposition rates vary significantly with crystallite size (Fig. 7). The fine crystallites (0.1 μm) of α-AlH3 (lower inset of Fig. 7) exhibit a decomposition temperature below 60 °C while the material composed of 50 μm crystallites (upper inset of Fig. 7) decomposes at temperatures above 130 °C with a H2 evolution rate more than two orders of magnitude slower. These results demonstrate that the temperature-dependent H2 rates can be tuned by controlling the crystallite size. A better understanding of the role of size and morphology will be critical for the development of a kinetically stabilized hydrogen storage system.In addition to meeting the rapid H2 rates at low temperature a suitable material must also exhibit controlled decomposition. Isothermal and intermittent isothermal decomposition from α-AlH3 are shown in Fig. 8(a) and (b), respectively. This plot shows the total H2 evolved (circles) and the first derivative of the total H2 evolved (line), which corresponds to the H2 rate for 100 kg of material. The H2 evolution rate can be slowed and even stopped by reducing the reactor temperature to room temperature. When the reactor is heated back to the decomposition temperature (120 °C), the rate returns to the rate before the temperature was reduced. In fact, regardless of whether the decomposition occurs in one step (Fig. 8(a)) or after multiple starts and stops (Fig. 8(b)) the general Lorentzian shape of the rate curves are strikingly similar. This suggests that the full spectrum of H2 evolution rates (e.g. 0.0–1.0 g H2 s−1) can likely be obtained using a 100 kg α-AlH3 with a variable temperature hydride bed operating at 23 ≤T≤ 115 °C. However, heating and cooling a large thermal mass such as a hydride bed is unreasonable. A more sensible approach may be to use a pumpable system (possibly a slurry) where the AlH3 is transferred to a reactor, decomposed, and returned to a separate region of the tank for spent fuel. Separation of spent and unreacted fuel also simplifies the refuelling process.
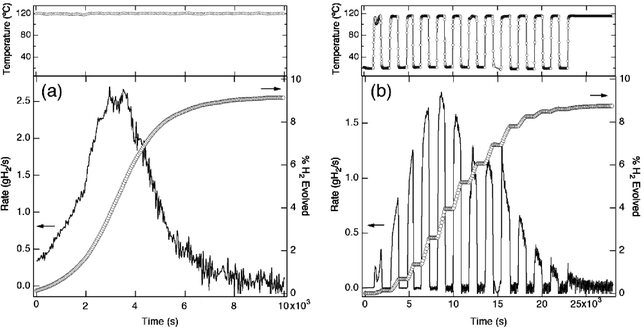 | ||
| Fig. 8 (a) Isothermal and (b) “on/off” decomposition at 120 °C showing the percent H2 evolved, the rate of H2 evolution based on 100 kg of AlH3 and the sample temperature. | ||
7.2 Regeneration
Despite the recent promising results on the decomposition of AlH3, the challenge of regeneration remains a major obstacle. This problem is clearly illustrated in the pressure-temperature phase diagram for α-AlH3 shown in Fig. 9. Thermal stability studies of α-AlH335 were used to calculate the equilibrium pressures between α-AlH3 and the elements (Al + H2) at various temperatures. These data were combined with high-pressure experimental results from Konovalov and Bulychev.39 The H2 equilibrium pressure at room temperature is approximately 7 kbar,40 suggesting that extremely high pressures are required to form this material directly from aluminum and H2 gas. The equilibrium pressures of many other kinetically-stabilized hydrides (e.g. Mg(AlH4)2) are likely to be even greater than 7 kbar at room temperature. | ||
| Fig. 9 Pressure–temperature phase diagram for α-AlH3. Equilibria were determined from high-pressure experiments (≥300 °C)39 and thermodynamic data (≤25 °C).35 | ||
Although direct hydrogenation of aluminum is extremely difficult it may be possible to regenerate AlH3 through other routes. Currently, the most common method of preparing AlH3 is by reacting lithium alanate (LiAlH4) and aluminum chloride (AlCl3) in an ethereal reaction. This solution is filtered and dried (desolvated) to form crystalline AlH3.38 Although there are a number of different possible methods to regenerate the kinetically stabilized hydrides, one of the most obvious is to simply recycle the byproducts (Al + metal hydride + metal chloride). However, the metal chlorides are extremely stable and will require a substantial energy input to break apart. Even if the recycling reaction were to make use of the formation energies of the precursors (LiAlH4 + AlCl3) the energy cost would still be significant. A perfectly efficient recycling reaction for AlH3 would require an energy input of 167 kJ/mol H2, which is equivalent to approximately 70% of the fuel energy contained within AlH3 (based on the lower heating value of H2).
Other approaches to regenerate the kinetically stabilized hydrides such as AlH3 include electrochemical hydrogenation. In this process an electrolytic potential is used to increase the hydrogen activity and drive the hydrogen into the host material. Since the electrolytic potential scales as the natural log of the pressure (V∝ ln[P]) the thermodynamic requirement can be met by a relatively low voltage. Zidan et al. at Savannah River National Laboratory are actively using this approach to regenerate AlH3.
