Biology and technology for photochemical fuel production†
Michael Hambourgera, Gary F. Moorea, David M. Kramerb, Devens Gust*a, Ana L. Moore*a and Thomas A. Moore*a
aCenter for Bioenergy and Photosynthesis, and Department of Chemistry and Biochemistry, Arizona State University, Tempe, Arizona 85287-1604, USA. E-mail: gust@asu.edu; amoore@asu.edu; tmoore@asu.edu; Fax: 001 (480) 965-2747; Tel: 001 (480) 965-3461
bInstitute of Biological Chemistry, 289 Clark Hall, Washington State University, Pullman, Washington 99164-6340, USA
First published on 4th November 2008
Abstract
Sunlight is the ultimate energy source for the vast majority of life on Earth, and organisms have evolved elegant machinery for energy capture and utilization. Solar energy, whether converted to wind, rain, biomass or fossil fuels, is also the primary energy source for human-engineered energy transduction systems. This tutorial review draws parallels between biological and technological energy systems. Aspects of biology that might be advantageously incorporated into emerging technologies are highlighted, as well as ways in which technology might improve upon the principles found in biological systems. Emphasis is placed upon artificial photosynthesis, as well as the use of protonmotive force in biology.
 From left to right: back row: Ana L. Moore, Thomas A. Moore, Gary F. Moore, Devens Gust; front row: Michael Hambourger, David M. Kramer | Michael Hambourger has been at ASU since 2003, where he received his PhD, and continues as a post-doctoral researcher working under Thomas A. Moore. Mike is a bird watcher and recreational botanist. |
Gary F. Moore has been at ASU since 2004, where he is completing his PhD working under Ana L. Moore. Gary enjoys the art of organic synthesis. |
David M. Kramer has been at Washington State University since 1995, where he is a professor and fellow of the Institute of Biological Chemistry. Dave enjoys kayak camping in grizzly country and skiing on steep slopes through thick forests. |
Devens Gust has been at ASU since 1975, where he is Foundation Professor of chemistry and biochemistry. Devens would like to pursue interests in a family cattle ranch and woodworking, but has little time to do so. |
Ana L. Moore has been at ASU since 1976, where she is a professor of chemistry and biochemistry. Ana provides a supportive environment for retired greyhounds. |
Thomas A. Moore has been at ASU since 1976, where he is a professor of chemistry and biochemistry, and interim director of the Center for Bioenergy and Photosynthesis. Tom maintains an interest in classical mechanics of the motorcycle type and longs to be a skilled pilot. |
Introduction: global scale energy
Energy is fundamental to civilization. Since the taming of fire and the advent of agriculture, human advancement has largely tracked our ability to convert ever larger quantities of energy into increasingly useable forms. Over the past 150 years, the combustion of fossil fuels has powered innovations in agriculture, medicine, transportation, and communication, improving the standard of living for a sizeable portion of the global population. However, global supplies of fossil fuels are not limitless, and their combustion is clearly linked with ongoing changes to our planet’s climate.1 Indeed, given the cumulative anthropogenic warming since 1750, it would take an increase in the radius of Earth’s orbit of about 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km to rebalance the planetary energy budget.‡ Viewed in this way, human activity since the industrial revolution is of a scale that would be discernible at the solar system level.
000 km to rebalance the planetary energy budget.‡ Viewed in this way, human activity since the industrial revolution is of a scale that would be discernible at the solar system level.Fueled by growth in both population and per-capita gross domestic product (and offset modestly by the more efficient use of energy),2,3 global energy demand is projected to increase by 57% in 26 years (from 14.9 terawatt (TW) in 2004 to 23.4 TW in 2030).4 Satisfying this projected demand will require infrastructure investments of unprecedented scale. Moreover, environmental concerns require that we quickly transition to carbon-neutral energy sources. Towards these ends, numerous technologies will have valuable application within the local context. However, at the global scale, sunlight is one of the few energy sources that can meet projected human energy demands in an environmentally and socially responsible manner. Solar energy reaches the surface of the earth at a rate of ∼120000 TW,2 far exceeding the global rate of human energy consumption.4 Clearly, solar energy has the capacity to provide for human energy needs if efficient, cost-effective systems of solar energy capture, conversion, storage, and utilization can be developed.
Nature provides us with the assurance that solar energy conversion can be performed on such a vast scale. Sunlight is the primary energy input for the majority of life on the planet. Globally, photosynthesis stores solar energy in reduced carbon compounds at a rate of ∼120 TW.5 Likewise, nature uses the process of cellular respiration to efficiently oxidize these ‘solar fuels’, with oxygen typically acting as the terminal electron acceptor. Taken together, photosynthesis and cellular respiration provide the blueprints for human energy infrastructures based upon the conversion of solar to stored chemical energy, which is subsequently released in reaction with molecular oxygen. The requirement for fuels is fundamental given the temporal mismatch between solar irradiance and human energy demands. Indeed our current energy infrastructure is predicated upon the long-term energy storage afforded by solar fuels produced in the distant past (i.e. fossil fuels). Fuel production is also a means of concentrating diffuse solar energy for transportation needs, and affords higher energy densities than are attainable with battery or mechanical storage. Satisfying future human energy demands will require the widespread conversion of contemporary sunlight into solar fuels, using only inexpensive earth-abundant materials in the process. Doing so is one of the few viable means of providing for human needs in an egalitarian and carbon neutral manner.
A basic premise underlying this tutorial review is that over more than 3 billion years nature has evolved elegant solutions to the problem of providing the energy required by biological organisms. However, throughout evolutionary history, nature has always selected for the most reproductively fit individuals, not necessarily those with the attributes most readily adapted to fulfilling human energy needs. In this article we explore aspects of natural energy systems that may enable attainment of the high rates of solar energy transduction needed to power society. We begin with a brief comparison between the natural processes of photosynthesis and cellular respiration and their human-engineered analogs. The remainder of the article focuses primarily on artificial photosynthesis, illustrating its promise for new methods of solar energy conversion, and for the synergistic development of improved fuel cells and related technologies. Lastly, we emphasize some of the lessons that remain to be learned from nature, as well as ways in which human ingenuity can modify the principles of natural systems for enhanced utility.
Biological and technological energy transduction
Photosynthesis and cellular respiration
At heart, photosynthesis depends upon the generation of a charge-separated state using light.6 Actinic photons are absorbed by an antenna network primarily comprised of chlorophylls, carotenoid polyenes, and sometimes linear tetrapyrrole molecules. The resulting excitation energy is transferred to the chlorophyll-based primary electron donor. Here an excited-state electron transfer initiates a cascade of dark electron transfers, resulting in the vectorial movement of an electron and a ‘hole’, the vacancy left by the absence of an electron, to opposite sides of a biological membrane. This high-energy charge-separated state preserves some of the photon energy as chemical potential.In bacterial photosynthesis, this chemical potential gives rise to transmembrane proton pumping, as cyclic electron flow occurs between the bacterial reaction center and the cytochrome bc1 complex. The result of proton pumping is the generation of protonmotive force (pmf), vide infra, the energetic ‘currency’ of biological organisms. Bacteria use pmf to synthesize the ubiquitous energy carrier adenosine triphosphate (ATP). Given the necessity of coupling redox poise with pmf, some organisms also have NAD-linked dehydrogenase enzymes that use pmf directly to drive thermodynamically unfavorable redox processes.
