Dynamic electrochemical investigations of hydrogen oxidation and production by enzymes and implications for future technology†
Fraser A.
Armstrong
*,
Natalie A.
Belsey
,
James A.
Cracknell
,
Gabrielle
Goldet
,
Alison
Parkin
,
Erwin
Reisner
,
Kylie A.
Vincent
and
Annemarie F.
Wait
Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, UK OX1 3QR. E-mail: fraser.armstrong@chem.ox.ac.uk; Fax: +44 1865 272690; Tel: +44 1865 272647
First published on 1st December 2008
Abstract
This tutorial review decribes studies of hydrogen production and oxidation by biological catalysts—metalloenzymes known as hydrogenases—attached to electrodes. It explains how the electrocatalytic properties of hydrogenases are studied using specialised electrochemical techniques and how the data are interpreted to allow assessments of catalytic rates and performance under different conditions, including the presence of O2, CO and H2S. It concludes by drawing some comparisons between the enzyme active sites and platinum catalysts and describing some novel proof-of-concept applications that demonstrate the high activities and selectivities of these ‘alternative’ catalysts for promoting H2 as a fuel.
 The authors | From left to right: Gabrielle Goldet graduated with an MChem from Durham University in 2004 and is now completing her DPhil with Fraser Armstrong at St John’s College, Oxford. Dr Kylie Vincent is Royal Society University Research Fellow and RCUK Academic Fellow in the Inorganic Chemistry Laboratory at the University of Oxford, and is a Fellow of Jesus College, Oxford. She is a graduate of the University of Melbourne, Australia, where she completed a BA/BSc (Hons), and a PhD jointly with Stephen Best (Melbourne) and Chris Pickett (Norwich, UK). She carried out postdoctoral work with Fraser Armstrong from 2002–2007 and was a Research Fellow at Wadham College during this time. Her research interests include the application of electrochemical and spectroelectrochemical methods to biological systems, in particular enzymes involved in energy cycling. James Cracknell graduated with an MChem from Corpus Christi College, University of Oxford in 2005, and is now completing his DPhil with Fraser Armstrong at St John’s College, Oxford. Fraser Armstrong is Professor of Chemistry at Oxford University and is Fellow of St. John’s College. His interests are in biological redox chemistry, in particular the application of dynamic electrochemical techniques in studies of complex electron-transfer and catalytic reactions in proteins, and most recently the mechanisms and exploitation of biological hydrogen cycling. He was elected a Fellow of the Royal Society in 2008. Dr Alison Parkin completed her doctoral studies under the supervision of Fraser Armstrong in 2008 and is now a Junior Research Fellow at Merton College, Oxford. Natalie Belsey graduated with an MChem from Lincoln College, University of Oxford in 2005 and is now completing her DPhil with Fraser Armstrong at Lincoln College, Oxford. Dr Erwin Reisner performed his doctorate work as a joint project between the University of Vienna (Professor Bernhard Keppler) and the Technical University of Lisbon (Professor Armando Pombeiro). He then joined the laboratory of Professor Stephen Lippard at MIT as a Schrödinger Postdoctoral Fellow. Currently, he is a Research Assistant with Fraser Armstrong interested in solar H2 production. Annemarie Wait graduated with an MChem from St Hilda’s College, University of Oxford in 2007, and is now completing her DPhil with Fraser Armstrong at Merton College, Oxford. |
Introduction
One of the most promising directions for chemistry today is the development of ways to capture and store renewable energy (meaning, most obviously, sunlight). Hydrogen is one answer as there are infinite resources of water available to be ‘energised’ to form H2 (energy content over 100 kJ per gram) using direct solar energy or surplus electricity. Generation of H2 from water and its reoxidation are (at first glance) simple reactions, but H2 is a rather stable and inert molecule (the H–H bond energy is 436 kJ mol–1) and it is difficult to produce efficiently. In practice, many aspects of the clean and efficient production, storage and oxidation of H2 are totally unresolved at the molecular level. Regarding almost insurmountable scale-up demands, we should be reminded that the nuclear age grew out of the serendipitous detection of trace BaSO4 precipitates in neutron-irradiated U(VI) solutions by Meitner and Hahn in 1938. In this tutorial review we describe how to extract detailed information on how the H2 molecule is manipulated very efficiently in biology, at the active sites of metalloenzymes. In ‘learning from