Another route to forming the kinetically-stabilized hydrides under mild conditions involves a two-step process: first a stable intermediate hydride phase, such as an adduct, is formed by direct hydrogenation, then the intermediate phase is carefully decomposed (separated) to recover the pure hydride. Complex metal hydrides and aluminum hydride form stable amine complexes (e.g. AlH3·C6H12N2) and solvated phases (AlH3·nTHF). These stabilized phases form under much more mild pressure and temperature conditions than the pure hydride. There are also a few examples of solvated hydrides or adducts (e.g. AlH3·N(C2H5)3) that can be separated to recover the metastable hydride.
Recently, Reilly et al. discovered a reversible hydrogenation reaction using Ti-catalyzed Al (Al*) powder and triethylenediamine (TEDA or C6H12N2) in tetrahydrofuran (THF) to form the alane adduct (AlH3·TEDA) at low pressure (<20 bar) and room temperature:41
| Al* + TEDA + 3/2H2↔ AlH3·TEDA | (7) |
Reaction (7) is a useful step in the regeneration of AlH3, but is also an interesting example of a unique hydrogen storage compound, an organic-metal hydride. In this system an organic ligand (TEDA) stabilizes an unstable hydride (AlH3). The hydrogenation reaction is completely reversible with a theoretical capacity of 2.1 wt% and a plateau pressure of 1.5 bar at 72 °C.41 During cycling the hydrogen atoms are reversibly added or removed from the metal atom, similar to a conventional metal hydride, and the organic molecule remains relatively unchanged. Despite the low capacity of this particular reaction, a number of potentially high capacity reactions can be envisioned. For example, multiple hydrides may be attached to a single ligand or it may be possible to cycle hydrogen from both the metal and the stabilizing organic ligand. Although these examples are considerably more complex and may be somewhat far-fetched, the proof of concept has been demonstrated and it is possible that a organic-metal hydride could yield reversible capacities greater than 6 wt%.
Conclusions
Metal hydrides have the most potential for meeting the US DOE system targets for automotive hydrogen storage. Recent materials development and basic hydride research has led to significant improvements in the metal hydride properties (thermodynamics and kinetics). However, there are currently no materials that can adequately meet the demands of a low-temperature automotive fuel cell. Despite the rapid advances with the reversible complex hydrides, onboard hydrogenation will likely remain a challenge due to the heat problem associated with extremely fast charging times (3–5 minutes). The kinetically stabilized metal hydrides with their low decomposition enthalpy and rapid low temperature kinetics offer some new hope, but also present a number of new challenges such as limiting unwanted H2 evolution and regeneration.The development of a high capacity hydride with suitable properties is simply the first step to solving the automotive hydrogen storage problem. The utility of any new hydride will ultimately be determined not by its material performance, but by its system performance. Although this area is still emerging, there is a need for more life cycle analysis, safety, environmental and engineering studies of new “candidate materials” to evaluate the performance of these new hydrides on a system level. New concepts are needed for systems based on kinetically stabilized hydrides and new studies are needed to investigate hydride slurries, off-board refueling, low-temperature decomposition (shelf life) and safety.
Acknowledgements
This work was supported through the Metal Hydrides Center of Excellence, Office of Energy Efficiency and Renewable Energy, and the Hydrogen Fuel Initiative, Office of Basic Energy Sciences, US Department of Energy under Contract No. DE-AC02-98CH1-886.References
- Basic Research Needs for Electrical Energy Storage—a report of the Basic Energy Sciences Workshop on Electrical Energy Storage, April 2–4, 2007. U.S. Department of Energy Report, July 2007.
- DOE Hydrogen, Fuel Cells & Infrastructure Technologies Program Multi-Year Research, Development, and Demonstration Plan, Hydrogen Storage Technical Plan, 2007 http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/.
- L. Schlapbach, MRS Bull., 2002, 27, 675 CAS; R. S. Irani, MRS Bull., 2002, 27, 680 CAS; J. Wolf, MRS Bull., 2002, 27, 684 CAS; R. C. Bowman, Jr and B. Fultz, MRS Bull., 2002, 27, 688; J. M. Joubert, M. Latroche and A. Percheron-Guegan, MRS Bull., 2002, 27, 694 CAS; E. Akiba and M. Okada, MRS Bull., 2002, 27, 699 CAS; A. Zuttel and S. Orimo, MRS Bull., 2002, 27, 705; B. Bogdanovic and G. Sandrock, MRS Bull., 2002, 27, 712 CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS.
- M. Hirscher and B. Panella, Scr. Mater., 2007, 56, 809 CrossRef CAS.
- G. Sandrock, J. Alloys Compd., 1999, 293–295, 877 CrossRef CAS.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS.
- B. C. Hauback, Z. Kristallogr., 2008 Search PubMed , in press.
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 253–254, 1 CrossRef CAS.
- S. S. Srinivasan, H. W. Brinks, B. C. Hauback, D. Sun and C. M. Jensen, J. Alloys Compd., 2004, 377, 283 CrossRef CAS.
- S. Chaudhuri, J. Graetz, A. Ignatov, J. J. Reilly and J. T. Muckerman, J. Am. Chem. Soc., 2006, 128, 11404 CrossRef CAS.