In oxygenic photosynthesis, photochemical charge separation occurs in an analogous manner. Two photons are required for the linear transport of one electron from photosystem II (PSII), through the cytochrome b6f complex, and then through photosystem I (PSI). At the donor side of PSII, the hole is passed to a Mn4O4Ca cluster, the oxygen evolving complex (OEC), which ultimately accumulates four oxidizing equivalents and catalyzes the dissociation of two water molecules to form molecular oxygen and protons. From the reducing side of PSI, the electrons liberated from water are ultimately passed to NADP+, producing NADPH. Concomitant with this linear electron flow, proton pumping generates pmf. Thus a fraction of the absorbed solar energy is stored as redox potential in NADPH, and a fraction as pmf: forms of energy which are both used in the Calvin–Benson–Bassham cycle to produce sugar (a reduced fuel) from CO2. In this simplified ‘Z-scheme’ of oxygenic photosynthesis, light energy is converted into chemical energy and stored in the chemical bonds of biomass. Four photoexcitations each for PSI and PSII are required to produce one O2, two NADPH, and three ATP molecules.7
The theoretical maximum overall solar energy conversion efficiency of oxygenic photosynthesis has been estimated to be ∼9%, based upon factors including the solar spectrum, the stoichiometry of the reaction, and the free energy change associated with the production of glucose from carbon dioxide and water.8 This instantaneous efficiency would only be achievable under low light conditions, where essentially every incident photon of appropriate wavelength can be absorbed and utilized for productive electron transfers. Under full sunlight, natural photosynthesis uses only a portion of these incident photons. Downstream inefficiencies during carbon fixation further reduce the attainable efficiency. Additionally, many photosynthetic organisms have seasonal variations in their photosynthetic rates. Thus, on an annual basis photosynthesis is remarkably inefficient, with energy conversion efficiencies <1% for land-based organisms, and <5% for microalgae.9 Even with these low efficiencies, solar energy converted into fuels via photosynthesis provides essentially 100% of the energy input for life on this planet.
The oxidation of photosynthetically produced biomass, in a process known as aerobic respiration, provides energy for both photosynthetic and non-photosynthetic organisms. In this process, electrons derived from biomass are collected in the coenzyme NADH, which is subsequently oxidized by molecular oxygen. The oxidation of NADH is carried out stepwise at three coupling sites across the inner mitochondrial membrane. The last step of this electron transport chain occurs at cytochrome c oxidase, where molecular oxygen is reduced to water. The large, negative free energy change associated with the oxidation of NADH and reduction of O2 is coupled to charge-uncompensated transmembrane proton pumping at the three coupling sites. ATP synthase uses the resulting pmf to drive the synthesis of ATP. The oxidation of NADH, proton pumping at the coupling sites, and synthesis of ATP are collectively referred to as oxidative phosphorylation. Oxidative phosphorylation occurs with very high energy conversion efficiencies, approaching 90% in some organisms.10 In both photosynthesis and respiration, pmf is used for myriad energy-linked processes, chief of which is ATP synthesis, and thereby powers the metabolic, transport, mechanical, informational, and structural processes of life.
Biochemical half reactions
The electron transfer processes carried out by photosynthesis and cellular respiration can be described in terms of various biochemical half reactions, by convention written as reductions.11 For the conversion of sunlight to fuels (compounds whose oxidation, generally with oxygen, yields useful energy), a fundamental requirement is the oxidation of a low energy electron source, with the production of a high energy reduced chemical species. In oxygenic photosynthesis, water is the ultimate electron donor. In applying the principles of photosynthesis to human systems, water remains an ideal source of electrons due to its low energy content, abundance, and the production of O2 which can be allowed to react on demand with the reduced fuel for the release of energy.12The interconversion between oxygen and water is described by eqn (1), where E°′ is the formal reduction potential.
| O2 + 4H+ + 4e−⇆ 2H2O | (1) |
| E°′ = (1.23 – 0.0591·pH) V vs. NHE |
| 2H+ + 2e−⇆ H2 | (2) |
| E°′ = (0.00 – 0.0591·pH) V vs. NHE |
| 2H2O ⇆ 2H2 + O2 | (3) |
| ΔE°′ = −1.23 V |
| ΔG°′ = 474 kJ mol−1 |
Photovoltaics, electrolyzers, and fuel cells
The same electrochemical processes that underlie biological energy transduction can be replicated in technological energy conversion systems. For the production of solar fuels, first steps are the collection of solar energy and its conversion into a charge-separated state. This process occurs in photovoltaic modules, most commonly made of silicon.13 Photon absorption in the bulk silicon semiconductor leads to exciton migration to a p–n junction where charge separation occurs. The intrinsic electric field of the p–n junction helps to prevent charge recombination, and the separated charges generate electromotive force (emf), resulting in current flow through a wire. Commercially available silicon photovoltaics convert sunlight to electricity with ∼18% energy conversion efficiency, while prototype cells have even higher efficiencies. Aside from silicon photovoltaics, alternative materials have been developed, such as dye-sensitized solar cells14 and organic photovoltaics.15 Compared to silicon photovoltaics, these systems utilize lower cost materials to generate emf, and may be of considerable future utility provided that questions of efficiency and stability can be addressed.In order to store the converted solar energy, photovoltaics can be wired to a water electrolyzer for fuel (H2) production (eqn (3)). Typical alkaline electrolyzers operate in concentrated KOH, utilizing nickel or nickel-coated steel electrodes, and convert electricity to chemical energy in hydrogen with ∼70% energy conversion efficiency. Alternatively, proton exchange membrane (PEM) electrolyzers operate near neutral pH, utilizing noble metal catalysts. Using photovoltaic–electrolyzer technology, it is readily possible to convert sunlight to chemical energy in a fuel with an energy conversion efficiency exceeding 12%. However, at present, the cost and efficiency of these systems limit their applicability for practical, large-scale fuel production.16
Energy release through the oxidation of fuels is equally achievable using current technologies. Fuels can be allowed to react with oxygen via combustion, although the energy conversion efficiency for fuel to electricity conversion in this manner is on average only ∼34%. Alternatively, the fuel and oxygen may react electrochemically in a fuel cell. Using H2 as the fuel, the PEM electrolyzer can function in reverse to oxidize H2 and reduce O2. These two half reactions are mediated via a metallic conductor, generating emf as the energy output. Modern PEM hydrogen fuel cells operate with ∼60% energy conversion efficiency. However, the availability and price of noble metal catalysts and other components, as well as questions of long-term stability, currently limit the widespread use of fuel cell technologies.17
Comparison of biology and technology
As depicted in Fig. 1, many parallels can be drawn between biological and technological energy transduction. Photochemical charge separation is functionally replicated in both solid-state and molecular photoconversion devices. Similarly, water electrolyzers can be used to store this converted solar energy in a fuel, as occurs in oxygenic photosynthesis. Conversely, the oxidation of fuels in a fuel cell is functionally analogous to the mitochondrial process of oxidative phosphorylation. Nevertheless, there are many crucial differences between biological and technological energy transduction, and much that humans can still learn from nature.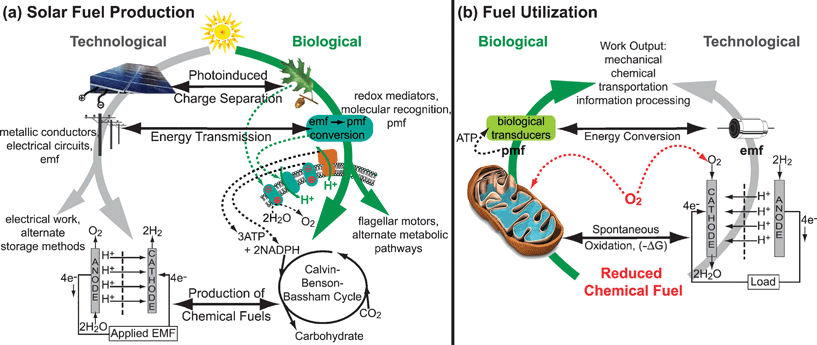 | ||
| Fig. 1 Comparison of biological and technological systems for (a) solar energy conversion to fuels and (b) the utilization of this stored chemical energy. Both biology and technology use functionally analogous steps, indicated by horizontal arrows, during fuel production and consumption. However, a key difference is the use of molecular recognition and pmf in biology, as compared to the electrical circuits and emf used in technological systems. | ||
In photosynthesis, energy is collected as molecular excited states. These excited states migrate among molecules, finally reaching reaction centers where the exciton is split into a hole and an electron by electron transfer to a discrete molecular electron acceptor. The energy loss in splitting the exciton in this way is at most ∼200 mV, considerably less than the cost of splitting the exciton in solid state silicon-based devices.