biology’ we believe it would be hard to find a better example of a ‘bottom up’ approach (from the molecular level) to such a large and daunting problem.As a fuel, H2 was familiar as a 50% component of coal gas, once piped to homes in many industrialised nations. More than 99% of the world’s H2 is still produced by steam reforming of fossil fuels, and most is used directly by the chemical industry, rather than as a fuel. Currently, production of H2 by electrolysis at Fe and Ni electrodes is costly in terms of energy as it involves high temperatures and a voltage of 2 V or more. Most fuel cells operating at temperatures below 100 °C use Pt as electrocatalysts, but Pt is not an unlimited resource and even the best efforts to maximise the efficiency of its use will not be enough. Neither is Pt the perfect catalyst for H2 cycling, because it is poisoned by H2S and CO which are contaminants of H2 from steam reforming. Alternative catalysts are therefore highly desirable, both to produce and oxidise H2, and one important question is: ‘what can we learn from biology?’. The reason for the interest in biology is that H2 is cycled and used efficiently by microbial organisms, in processes such as photosynthesis and respiration. The enzymes responsible are known as hydrogenases1 and they were first discovered in the 1930s. Hydrogenases in certain organisms must have high affinity for H2 and scavenge it from the atmosphere or local zones, whereas other hydrogenases are more active in producing H2. Hydrogenases are found in pathogens, and H2 may be a useful disease marker.2
Although hydrogenases are among the most ancient of enzymes, they represent a ‘new’ class of catalyst—complexes of abundant first-row transition elements that are potentially as active as Pt. The structures of the main classes of hydrogenases were solved in the 1990s,3 and we describe these briefly below. In our studies4–6 of hydrogenases by electrochemical methods, our aim is to develop a good understanding at the atomic-mechanistic level. We measure their performance as catalysts, not only in simple terms of rates but also in terms of their abilities to fend off inactivation by molecules such as O2, CO, H2S, and even H2 itself which is often an inhibitor of its own production. The information obtained is helpful in assessing the viability of organisms for ‘H2 farms’, new alternative catalysts for H2 production and fuel cells, and niche applications for the enzymes themselves. We and others have studied hydrogenases from a range of bacterial sources, and Table 1 summarises details and abbreviations used in this review.
| Bacterium | Type of organism | Hydrogenase studied by PFV (Abbreviation) |
|---|---|---|
| Allochromatium vinosum | Purple photosynthetic sulfur metabolizer, facultative anaerobe | Membrane-bound [NiFe]-hydrogenase |
| (Av [NiFe]-MBH) | ||
| Clostridium acetobutylicum | Fermentative anaerobe | HydA—[FeFe]-hydrogenase |
| (Ca [FeFe]-H) | ||
| Desulfomicrobium baculatum | Sulfate reducer, facultative anaerobe | Periplasmic [NiFeSe]-hydrogenase |
| (Db [NiFeSe]-H) | ||
| Desulfovibrio desulfuricans | Sulfate reducer, facultative anaerobe | Periplasmic [FeFe]-hydrogenase |
| (Dd [FeFe]-H) | ||
| Desulfovibrio fructosovorans | Sulfate reducer, facultative anaerobe | Periplasmic [NiFe]-hydrogenase |
| (Df [NiFe]-H) | ||
| Desulfovibrio gigas | Sulfate reducer, facultative anaerobe | Periplasmic soluble [NiFe]-hydrogenase |
| (Dg [NiFe]-H) | ||
| Desulfovibrio vulgaris Miyazaki F | Sulfate reducer, facultative anaerobe | Membrane-bound [NiFe]-hydrogenase |
| (Dv [NiFe]-MBH) | ||
| Ralstonia eutropha H16 | Knallgas bacterium, aerobe | Membrane-bound [NiFe]-hydrogenase |
| (Re [NiFe]-MBH) | ||
| Ralstonia metallidurans CH34 | Knallgas bacterium, aerobe | Membrane-bound [NiFe]-hydrogenase |
| (Rm [NiFe]-MBH) |
Key features of the active sites of hydrogenases are shown in Fig. 1, which includes the structure of an entire enzyme molecule.3 The two main classes of enzyme are known as [FeFe]- or [NiFe]-hydrogenases, depending on the metal content of the active site. Some, known as membrane-bound hydrogenases (MBHs), are components of energy chains, although they are easily solubilised by detergents or by loss of the hydrophobic membrane anchor domain. The minimum common factor in hydrogenases studied to date is a [Fe(CO)(CN)(RS)] unit, bridged to either Ni or a second Fe by thiolate ligands. These fragile active sites are protected by the surrounding protein and serviced by a chain of Fe–S clusters that transfer electrons to and from the enzyme surface. It is widely accepted that H2oxidation and production involve heterolytic mechanisms in which H– and H+ are stabilised by metal coordination and Brønsted base, respectively. Different cycles may be used, depending on the direction of catalysis.4,5