- T. Matsunaga, F. Buchter, K. Miwa, S. Towata, S. Orimo and A. Zuttel, Renewable Energy, 2008, 33, 193 CrossRef CAS.
- E. Ronnebro and E. H. Majzoub, J. Phys. Chem. B, 2007, 111, 12045 CrossRef.
- Y. Nakamori, H. W. Li, K. Miwa, S. Towata and S. Orimo, Mater. Trans., 2006, 47, 1898 CrossRef CAS.
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, Nature, 2002, 420, 303.
- W. Luo and S. Sickafoose, J. Alloys Compd., 2006, 407, 274 CrossRef CAS.
- J. Graetz, Y. Lee, J. J. Reilly, S. Park and T. Vogt, Phys. Rev. B, 2005, 71, 184115 CrossRef.
- E. Ronnebro and E. H. Majzoub, J. Phys. Chem. B, 2006, 110, 25686 CrossRef.
- H. W. Brinks, A. Fossdal and B. C. Hauback, J. Phys. Chem. C, 2008, 112, 5658 CrossRef CAS.
- M. Mamatha, B. Bogdanovic, M. Felderhoff, A. Pommerin, W. Schmidt, F. Schuth and C. Weidenthaler, J. Alloys Compd., 2006, 407, 78 CrossRef CAS.
- X. Tang, S. M. Opalka, B. L. Laube, F. J. Wu, J. R. Strickler and D. L. Anton, J. Alloys Compd., 2007, 446–447, 228 CrossRef CAS.
- Y. Nakamori, H. W. Li, K. Kikuchi, M. Aoki, K. Miwa, S. Towata and S. Orimo, J. Alloys Compd., 2007, 446–447, 296 CrossRef CAS.
- E. A. Nickels, M. O. Jones, W. I. F. David, S. R. Johnson, R. L. Lowton, M. Sommariva and P. P. Edwards, Angew. Chem., Int. Ed., 2008, 47, 2817 CrossRef CAS.
- B. Bogdanovıc, R. A. Brand, A. Marjanovic, M. Schwickardi and J. Tolle, J. Alloys Compd., 2000, 302, 36 CrossRef CAS.
- P. A. Chater, W. I. F. David, S. R. Johnson, P. P. Edwards and P. A. Anderson, Chem. Commun., 2006, 2439 RSC.
- J. J. Vajo, F. Mertens, C. C. Ahn, R. C. Bowman, Jr and B. Fultz, J. Phys. Chem. B, 2004, 108, 13977 CrossRef CAS.
- J. J. Vajo and G. L. Olson, Scr. Mater., 2007, 56, 829 CrossRef CAS.
- S. V. Alapati, J. K. Johnson and D. S. Sholl, Phys. Chem. Chem. Phys., 2007, 9, 1438 RSC.
- C. Wolverton, D. J. Siegel, A. R. Akbarzadeh and V. Ozolins, J. Phys.: Condens. Matter, 2008, 20, 064228 CrossRef.
- G. Sandrock, J. Reilly, J. Graetz, W.-M. Zhou, J. Johnson and J. Wegrzyn, Appl. Phys. A, 2005, 80, 687 CrossRef CAS.
- M. H. Matus, K. D. Anderson, D. M. Camaioni, S. T. Autrey and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 4411 CrossRef CAS.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613 RSC.
- H. W. Langmi and G. S. McGrady, Coord. Chem. Rev., 2007, 251, 925 CrossRef CAS.
- P. Claudy, B. Bonnetot and J. M. Letoffe, J. Therm. Anal., 1979, 15, 119 CrossRef CAS.
- J. Graetz and J. J. Reilly, J. Alloys Compd., 2006, 424, 262 CrossRef CAS.
- G. S. Sandrock, L. Suda and L. Schlapbach, in Hydrogen in Intermetallic Compounds II, ed. L. Schlapbach, Springer-Verlag, Berlin, 1992, pp. 197–246 Search PubMed.
- J. Graetz, J. J. Reilly, J. G. Kulleck and R. C. Bowman, Jr, J. Alloys Compd., 2007, 446–447, 271 CrossRef CAS.
- J. Graetz and J. J. Reilly, J. Phys. Chem. B, 2005, 109, 22181 CrossRef CAS.
- A. K. Konovalov and B. M. Bulychev, Inorg. Chem., 1995, 34, 172 CrossRef CAS.
- J. Graetz, S. Chaudhuri, Y. Lee, T. Vogt and J. J. Reilly, Phys. Rev. B, 2006, 74, 214114 CrossRef.
- J. Graetz, S. Chaudhari, J. Wegrzyn, Y. Celebi, J. R. Johnson, W. Zhou and J. J. Reilly, J. Phys. Chem. C, 2007, 111, 19148 CrossRef CAS.
Footnotes |
| † Part of the renewable energy theme issue. |
| ‡ Thermal nitrous oxides (NOx) are an indirect byproduct and are produced when the combustion reaction occurs in the presence of N2. |
| § These precursors are often formed by an initial reaction between 2LiNH2 and MgH2. |
| This journal is © The Royal Society of Chemistry 2009 |