In biological systems, electrons are carried by discrete molecular species, such as the quinone and cytochrome shuttles operating in the electron transport chains of photosynthetic and mitochondrial membranes. In both photosynthesis and oxidative phosphorylation, precise molecular recognition is used to avoid undesired side reactions and ‘short circuits’, placing electrons exactly where they are needed to achieve the desired biochemical process. Indeed, molecular recognition is a fundamental characteristic of biological systems, effectively replacing the wires of human-engineered systems. At transmembrane sites, proton translocation is tightly coupled to electron flow such that electrochemical energy is translated into a concentration gradient of protons, ΔpH, across the membrane. Moreover, electrical neutrality is not maintained so that an electrical potential, ΔΨ, is also generated. The protonmotive force (pmf) across a membrane is comprised of both the ΔΨ and ΔpH terms. In biology a current of protons, known as proticity, driven by pmf is available to the transducers that span energy-coupling membranes. This pmf is the common denominator underlying all bioenergetic processes in cells. Spectacular molecular motors, such as the bacterial flagella, and ATP synthase which is ubiquitous in cells, are driven by pmf.
A key to the utilization of pmf in biological systems is membrane compartmentalization, allowing the accumulation of ΔΨ and ΔpH. Within these membranes, nature uses discrete sites of catalytic activity. These membrane assemblies, as well as nature’s soluble protein-based catalysts, exhibit a highly organized architecture. Within these constructs factors such as electron flow, proton activity, reactive intermediates, substrate access, product release, and the local dielectric environment are rigorously controlled in order to achieve the desired chemical transformation with high efficiency. Moreover, these natural constructs are often modular, facilitating the self-assembly and self-repair which are some of the most appealing aspects of biological systems. Further, biological systems typically operate at ambient temperatures and pressures and near neutral pH, using only earth-abundant materials. It is these remarkable features of biology that will inspire future innovations in technological energy transduction.
In contrast to biology, technological systems make use of electromotive force (emf) to conduct electrons through a wire. Rather than using molecular recognition, the pathway of electron flow is dictated by the electrical circuit. As has been demonstrated in the electronics industry, human technologies are capable of extremely intricate circuits, directing electrons to precise locations. However, semiconductor fabrication is only achieved with considerable effort, and is (thus far) unable to attain the atomic-scale precision found in biological ‘circuits’. Similarly, technological systems remain unable to take advantage of some of the most extraordinary aspects of biology, such as reactions driven by pmf, precise control of proton activity, and three-dimensional catalyst architectures. In many photovoltaics, efficient exciton migration and exciton splitting remain fundamental challenges to highly efficient devices. In both fuel cells and water electrolyzers, compartmentalization of anodic and cathodic reactions is only achieved by macroscopic separation, and ion-conducting membranes remain an expensive component of these devices.16 Further, many of the most effective technological catalysts rely upon exotic materials, which constrains efforts to scale-up these devices to the terawatt level.
Clearly, nature still has much to teach us. Recent advances in nanotechnology are allowing humans to control matter on the scale of the biological machinery of life.18 As this field progresses, and we continue to turn to nature for inspiration, humans will have the opportunity to develop complex, self-assembling, self-repairing constructs that previously have been the secrets of biological systems. In these efforts, detailed knowledge of natural systems will provide fundamental design principles for the development of artificial systems. Conversely, knowledge gained from the study of artificial systems will improve our understanding of, and ultimately control over, complex biological machinery.
Legacy biochemistry
As humans endeavor to mimic the processes of nature, it is important to recognize the limitations of biological systems. These limitations largely arise from the evolutionary history of certain biochemical pathways. In particular, selection for maximal biological fitness, including the flexibility to adjust to disparate environmental conditions, is not the same as selection for maximal energy conversion efficiencies. Evolution involves incremental alterations to existing biological machinery, and thus aspects of modern biology likely reflect adaptational holdovers from selective pressures in ancient environments. Through billions of years of fierce evolution, organisms have become finely tuned to prevail in their local environment. However, it is likely that in the stochastic search for optimal fitness evolution has taken ‘missteps,’ and also failed to explore some of the sequence space most relevant to human endeavors. By way of example, we briefly explore instances of legacy biochemistry in the photosynthetic machinery.An initial observation is that photosynthetic organisms evolved from earlier life forms that had already developed key energy carriers (i.e. NAD(P)H and ATP). This early life evolved under anaerobic conditions, with a limited redox span in the environment (i.e. CO was one of the most reducing species [CO2/CO, E°′ = −0.517 V vs. NHE] and NO3− or perhaps Fe3+ was one of the most oxidizing [NO3−/NO2−, E°′ = 0.433 V vs. NHE; Fe3+/Fe2+, E°′ = 0.771 V vs. NHE]).11,19 Clearly, the photosynthetic machinery was selected to interface with the existing bioenergetics of the cell, but the evolution of oxygenic photosynthesis gave rise to large redox spans (at least 2.4 V between the P680˙+/P680 and P700˙+/P700* redox couples). This energy mismatch led to inefficiency, with electrons in PSI dropping ∼1.1 V from the potential of the P700˙+/P700* couple before equilibrating with the NADP+/NADPH couple in solution. Some portion of this apparent ‘overpotential’ is required to drive the forward reaction at an appreciable rate, but some portion of this large voltage drop appears to be a wasteful consequence of the evolutionary history of the photosynthetic apparatus.9
A second example of legacy biochemistry in photosynthetic organisms involves the Calvin–Benson–Bassham cycle, in which the immediate products of photosynthesis (NADPH and ATP) are used to drive the production of carbohydrates from CO2. The key enzyme of this pathway, RuBisCO, exhibits a low turnover rate and poor specificity for the CO2 substrate. Consequently, CO2 fixation is often a slow step in the overall photosynthetic process. As a result, under full sunlight, plants have evolved complex photoprotection mechanisms to dissipate the majority of incident solar energy as heat, rather than using this energy for the production of carbohydrates. This adaptation is advantageous to the organism, minimizing the production of singlet oxygen and other reactive and deleterious species.6 However, this mechanism of avoiding oxidative damage to the photosynthetic machinery comes with a substantial loss of energy conversion efficiency, up to 80% under high solar irradiance.