![Hydrogenases and their active sites and relays. Left: Ribbon representation of the X-ray determined structure of Dg [NiFe]-H (PDB code 1YQ9). Right: The structures of the active sites. A shows the active site of Db [NiFeSe]-H in its active state (PDB code 1CC1), B shows the active site of Dg [NiFe]-H in the oxidised inactive ‘Ni-A’ state (PDB code 1YQ9) in which an inhibitory peroxide is present in the bridging position, and C shows the active site (the ‘H-cluster’) of Dd [FeFe]-H (PDB code 1HFE) in its active form.](/image/article/2009/CS/b801144n/b801144n-f1.gif) | ||
| Fig. 1 Hydrogenases and their active sites and relays. Left: Ribbon representation of the X-ray determined structure of Dg [NiFe]-H (PDB code 1YQ9). Right: The structures of the active sites. A shows the active site of Db [NiFeSe]-H in its active state (PDB code 1CC1), B shows the active site of Dg [NiFe]-H in the oxidised inactive ‘Ni-A’ state (PDB code 1YQ9) in which an inhibitory peroxide is present in the bridging position, and C shows the active site (the ‘H-cluster’) of Dd [FeFe]-H (PDB code 1HFE) in its active form. | ||
Dynamic electrochemical techniques in hydrogenase research
Equipment
In protein film voltammetry (PFV), a minuscule sample of protein is adsorbed directly on an electrode to give a mono- or submonolayer film.6–8 In this state the enzyme’s catalytic properties are fully expressed, and facile, reversible electron transfer occurs with the electrode. The enzyme’s catalytic activity is now controlled by the electrode potential and simultaneously monitored through the current that is observed.5,7,8 The steady-state current is a direct measure of the catalytic turnover rate at any given electrode potential. Because the enzyme is immobilised on the electrode, the environment (a few mL of solution) is easily switched from one composition to another. Precise hydrodynamic control is achieved by rotating the electrode at variable high speeds. Rotation draws substrate to the electrode and it is easy to see if a reaction is diffusion controlled; likewise, products are swept away, allowing product inhibition to be measured and controlled. Contrary to expectation, in our experience, rotating at high speeds does not normally cause problems due to increased instability or loss of enzyme from the electrode surface. The technique is sensitive to trace reagents (<nM) because the electrochemical response at a typical electrode (area <0.1 cm2) reflects the action of much less than a picomole of active enzyme. For experiments on hydrogenases (and other enzymes acting on gaseous substrates) we use an all-glass electrochemical cell, fitting snugly against the electrode rotator.6 A precise mixture of gases can be passed through and solutions can be injected through a septum; gases can subsequently be removed by flushing with another gas. The experimental reference electrode we have used is the saturated calomel electrode (SCE): potentials are quoted relative to the standard hydrogen electrode (SHE) by adding +242 mV at 25 °C. The sealed apparatus, illustrated in Fig. 2, gives much improved results compared to open cells in which gases are introduced simply by bubbling into the solution.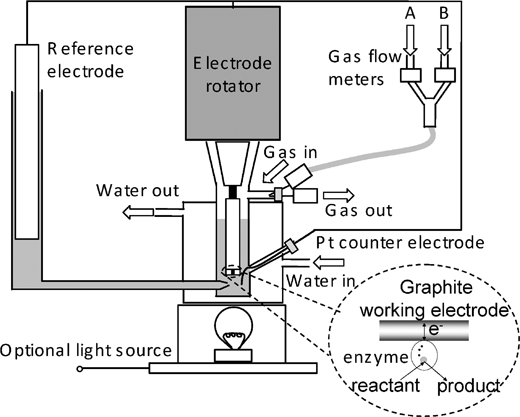 | ||
| Fig. 2 A schematic diagram of the apparatus used for protein film voltammetry experiments. The cell fits tightly against an electrode rotator and is equipped with gas flow controllers for providing accurate mixtures of gases. | ||
So far, most PFV studies on hydrogenases have used a carbon electrode, particularly pyrolytic graphite cut so that the ‘edge’ surface rather than the hydrophobic basal plane contacts the solution. This is known as a PGE electrode. Polishing produces a rough, oxidised surface that is effective for non-covalent hydrogenase adsorption. In some cases, polyions such as polymyxin (a polycation) pre-adsorbed on the electrode or mixed with enzyme solution, increase both current and stability.9 Efforts are being made to establish procedures for achieving more permanent, covalent links between hydrogenases and electrode surfaces.10Hydrogenase electrocatalysis has been observed on carbon microelectrodes, nanotubes and even Au modified with polymyxin.9 Semiconductors such as TiO2 have also been successfully tested and are now under investigation for photo H2 production.11
Interpreting data
For hydrogenases, two main types of experiment are employed to measure catalytic electron transport. The first of these is cyclic voltammetry, which involves cycling the electrode potential between two limiting values and monitoring the resulting current. This is useful for gaining an overall picture of the catalytic activity of the enzyme in each direction and for identifying potentials at which there is an interesting change in catalytic current, caused, for example, by a transition between active and inactive states. The second method is chronoamperometry, in which current is monitored as a function of time, after initiating the reaction, for instance by stepping the electrode potential to a particular value or changing the composition of the gas entering the cell.Oxidation processes produce a positive current whereas reduction processes give a negative current. The voltammetry of a hydrogenase usually shows three distinct regions, as shown in Fig. 3. At low potentials, the region of negative current (zone 1) corresponds to H+ reduction (H2 production) whereas at higher potential there are typically two regions of positive current (zones 2 and 3) each corresponding to net H2oxidation activity. Under conditions where the enzyme catalyses both H2oxidation and H2 production, the current trace cuts the zero-current line cleanly at the thermodynamic potential of the 2H+/H2 couple. This observation shows that hydrogenases operate ‘reversibly’ and require only the smallest overpotential to work in either direction (as with Pt, see later). The magnitudes of the currents achieved, once a sufficiently high overpotential is applied, reflect the inherent activities of the active sites. Taking pH and H2 partial pressure into consideration, comparisons of the currents achieved in zones 1 and 2 quantify the catalytic bias of a hydrogenase for catalysing H2 production relative to H2oxidation. At high potential, zone 3, hydrogenases undergo oxidative inactivation;5,8,12,13 and although this occurs over a large range of rates (depending on the hydrogenase) it is usually reversible and results in an oxidised ‘resting’ state. This is considered in greater detail below.
![Cyclic voltammograms comparing the catalytic activities, catalytic bias and extent of anaerobic inactivation of different hydrogenases as a function of potential. Experiments were conducted on films of Dd [FeFe]-H (10 °C, pH 6.0, scan rate 50 mV s−1), Rm [NiFe]-MBH (30 °C, pH 5.5, scan rate 20 mV s−1) and Db [NiFeSe]-H (25 °C, pH 6.0, scan rate 10 mV s−1) under Ar (grey lines) and H2 (black lines). The electrode was rotated at a constant rate ≥2500 rpm in each case. The results show that the [FeFe]-H is the most biased towards H2 production and that the [NiFeSe]-H is the least prone to anaerobic inactivation.](/image/article/2009/CS/b801144n/b801144n-f3.gif) | ||
| Fig. 3 Cyclic voltammograms comparing the catalytic activities, catalytic bias and extent of anaerobic inactivation of different hydrogenases as a function of potential. Experiments were conducted on films of Dd [FeFe]-H (10 °C, pH 6.0, scan rate 50 mV s−1), Rm [NiFe]-MBH (30 °C, pH 5.5, scan rate 20 mV s−1) and Db [NiFeSe]-H (25 °C, pH 6.0, scan rate 10 mV s−1) under Ar (grey lines) and H2 (black lines). The electrode was rotated at a constant rate ≥2500 rpm in each case. The results show that the [FeFe]-H is the most biased towards H2 production and that the [NiFeSe]-H is the least prone to anaerobic inactivation. | ||
In addition to catalysis, it is sometimes possible to observe discrete ‘non-turnover’ signals due to reversible electron transfer at redox centres. This requires a high electroactive coverage (above a few picomoles per square cm) and an inhibitor (such as CO) to block catalytic electron flow. Non-turnover signals consist of a pair of peaks, corresponding to oxidation and reduction, centred at the reduction potential of the redox centre, in this case, one or more of the FeS-clusters of the intramolecular relay.14 The signals can be analysed to obtain estimates of electron-transfer rates and electroactive coverage, the latter being necessary to convert current into turnover frequency per molecule (kcat).5
Some non-idealities must be mentioned. First, long-term stability is rarely achieved. The problem we refer to as ‘film loss’ involves enzyme molecules desorbing from the electrode or even unfolding, and it is a major reason for efforts that are underway to attach hydrogenases to the electrode by covalent bonds.10 Second, an effect that influences the shape of voltammograms is dispersion—inhomogeneity among the environments and orientations of the enzyme molecules in the film. A range of orientations of the enzyme with respect to the electrode surface gives rise to a spread of interfacial (enzyme-electrode) electron-transfer rate constants. At sufficiently high scan rates, an ideal voltammogram should show a constant