RuBisCO can alternatively act as an oxygenase in reaction with O2. This process, termed photorespiration, results in the uptake of O2 and release of ‘fixed’ CO2, thereby wasting energy converted during the light reactions of photosynthesis. In C3 plants, this wasteful side reaction can account for a 30% to 50% energy loss during carbon fixation, and thus is a significant limitation to the photosynthetic energy conversion efficiency.6,20 In response to this loss mechanism, C4 plants have evolved an energy intensive mechanism to concentrate CO2 at the site of RuBisCO activity. While this adaptation gives C4 plants a selective advantage under favorable environmental conditions, it is a metabolically expensive ‘fix’ to limitations imposed by the legacy biochemistry of carbon fixation.
In mimicking photosynthesis for solar energy conversion, it is important to recognize that in many environments light is not the limiting factor for organism growth, and hence there may have been only minimal selective pressure to maximize the efficiency of solar energy conversion in natural photosynthesis. Also, in attempting to develop synthetic catalysts with the exquisite specificity of enzymes, it can be useful to separate, at least intellectually, that part of the protein responsible for biological functions (such as information content specifying location, assembly, control, turnover, etc.) from the purely catalytic part of the protein that provides a minimum energy reaction coordinate from reactant to product. In most cases the purely catalytic part is of primary interest for applications in human-engineered systems.
The inefficiencies of legacy biochemistry provide opportunities for human ingenuity to make transformational improvements in energy transduction. For example, human ingenuity over ∼7500 years of selective breeding has turned the small fruit of teosinte into the ear of corn grown around the world today.21 It is hoped that similar ingenuity, which can now be coupled with the almost limitless potential of synthetic biology, and a host of other advanced technologies, can quickly find novel solutions to limitations imposed by legacy biochemistry, meeting human energy demands in the process.
Artificial photosynthesis
In order to mimic photosynthetic energy conversion, it is necessary for a synthetic photosystem to (1) absorb incident photons, generating excited states (i.e. excitons), (2) transfer this excitation energy to a donor/acceptor interface, where photochemical charge separation takes place (the exciton is split), (3) transfer charge away from this interface, in order to limit the rate of wasteful recombination reactions, and (4) couple the photochemically generated charges to appropriate catalysts for the oxidation of a low-energy feedstock and production of a high-energy, reduced fuel. Synthetic photosystems allow ready manipulation of individual components at the molecular level, facilitating direct testing of theoretical considerations such as the effects of distance, orientation, linkage, driving force, solvent polarity, and reorganization energy upon the rate and yield of a photoinduced electron transfer, as well as subsequent charge shift and charge recombination reactions. Many of these parameters are not readily altered in the natural system, and over the past several decades experiments with molecular mimics have been helpful in understanding the principles of natural photosynthesis.22–25In these artificial photosynthetic reaction centers, an electron donor and acceptor are suitably organized for photoinduced electron transfer. The primary donor is typically a porphyrin (reminiscent of the chlorophyll primary donor in nature) or metal-polypyridyl complex, and the primary acceptor is often a porphyrin, viologen, quinone, perylene imide, or fullerene. Light excitation gives rise to a photoinduced electron transfer, functionally mimicking photochemical charge separation in natural photosynthesis. Secondary electron donors or acceptors can be attached to this reaction center, allowing subsequent charge shift reactions that further spatially separate the electron and hole, thereby increasing the lifetime of the charge-separated state. The rates of the charge separation, charge shift, and charge recombination reactions are controlled by thermodynamics, the electronic coupling between the initial and final states, and the reorganization energy needed to convert the initial into the final state. In many systems, the electronic coupling between donor and acceptor moieties is precisely controlled by covalent attachment. The energetics of the electron transfer processes and the reorganization energies are controlled by the choice of donors, acceptors, and medium. Aside from photoinduced electron transfer, other photosynthetic processes such as energy transfer from an antenna system to the reaction center, and photoprotection at high light intensity have been replicated in molecular systems.26,27
Such synthetic reaction centers can be used to convert photon energy to pmf, as demonstrated using a molecular triad (containing a carotenoid, free base porphyrin, and carboxylate-bearing naphthoquinone) inserted into a liposomal membrane containing a lipid-soluble quinone shuttle (Fig. 2). Due to the amphiphilic character of the triad, the polyene tail inserts preferentially into the lipid membrane, providing vectorial orientation for the array of reaction centers. Photon absorption by the porphyrin leads to excited state electron transfer to the covalently attached quinone, followed by hole transfer from the oxidized porphyrin to the carotenoid secondary donor. The final charge-separated state is sufficiently long-lived to allow electron transfer from the reducing side of the triad (the quinone) to the lipid-soluble quinone shuttle. Along with reduction of this quinone, a proton is extracted from the external aqueous solution, creating a neutral semiquinone that diffuses across the lipid membrane to the oxidizing side of the triad (the carotenoid). Here the semiquinone donates an electron to the oxidized carotenoid, in the process expelling a proton into the internal aqueous solution, generating pmf. Such a system can drive ATP production from ADP and inorganic phosphate, when ATP synthase is incorporated into the liposomal membrane.28 These liposomal systems serve as proof of concept that photoinduced charge separation in molecular systems can be used to store incident solar energy by generating pmf in a manner analogous to bacterial photosynthesis. Alternatively, recent results have demonstrated the conversion of emf to pmf across a planar lipid bilayer containing a redox-active proton shuttle.29 Developing constructs for emf–pmf interconversion is central to some approaches to artificial photosynthesis and will open the door to employing biological energy-transducing catalysts, coupled to pmf, in hybrid devices capable of the synthesis of energy-rich compounds.