current plateau corresponding to the enzyme turning over at its maximum possible rate. In real experiments, with the electrode rotating rapidly to remove mass-transport control, the voltammograms at high overpotential rarely reach a plateau but instead show a residual slope (a linear potential dependence), as is most evident in Fig. 3 (right-hand panel). Detailed studies15 of this potential dependence for some [NiFe]-hydrogenases show that the gradient of the residual slope is related to the degree of dispersion and to the ratio between the rate of enzyme turnover (kcat) and the (exchange) rate of interfacial electron transfer (k0)—a high ratio leading to greater slope. Raising the temperature results in a more linear waveform because chemical transformations at the active site (kcat) should have a higher activation energy (rate-determining at low temperature) than long-range interfacial electron transfer (k0) (rate limiting at high temperature). In hydrogenases, intramolecular electron transfer along the FeS relay is fast, yet can be slowed down when individual FeS clusters are altered by site directed mutagenesis.16
Reactions of hydrogenases with small molecules
Quantifying the affinity of a hydrogenase for H2
The affinity of hydrogenases for H2 is expressed most simplistically by the Michaelis constant (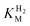 ). A useful electrochemical method for determining
). A useful electrochemical method for determining 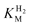 reported by Léger et al.17 is based upon the fact that when a gas is introduced into the cell solution, it can (unlike non-volatiles) be removed simply by flushing with an inert gas. The key point is that removal of the gas from solution follows a simple exponential time course. By measuring current as a function of time during the gas exchange, it is possible to extract not only KM values but also (as described later) inhibition constants (KI) for gaseous inhibitors such as CO.
reported by Léger et al.17 is based upon the fact that when a gas is introduced into the cell solution, it can (unlike non-volatiles) be removed simply by flushing with an inert gas. The key point is that removal of the gas from solution follows a simple exponential time course. By measuring current as a function of time during the gas exchange, it is possible to extract not only KM values but also (as described later) inhibition constants (KI) for gaseous inhibitors such as CO.
Results of a typical experiment to determine 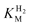 for Re [NiFe]-MBH are shown in Fig. 4. A PGE electrode modified with the enzyme was placed in a sealed glass electrochemical cell containing 2 mL buffer solution. The electrode, rotating at high speed, was poised at a H2-oxidising potential and the cell was flushed with N2 to obtain a ‘background’ current. At time t = 0, 2 mL of H2-saturated buffer (of identical composition and temperature to that already in the cell) was injected into the cell, causing a steep rise in current due to H2oxidation. The H2 concentration in the solution immediately begins to fall from its initial concentration of 0.4 mM as N2 is flushed through the headspace of the cell (the H2 concentration as a function of time is also indicated in Fig. 4), and the current decreases in a sigmoidal manner. The early part of the trace corresponds to the activity remaining fairly constant so long as the concentration of H2 is close to the level needed to saturate the active site; thus, even at the qualitative level, this experiment provides a rapid appraisal of the magnitude of
for Re [NiFe]-MBH are shown in Fig. 4. A PGE electrode modified with the enzyme was placed in a sealed glass electrochemical cell containing 2 mL buffer solution. The electrode, rotating at high speed, was poised at a H2-oxidising potential and the cell was flushed with N2 to obtain a ‘background’ current. At time t = 0, 2 mL of H2-saturated buffer (of identical composition and temperature to that already in the cell) was injected into the cell, causing a steep rise in current due to H2oxidation. The H2 concentration in the solution immediately begins to fall from its initial concentration of 0.4 mM as N2 is flushed through the headspace of the cell (the H2 concentration as a function of time is also indicated in Fig. 4), and the current decreases in a sigmoidal manner. The early part of the trace corresponds to the activity remaining fairly constant so long as the concentration of H2 is close to the level needed to saturate the active site; thus, even at the qualitative level, this experiment provides a rapid appraisal of the magnitude of 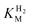 .
.