![An artificial photosynthetic membrane converting light energy into pmf, thereby driving ATP synthesis. Molecular triad (C–P–Q) molecules are inserted into a liposome containing the lipid soluble quinone shuttle (QS). Photoinduced charge separation gives rise to proton pumping via QS. With ATP synthase incorporated into the membrane, the resulting pmf is utilized for the synthesis of ATP. The graph shows the amount of ATP produced as a function of irradiation time for [ATP] = [ADP] = 0.2 mM and [Pi] = 5 mM, open triangles and [ATP] = 0.2 mM, [ADP] = 0.02 mM and [Pi] = 5 mM, filled circles, as well as for control experiments. The figure is adapted from ref. 28.](/image/article/2009/CS/b800582f/b800582f-f2.gif) | ||
| Fig. 2 An artificial photosynthetic membrane converting light energy into pmf, thereby driving ATP synthesis. Molecular triad (C–P–Q) molecules are inserted into a liposome containing the lipid soluble quinone shuttle (QS). Photoinduced charge separation gives rise to proton pumping via QS. With ATP synthase incorporated into the membrane, the resulting pmf is utilized for the synthesis of ATP. The graph shows the amount of ATP produced as a function of irradiation time for [ATP] = [ADP] = 0.2 mM and [Pi] = 5 mM, open triangles and [ATP] = 0.2 mM, [ADP] = 0.02 mM and [Pi] = 5 mM, filled circles, as well as for control experiments. The figure is adapted from ref. 28. | ||
Photochemical charge separation in molecular systems has also been coupled to the oxidation of coordinated manganese ions. In one example, excited-state electron transfer from a ruthenium-polypyridyl dye to a soluble viologen acceptor leads to the generation of Ru3+. This species withdraws an electron from a covalently attached manganese center. In some of the most successful compounds, three rounds of light excitation are able to accumulate three oxidizing equivalents on a manganese dimer, transforming the initial Mn2+–Mn2+ cluster into Mn3+–Mn4+.30 In the process, an acetate to aquo ligand exchange occurs, with possible deprotonation of an aquo ligand concomitant with accessing the Mn3+–Mn4+ state. Such work has many similarities with the donor side of PSII, and is a significant step towards interfacing synthetic reaction centers with functional water-oxidation catalysts. Both the generation of pmf and the multielectron oxidation of water are research areas requiring a detailed understanding of how proton motion can be coupled to electron transfer, a topic discussed below.
Controlling proton activity
Both biological and technological systems for the production of solar fuels inevitably have to interface single-electron photochemistry with multielectron catalytic reactions, such as the oxidation of water and the reduction of CO2. In biological systems such reactions involve the coupling of electron transfer to proton motion, aiding the accumulation of multiple redox equivalents. Proton coupled electron transfer (PCET),31–34 a term used here to denote all regimes of coupling from stepwise to concerted, is involved in the basic mechanisms of myriad bioenergetic schemes including redox-driven proton pumps, small molecule activation, and radical initiation and transport. A mechanistic understanding of how electrons and protons are transferred in these reactions is fundamental to the design of successful energy conversion systems.PCET can allow reaction coordinates in which electrons and protons are transferred in a concerted fashion, thus avoiding high-energy intermediates (Fig. 3). PCET mechanisms involving a single transition state and no intermediate are referred to as concerted proton electron transfers (CPET). The concerted mechanism offers a thermodynamic advantage over the initial proton transfer or initial electron transfer in stepwise PCET processes involving proton transfer followed by electron transfer (PTET) or electron transfer followed by proton transfer (ETPT). However, due to the inherent difference in mass, protons can only tunnel over limited distances of a few angstroms, while electrons can tunnel over tens of angstroms. Nature appears to work within this constraint by coupling long-range electron transport to relatively short distance proton transfer within hydrogen bonds.
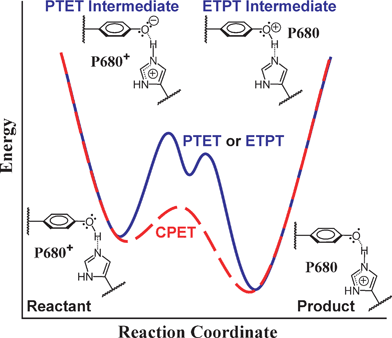 | ||
| Fig. 3 Conceptual reaction coordinate diagram for the oxidation of TyrZ by P680˙+, showing the relationship between the potential energy surfaces for concerted and stepwise processes. The concerted proton electron transfer (CPET), red dashed line, has a single transition state and no intermediate state. Both of the stepwise mechanisms (PTET or ETPT), for simplicity represented by a single blue line, involve a high-energy intermediate. A putative hydrogen bond between TyrZ and His190 is shown, and His190 likely acts as a proton acceptor upon deprotonation of TyrZ. | ||
One of the most discussed examples of PCET in natural energy systems is the conversion of tyrosine to the corresponding tyrosyl radical in PSII. In this case, tyrosine Z (TyrZ) functions as a redox mediator between the photo-oxidized primary donor (P680˙+) and the Mn-containing oxygen evolving complex (OEC). Important aspects of the electron transfer from TyrZ to P680˙+ are the protonation states involved in the reaction.35–37
Under most conditions the oxidation of phenols (e.g., tyrosine) is coupled to deprotonation (i.e. the phenoxyl radical is a strong acid, pKa≈−2),36 and phenol electrochemistry is often irreversible. The coupling of oxidation to deprotonation is reflected in the pH dependence of the formal potential (eqn (4)), which shows a 59 mV per pH unit slope within the −2 to 10 pH range (with −2 corresponding to the pKa of the oxidized tyrosine species and 10 to the pKa of reduced tyrosine).38
| TyrO˙ + H+ + e−⇄ TyrOH | (4) |
| E°′ = (1.34 − 0.0591·pH) V vs. NHE |
It is interesting to compare these values to estimates for the P680˙+/P680 couple in PSII (E°′ = 1.26 V vs. NHE)37 which occurs in the relatively low dielectric medium of a protein, as well as to the formal reduction potential for the water oxidation half reaction (eqn (1)) at pH 5 (E°′ = 0.93 V vs. NHE), which approximates the acidic limit for the lumen pH.35 Based on these values, it is apparent that the protonated tyrosine species is not poised to reduce P680˙+ without the aid of deprotonation. Conversely, the deprotonated tyrosine redox couple is not thermodynamically capable of water oxidation. While the electrochemical properties of a species will vary between an aqueous solution and a protein environment, this example illustrates the importance of the protonation state of TyrZ in controlling the redox potential of this mediator.
In PSII the oxidation of TyrZ by P680˙+ likely occurs with transfer of the tyrosyl proton to the hydrogen-bonded histidine residue (His190), and the TyrZ˙/TyrZ couple is estimated to operate at ∼0.97 to 1.20 V vs. NHE.35,36 It appears likely that the TyrZ–His190 hydrogen bond tunes the reduction potential of the TyrZ˙/TyrZ couple to an appropriate value between the potentials for P680˙+ and the OEC, while also bestowing chemically reversible protonation/deprotonation upon this essential redox mediator.37 Following four consecutive photoinduced turnovers, the oxidizing equivalents accumulated in the OEC are used to carry out the four-electron oxidation of two water molecules.
Experimental and theoretical interest in the role of PCET in biological and chemical processes has led to the synthesis and investigation of molecular systems containing phenoxyl radicals stabilized by intramolecular hydrogen bonds.39 Synthetic models of PSII have also been explored, in which photoinduced electron transfer occurs from a modified tyrosine residue to a covalently attached ruthenium-polypyridyl complex.30 Aukauloo and co-workers have prepared a biomimetic model for the TyrZ-His190 pair consisting of an intramolecular hydrogen-bonded phenol attached to a ruthenium-based chromophore.40 The photo-oxidation of ruthenium is followed by a secondary charge shift, forming the phenoxyl radical. However, in this system the potential of the resulting phenoxyl radical is insufficient to oxidize water at a biologically relevant pH.
Recently, Moore et al. have designed a photochemical system in which a modified bis-pentafluorophenyl porphyrin is attached to colloidal TiO2 (Fig. 4).41 The TiO2 conduction band serves as the primary electron acceptor from the photoexcited porphyrin moiety. Hole transport, facilitated by PCET, from the photo-oxidized porphyrin to a covalently attached tyrosine-histidine mimic (a phenol-benzimidazole pair) produces a phenoxyl radical (E°′ = 1.24 V vs. NHE) that is chemically reversible, an imperative for a mediator, and thermodynamically poised to oxidize water. The chemical reversibility of the phenoxyl/phenol couple associated with this construct is attributed to the ability of the proton to shuttle between the oxygen of the phenol and the nitrogen lone pair electrons of the benzimidazole in the corresponding reduced and oxidized forms with minimal nuclear motion, effectively trapping the proton at the site of electrochemical activity.
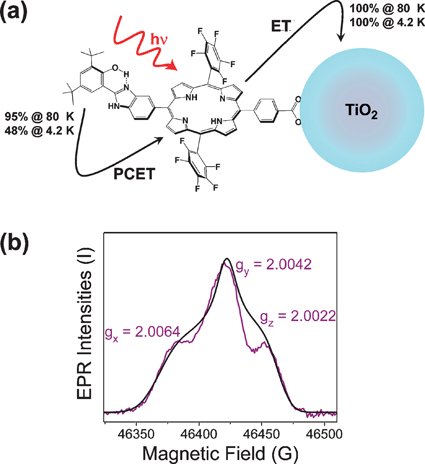 | ||
| Fig. 4 A photochemically active mimic of the chlorophyll-TyrZ-His190 complex of PSII. (a) A modified bis-pentafluorophenyl porphyrin is adsorbed to colloidal TiO2. Light excitation gives rise to a photoinduced electron transfer from the excited porphyrin to the TiO2 conduction band. A secondary, temperature-dependent hole transfer from the photo-oxidized porphyrin to the hydrogen-bonded phenol moiety is observed. (b) A photoinduced D-band (130 GHz) EPR difference spectrum at 13 K, purple line, along with a simulated phenoxyl radical spectrum, black line, demonstrating the formation of the phenoxyl radical. The figure is adapted from ref. 41. | ||
These model systems facilitate a deeper understanding of the electrochemical and photochemical aspects of PCET reactions. Such insight is helpful in elucidating the mechanisms of biological catalysts and energy transduction schemes. This knowledge is also critical for the development of artificial constructs that mimic key aspects of biological energy conversion. The coupling of electron and proton motions is a fundamental aspect of biological energy transduction, and will likely be of equal importance for the success of biomimetic devices.
Applying the lessons of nature
In the long term, bioinspired technological approaches to the production and use of solar fuels provide the opportunity to mimic the essential components of biological systems while leaving behind the sub-optimal aspects that result from legacy biochemistry.42 By learning from nature, humans may be able to produce efficient, stable energy conversion technologies, utilizing only earth-abundant materials and operating under mild conditions. Further, human ingenuity has the ability to discover novel solutions, using chemistry unexplored during the evolutionary history of biological organisms.The conversion of sunlight to chemical fuels at the rate of tens of terawatts is a critical aspect of our future energy infrastructure. Towards this end, significant advances have been made in the field of artificial photosynthesis. Synthetic molecular systems are able to capture incident photons and transfer excitation energy to artificial reaction centers, which perform the fundamental process of photochemical charge separation. Ongoing research in many laboratories is focused upon coupling these long-lived charge-separated states to appropriate anodic and cathodic catalysts, one day driving the production of fuels using only sunlight, water, and possibly CO2. However, much remains to be done. Key features of biological energy transduction, such as pmf, self-assembly, and self-repair, are only beginning to be explored in technological systems. For instance, humans are learning how to embed proteins into mechanically stable membranes with a thickness of similar dimension to the protein itself, an important prerequisite for systems utilizing pmf.43 Likewise, work with synthetic peptide maquettes is exploring three-dimensional organization within the peptide matrix for orienting chromophores, redox cofactors, and catalytic sites.44 We are only at the initial stages of developing synthetic enzymes. However, it is easy to envision future advances leading to synthetic protein scaffolds housing intricate architectures for efficient photochemical charge separation coupled to catalytic fuel production. Such constructs could be designed with tight control of proton activity, allowing photochemical proton pumping when vectorially oriented within an artificial membrane. Due to the small size of protein maquettes, it is feasible that artificial reaction centers could be packed in close proximity in such a membrane, resulting in higher power densities than are achieved in biological membranes. It is also interesting to consider techniques for the electrodeposition of catalytic films,45 which could allow in situ formation of anodic and cathodic catalysts on opposite sides of an artificial photosynthetic membrane.
As human engineering becomes better able to couple proton motion to electron motion, generating pmf, a question that arises is how can pmf be converted into useful work in the absence of biological transducers such as flagella and ATP synthase. One possibility involves the pH dependence of many key biological half-reactions, including water oxidation (eqn (1)), hydrogen production (eqn (2)), and the reduction of carbon dioxide. As shown schematically in Fig. 5a, one can envision a photoelectrochemical cell in which ‘blue’ photons are used to drive photolytic water splitting (eqn (3)), while ‘red’ photons passing through this top light-absorber, are used to generate pmf, facilitating the water splitting reaction.