![Experiments to measure for wild-type Re [NiFe]-MBH at a potential of –108 mV vs.SHE. At time = 0 s, 2 mL of H2-saturated buffer (pH 5.5, 30 °C) was injected into the cell, which originally contained 2 mL of N2-saturated electrolyte of the same composition and temperature. Current vs. time traces are shown (bold trace), and overlaid on these are the H2 concentration which decreases exponentially (thin trace) and the sigmoidal fit (dotted trace). The electrode was rotated at a constant rate of 4500 rpm. The extended ‘plateau region’ at the start of the experiment typifies an enzyme with a high affinity for H2. Figure reproduced from ref. 18 with permission (Copyright 2008, American Society for Biochemistry and Molecular Biology).](/image/article/2009/CS/b801144n/b801144n-f4.gif) | ||
Fig. 4 Experiments to measure 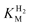 for wild-type Re [NiFe]-MBH at a potential of –108 mV vs.SHE. At time = 0 s, 2 mL of H2-saturated buffer (pH 5.5, 30 °C) was injected into the cell, which originally contained 2 mL of N2-saturated electrolyte of the same composition and temperature. Current vs. time traces are shown (bold trace), and overlaid on these are the H2 concentration which decreases exponentially (thin trace) and the sigmoidal fit (dotted trace). The electrode was rotated at a constant rate of 4500 rpm. The extended ‘plateau region’ at the start of the experiment typifies an enzyme with a high affinity for H2. Figure reproduced from ref. 18 with permission (Copyright 2008, American Society for Biochemistry and Molecular Biology). for wild-type Re [NiFe]-MBH at a potential of –108 mV vs.SHE. At time = 0 s, 2 mL of H2-saturated buffer (pH 5.5, 30 °C) was injected into the cell, which originally contained 2 mL of N2-saturated electrolyte of the same composition and temperature. Current vs. time traces are shown (bold trace), and overlaid on these are the H2 concentration which decreases exponentially (thin trace) and the sigmoidal fit (dotted trace). The electrode was rotated at a constant rate of 4500 rpm. The extended ‘plateau region’ at the start of the experiment typifies an enzyme with a high affinity for H2. Figure reproduced from ref. 18 with permission (Copyright 2008, American Society for Biochemistry and Molecular Biology). | ||
For a quantitative analysis, Léger and co-workers derived eqn (1) for the sigmoidal variation of current i(t) with time t following injection of H2 into the solution to give an initial concentration CH2(0). The initial current is imax and τ is the time constant for exponential gas removal (τ is typically in the range 30–100 s, depending on solution volume, rate of gas flow, and electrode rotation rate, which are constants for each experiment).
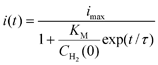 | (1) |
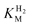 , which, from the average of multiple experiments, was determined as 6.1 µM at 30 °C, decreasing to 1.2 µM at 20 °C.18 These values are consistent with the requirement for hydrogenases from organisms that sequester low levels of H2 to have low
, which, from the average of multiple experiments, was determined as 6.1 µM at 30 °C, decreasing to 1.2 µM at 20 °C.18 These values are consistent with the requirement for hydrogenases from organisms that sequester low levels of H2 to have low 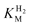 values.
values.
A number of factors, both experimental and fundamental, may cause the current vs. time trace to deviate from the sigmoidal shape. Trace levels of H2 diffusing back from the cell side arm or the glovebox itself hinder measurement of very low 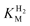 values because the assumption is that the H2 level in the cell tends to zero over time. Substrate transport limitation, unaccounted for in the assumptions, becomes unavoidable at extremely low levels of H2, even at high rotation rates and a low density of enzyme on the electrode. Care must also be taken to avoid measurements in a potential region corresponding to zone 3 (Fig. 3) because the current will decrease as the active site converts to an inactive form, and this transformation may become faster with decreasing H2 concentrations. The method is not useful for measuring high values of
values because the assumption is that the H2 level in the cell tends to zero over time. Substrate transport limitation, unaccounted for in the assumptions, becomes unavoidable at extremely low levels of H2, even at high rotation rates and a low density of enzyme on the electrode. Care must also be taken to avoid measurements in a potential region corresponding to zone 3 (Fig. 3) because the current will decrease as the active site converts to an inactive form, and this transformation may become faster with decreasing H2 concentrations. The method is not useful for measuring high values of 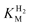 because if the initial H2 level is not sufficiently high relative to
because if the initial H2 level is not sufficiently high relative to 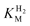 the current drops as soon as H2 begins to be removed, instead of following a sigmoidal trace, preventing determination of imax.
the current drops as soon as H2 begins to be removed, instead of following a sigmoidal trace, preventing determination of imax.