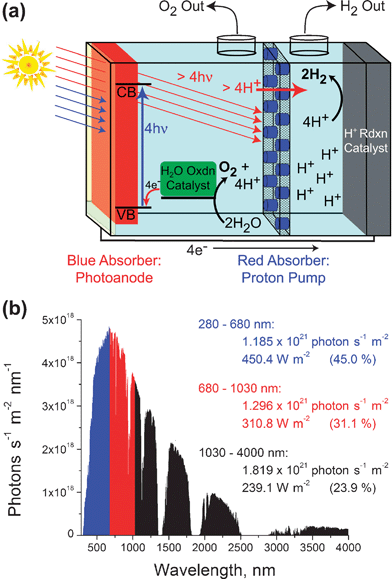 | ||
| Fig. 5 A proposed method of coupling pmf to redox chemistry in a human-engineered construct. (a) A schematic drawing of the proposed dual-photosystem device. ‘Blue’ photons are absorbed by the top absorber (colored red), driving charge separation which is coupled to O2 and H2 producing catalysts. ‘Red’ photons pass through this top light-absorber, and are absorbed by photosynthetic proton-pumps (colored blue) embedded in the ion-impermeable membrane separating the two solutions. Provided that the proton ‘current’ is greater than the electrical current, this device would generate ΔpH, thereby reducing the voltage required for the overall water splitting reaction. (b) The AM1.5G solar spectrum, divided into ‘blue’, ‘red’, and ‘far-red’ regions. For each wavelength range, the photon flux and energy flux are listed, along with the percentage of the total solar energy contained in that region of the spectrum (listed in parentheses). The particular wavelength ranges were chosen by analogy with natural photosynthesis, and do not represent the ideal theoretical absorption thresholds for a dual-photosystem photoconversion device. | ||
In Fig. 5a, sunlight strikes a photoanode, such as WO3 or a dye-sensitized semiconductor, generating electron–hole pairs. Holes are shuttled towards the anodic solution, driving water oxidation at appropriate catalytic sites. Electrons migrate toward the back-contact of this photoanode, and then through an external circuit to a cathode, driving the electroreductive synthesis of a fuel, depicted as H2. This process is driven with relatively high energy photons (λ≤ 680 nm). Because the blue photons are absorbed, this photosystem is colored red in Fig. 5a. The ‘red’ photons of the AM1.5G solar spectrum§ would not be absorbed, and hence would be available to drive proton pumping across an ion-impermeable membrane separating the anodic and cathodic solutions and placed in optical series behind the top light-absorber. This proton-pumping membrane could involve artificial reaction centers (colored blue in Fig. 5a) in a peptide maquette, as discussed above. If the rate of proton pumping exceeded the rate of electron flow, then charge neutrality would not be maintained, and pmf would be generated. With proper orientation of the proton pump it would be possible to acidify the cathodic solution, and make the anodic solution basic, thereby decreasing the cell voltage required to drive the overall process (eqn (3)).
Once again looking to nature, it is apparent that bacterial reaction centers using bacteriochlorophyll b are able to drive proton pumping with photons as ‘red’ as 1030 nm. Fig. 5b shows the AM1.5G solar spectrum, divided into ‘blue’ photons (280–680 nm) which would be absorbed by the photoanode, ‘red’ photons (680–1030 nm) which would be absorbed by the proton pump, and ‘far red’ photons which would not be active in the device. It should be noted that these wavelengths were selected by analogy with natural photosynthesis, and do not represent the ideal wavelengths for maximal solar energy conversion (which would have longer-wavelength absorption thresholds for both photosystems).46 Integration reveals a similar photon flux in the ‘blue’ and ‘red’ regions of the spectrum, suggesting that the envisioned approach to utilizing pmf is feasible. Further, if the proton pumping, ‘red’ photosystem could transport 2H+/e−, as in bacterial photosynthesis, then the contribution of pmf would be much more significant. Alternatively, the 1.2 eV excitation energy of a 1030 nm photon greatly exceeds the ∼0.2 eV required to pump one proton across a charged biological membrane, suggesting the possible use of even longer wavelength absorbers for proton pumping.
Surprisingly, photosynthetic organisms do not make use of dual reaction centers stacked in an optical series, with a short-wavelength photosystem placed in the light-path of a second photosystem with a longer-wavelength absorption threshold. From a theoretical perspective, such a true multi-threshold approach to oxygenic photosynthesis would allow greater utilization of incident solar energy, due to the collection of a larger number of incident photons, coupled with decreased energy losses resulting from thermalization of higher energy photons to the lowest-energy excited state of each photosystem.46 From this perspective, it seems clear that the structural arrangement of, and wavelengths of light utilized by, PSII and PSI in oxygenic photosynthesis are not optimal for maximum solar energy conversion. Within the thylakoid membrane, PSII and PSI act as optically parallel light-absorbers rather than a multi-threshold device, while the large overlap in redox span between these two photosystems diminishes the conversion of light to chemical energy.
Interestingly, within a photosynthetic community, different organisms do exploit regions of the solar spectrum not harvested by those organisms that live spatially above them. In a sense, the community can be viewed as a stacked optical series where the energy outcomes (life and biomass production) do sum. A good example is Acaryochloris marina, a water-oxidizing cyanobacteria that has evolved chlorophyll d-based photosystems to use light of 700 nm to 740 nm which is transmitted through Prochloron (chlorophyll a-based with a threshold of 700 nm) living above it.47 The peak solar photon flux occurs around 700 nm so Acaryochloris lives in a spectrally narrow, but quantum rich, solar radiation field. The red photon niche allowing this fascinating arrangement hints at the inefficiencies resulting from legacy biochemistry associated with the evolutionary history of oxygenic photosynthesis, exemplified in Prochloron.
Overall, the development of multiple photosystems in optical series, and ‘far-red’ light-absorbers, are examples of the myriad approaches for improving upon natural photosynthesis in artificial constructs.
Conclusions
Humanity faces an impending energy crisis, as growth in global population and per-capita consumption strain our existing energy infrastructure, while the environmental impacts of this infrastructure mandate that we severely curtail greenhouse-gas emissions within the next decade. Fortunately, sunlight is a clean, environmentally benign energy source of effectively limitless potential. Moreover, nature has shown us that solar energy conversion is feasible on the daunting scale required both for life on Earth and for the fulfilment of human energy demands. Technological systems have made significant advances toward mimicking the fundamental processes of photosynthesis and cellular respiration. The development of advanced techniques for the manipulation of matter on the nanoscale makes it possible for humans to achieve transformational energy systems, incorporating the most favorable aspects of natural systems. At the same time, technological approaches allow humans to selectively exclude undesirable aspects of natural energy conversion, providing the opportunity to improve upon natural systems. The road forward requires significant fundamental research, and the timelines of human energy demands and anthropogenic climate change require the rapid implementation of current and future scientific discoveries.Widespread solar energy conversion will require the use of inexpensive, earth-abundant materials that allow the efficient transduction of energy with excellent stability. Such systems can be achieved by applying the lessons learned from natural energy transduction. However, the most critical lessons to be learned from nature involve the efficient use of energy. Biological organisms have evolved to utilize unlikely sources of energy found in local niches around the planet. Where energy is available, some organism has evolved to make use of it. Humanity would do well to follow nature’s lead, and make more efficient use of our current energy supplies. Also, biological organisms are able to operate with only modest energy inputs. For instance, the 2000 kcal per day diet of a ‘typical’ human equates to a ‘burn rate’ of only 96.9 W. If we consider the remarkable achievements of individuals throughout human history, such as those of Galileo, Shakespeare, Michelangelo, or Da Vinci, each was achieved with essentially the energy consumption of a ‘classic’ 100 W lightbulb. Massive energy inputs are not a prerequisite for great achievements; a lesson that should be kept in mind as humans move to a sustainable energy future.
References
- IPCC 2007: Summary for policy makers, Cambridge University Press, Cambridge, UK, and New York, NY, USA, 2007 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS.
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quéré, J. G. Canadell, G. Klepper and C. B. Field, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288 CrossRef CAS.
- International energy outlook 2007, US Department of Energy, Washington, DC, May 2007 Search PubMed.
- M. D. Archer and J. Barber, Photosynthesis and photoconversion, in Molecular to global photosynthesis, ed. M. D. Archer and J. Barber, Imperial College Press, London, 2004 Search PubMed.
- R. E. Blankenship, Molecular mechanisms of photosynthesis, Blackwell Science, Ltd., Malden, MA, 2002 Search PubMed.
- D. M. Kramer, C. A. Sacksteder and J. A. Cruz, Photosynth. Res., 1999, 60, 151 CrossRef CAS.
- J. R. Bolton and D. O. Hall, Photochem. Photobiol., 1991, 53, 545 CrossRef CAS.
- D. Gust, D. Kramer, A. Moore, T. A. Moore and W. Vermaas, MRS Bull., 2008, 33, 383 CAS.
- G. P. Dobson and J. P. Headrick, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7317 CrossRef CAS.
- R. A. Alberty, Biochem. Ed., 2000, 28, 12 Search PubMed.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141 CrossRef CAS.
- A. Luque and S. Hegedus, Handbook of photovoltaic science and engineering, John Wiley and Sons, Hoboken, NJ, 2003 Search PubMed.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef CAS.
- F. Yang, K. Sun and S. R. Forrest, Adv. Mater., 2007, 19, 4166 CrossRef CAS.
- P. A. Lessing, Materials for Water Electrolysis Cells, in Materials for the Hydrogen Economy, ed. R. H. Jones and G. J. Thomas, CRC Press, Boca Raton, FL, 2008 Search PubMed.
- G. W. Crabtree and M. S. Dresselhaus, MRS Bull., 2008, 33, 421 CAS.
- BESAC Subcommittee on Grand Challenges for Basic Energy Sciences, Directing Matter and Energy: Five Challenges for Science and the Imagination, US Department of Energy, 2007.
- R. K. Thauer, K. Jungermann and K. Decker, Bacteriol. Rev., 1977, 41, 100 CAS.
- S. P. Long, X.-G. Zhu, S. L. Naidu and D. R. Ort, Plant, Cell Environ., 2006, 29, 315 CrossRef CAS.
- S. I. Wright, I. V. Bi, S. G. Schroeder, M. Yamasaki, J. F. Doebley, M. D. McMullen and B. S. Gaut, Science, 2005, 308, 1310 CrossRef CAS.
- T. A. Moore, D. Gust, P. Mathis, J.-C. Mialocq, C. Chachaty, R. V. Bensasson, E. J. Land, D. Doizi, P. A. Liddell, W. R. Lehman, G. A. Nemeth and A. L. Moore, Nature, 1984, 307, 630 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Mimicking Bacterial Photosynthesis, in Artificial Photosynthesis, ed. A. F. Collings and C. Critchley, Wiley-VCH, Weinheim, 2005 Search PubMed.
- G. Kodis, Y. Terazono, P. A. Liddell, J. Andréasson, V. Garg, M. Hambourger, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2006, 128, 1818 CrossRef CAS.
- S. D. Straight, G. Kodis, Y. Terazono, M. Hambourger, T. A. Moore, A. L. Moore and D. Gust, Nat. Nanotechnol., 2008, 3, 280 Search PubMed.
- G. Steinberg-Yfrach, J.-L. Rigaud, E. N. Durantini, A. L. Moore, D. Gust and T. A. Moore, Nature, 1998, 392, 479 CrossRef CAS.
- T. W. McBee, L. Wang, C. Ge, B. M. Beam, A. L. Moore, D. Gust, T. A. Moore, N. R. Armstrong and S. S. Saavedra, J. Am. Chem. Soc., 2006, 128, 2184 CrossRef CAS.
- R. Lomoth, A. Magnuson, M. Sjödin, P. Huang, S. Styring and L. Hammarström, Photosynth. Res., 2006, 87, 25 CrossRef CAS.
- J. Stubbe, D. G. Nocera, C. S. Yee and M. C. Y. Chang, Chem. Rev., 2003, 103, 2167 CrossRef CAS.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363 CrossRef CAS.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef CAS.
- C. Costentin, Chem. Rev., 2008, 108, 2145 CrossRef CAS.
- C. Tommos and G. T. Babcock, Biochim. Biophys. Acta, 2000, 1458, 199 CrossRef CAS.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284 CrossRef CAS.
- A. Harriman, J. Phys. Chem., 1987, 91, 6102 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2006, 128, 4552 CrossRef CAS.
- F. Lachaud, A. Quaranta, Y. Pellegrin, P. Dorlet, M.-F. Charlot, S. Un, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2005, 44, 1536 CrossRef.
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466 CrossRef CAS.
- A. W. Rutherford and T. A. Moore, Nature, 2008, 453, 449 CrossRef CAS.
- D. Ho, B. Chu, H. Lee, E. K. Brooks, K. Kuo and C. D. Montemagno, Nanotechnology, 2005, 16, 3120 CrossRef CAS.
- R. L. Koder and P. L. Dutton, Dalton Trans., 2006, 3045 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- M. Kühl, M. Chen, P. J. Ralph, U. Schreiber and A. W. D. Larkum, Nature, 2005, 433, 820 CrossRef.
Footnotes |
| † Part of the renewable energy theme issue. |
| ‡ A global average radiative forcing of +1.6 W m−2, due to cumulative human activities since 1750, is a best estimate obtained from the IPCC AR4 report.1 The solar insolation on a normal surface at a given distance from the sun equals the total power output of the sun divided by the surface area of a sphere centered on the sun and with a radius of that distance. Thus, the intensity of sunlight on a normal surface (I) depends upon the total power output of the sun (P) and the distance from the sun (r) as described by: I = P/(4πr2). Rearranging this equation gives: [(I1)/(I2)] = [(r2)2/(r1)2]. The annual average distance between the sun and the Earth is 1.49 × 108 km, and the solar insolation reaching a normal surface at the outer edge of Earth’s atmosphere is ∼1360 W m−2. Accounting for day and night, the global average solar insolation is then 340 W m−2. In order to counteract the current anthropogenic radiative forcing of +1.6 W m−2, the global average solar insolation would have to become 338.4 W m−2. Thus, [(340 W m−2)/(338.4 W m−2)] = [(r2)2/(1.49 × 108 km)2]; giving [(r2) − (r1)] = 3.5 × 105 km. Hypothetically, anthropogenic warming could be avoided if the radius of Earth’s orbit was increased by ∼350000 km; similar to the distance between the Earth and the moon. |
| § At the time of writing, the air-mass 1.5 global tilt (AM1.5G) solar spectrum, from the ASTM G173-03 data set, is available online at http://rredc.nrel.gov/solar/spectra/am1.5/. |
| This journal is © The Royal Society of Chemistry 2009 |