We have adapted Léger’s method to measure 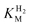 values for a series of mutants of Re [NiFe]-MBH and for the analogous Rm [NiFe]-MBH.18 All mutations of residues close to but not coordinating the [NiFe]-center have so far resulted in increases in
values for a series of mutants of Re [NiFe]-MBH and for the analogous Rm [NiFe]-MBH.18 All mutations of residues close to but not coordinating the [NiFe]-center have so far resulted in increases in 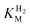 , consistent with the idea that these enzymes have become optimised for H2 scavenging. Similar results have recently been reported by Léger and co-workers for variants of Df [NiFe]-H.19
, consistent with the idea that these enzymes have become optimised for H2 scavenging. Similar results have recently been reported by Léger and co-workers for variants of Df [NiFe]-H.19
High potential anaerobic inactivation
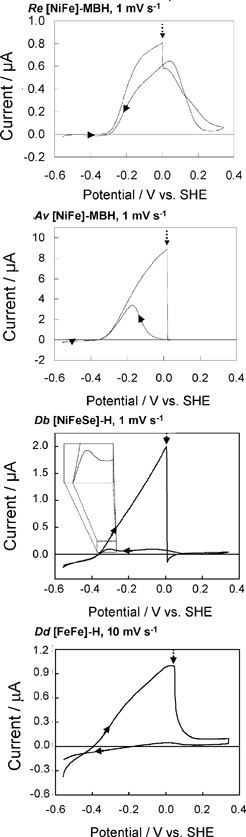
We consider next how to study the anaerobic inactivation reactions that are often manifest in the most oxidising region (zone 3) of the voltammograms. Reversible, anaerobic inactivation at high potential has been studied in detail for [NiFe]-hydrogenases, in particular Av [NiFe]-MBH.12 Anaerobic oxidation of [NiFe]-hydrogenases produces a Ni(III) state known as Ni-B, in which a hydroxide ligand is coordinated to the Ni in a bridging position with respect to the Fe(II).12,20 This state, which can also be formed upon reaction with O2, is rapidly re-activated by H2, hence it is also known as the ‘Ready’ (or Nir*) state. The [FeFe]-hydrogenases also undergo anaerobic inactivation, in this case resulting in a state known as Hoxinact in which the H-cluster valences are assigned as Fe(II)Fe(II)-[4Fe–4S]ox.21 Again, Hoxinact can be reactivated by H2. Based on our studies of Av [NiFe]-MBH and several other [NiFe]-hydrogenases, and the Dd [FeFe]-H, we take the following observations as a standard against which other hydrogenases can be compared. Some main comparisons are illustrated by the results shown in Fig. 5.![Comparison of anaerobic inactivation of hydrogenases under different conditions (the direction of potential cycling is indicated by the arrows); Re [NiFe]-MBH under atmospheres of 100% H2 and 10% H2 in N2 at pH 5.5 (30 °C, electrode rotation rate 4500 rpm, 10 mV s–1), Av [NiFe]-MBH under 100% H2 at pH 6.0 and pH 9.0 (50 °C, electrode rotation rate 2500 rpm, 0.3 mV s–1), Dd [FeFe]-H under 100% H2 at pH 4.8 and pH 8.4 (10 °C, electrode rotation rate 2500 rpm, 0.3 mV s–1). For Re [NiFe]-MBH, the response of an unmodified electrode recorded under the same conditions is overlaid. The hysteresis in zone 3 which is clearly evident in some experiments indicates where reactivation is much faster than inactivation.](/image/article/2009/CS/b801144n/b801144n-f5.gif) | ||
| Fig. 5 Comparison of anaerobic inactivation of hydrogenases under different conditions (the direction of potential cycling is indicated by the arrows); Re [NiFe]-MBH under atmospheres of 100% H2 and 10% H2 in N2 at pH 5.5 (30 °C, electrode rotation rate 4500 rpm, 10 mV s–1), Av [NiFe]-MBH under 100% H2 at pH 6.0 and pH 9.0 (50 °C, electrode rotation rate 2500 rpm, 0.3 mV s–1), Dd [FeFe]-H under 100% H2 at pH 4.8 and pH 8.4 (10 °C, electrode rotation rate 2500 rpm, 0.3 mV s–1). For Re [NiFe]-MBH, the response of an unmodified electrode recorded under the same conditions is overlaid. The hysteresis in zone 3 which is clearly evident in some experiments indicates where reactivation is much faster than inactivation. | ||
A unique feature of electrochemical methods is the ability to resolve different kinetic components of complex redox reactions. Cyclic voltammetry is supported here by chronoamperometry experiments in which transformations are initiated by stepping the electrode potential between different regions. This separates the potential domain from the time domain and allows us to distinguish between reaction steps that respond directly to electrochemical driving force and those that are controlled by other factors. Anaerobic inactivation, which is usually much slower than re-activation, is described in electrochemists’ terms as a ‘CE’ reaction: here, an initial chemical reaction or rearrangement (C), which may be coordination of OH– or H2O, is followed by an electron transfer (E) that traps the product. Viewed by chronoamperometry measurements, the rate of decrease in H2oxidation current does not depend on the electrode potential, in other words the chemical process controls the rate. Conversely, re-activation is an ‘EC’ reaction: the initial step is electron addition to give a reduced state that undergoes a chemical reaction to produce either active enzyme or a precursor that is finally activated by the binding of H2. Again, viewed by chronoamperometry, the rate of increase in H2oxidation activity now depends sharply on the electrode potential. As evident from Fig. 5, when comparing scans in the oxidising and reducing directions, re-activation is usually the better defined reaction in terms of reduction potential (this is sometimes called the ‘switch potential’ and it is obtained from the minimum of the first derivative of the current-potential plot).12 The entire cycle of inactivation and re-activation is a ‘CEEC’ sequence.
We have summarised the electrochemical interpretations of reversible anaerobic inactivation in Scheme 1. The electron-transfer (E) reactions are the simplest to assign: as determined for Av [NiFe]-MBH, formation of Ni-B involves oxidation of Ni(II) to Ni(III), the Fe remaining as Fe(II). For [FeFe]-hydrogenases, inactivation involves oxidation of the H-cluster to a state Hoxinact in which the binuclear center is formally Fe(II)Fe(II). The nature of the chemical steps is unclear. Our benchmark systems usually show a strong dependence on pH, with the [NiFe]- systems inactivating more rapidly at higher pH. Experiments on Dd [FeFe]-H have shown the opposite behaviour, i.e. more rapid inactivation at lower pH.22 For [NiFe]-hydrogenases, inactivation is also more extensive at lower H2 levels.5,12 This has not yet been established for the [FeFe]-hydrogenases.
![Simplest electrochemical representations of the conversions between active and inactive states of hydrogenases in the presence of H2. Note that in the case of [FeFe]-hydrogenases, we consider the active site to be just the [FeFe] unit and neglect redox changes at the FeS cluster.](/image/article/2009/CS/b801144n/b801144n-s1.gif) | ||
| Scheme 1 Simplest electrochemical representations of the conversions between active and inactive states of hydrogenases in the presence of H2. Note that in the case of [FeFe]-hydrogenases, we consider the active site to be just the [FeFe] unit and neglect redox changes at the FeS cluster. | ||
Inhibition by small molecules
The common inhibitors of hydrogenases are certain small gaseous molecules such as H2, CO, O2 and H2S, and electrochemical measurements reveal exquisite details of the rates and reversibility of the reactions involved. Inhibition can be quantified from steady-state current measurements for gas mixtures under constant flow or by using the procedure we have discussed earlier for measuring KM. Léger and co-workers derived an expression similar to eqn (1) to extract KI from the time dependence of the change in current as inhibitor is flushed out of solution.17Inhibition by H2
Hydrogenases exhibit widely differing behaviour in regard to how H2 production is inhibited by H2. Fig. 6 compares inhibition of H2 production by H2 for three different hydrogenases. Product inhibition is also evident in Fig. 3 where the [NiFe]-hydrogenases show a much lower H+ reduction current (zone 1) in the presence of H2 than in the presence of Ar. As a rough guide, KI values for H2 inhibition are of the order of µM for the [NiFe]-hydrogenases23 but may be much higher for the [FeFe]-hydrogenases. This is not surprising given that the physiological role of most [FeFe]-hydrogenases is production of H2 rather than its oxidation. Product inhibition is certainly one of the reasons why some hydrogenases are better H2 producers than others.![Experiments to compare the inhibition of H2 production by H2 and CO across a range of hydrogenases. All experiments were performed at –0.45 V vs.SHE at an electrode rotation rate of 3000 rpm. Experiments on Re [NiFe]-MBH and Dg [NiFe]-H were conducted at pH 5.5, 30 °C, whilst those on Dd [FeFe]-H were conducted at pH 6.0, 10 °C. All gas concentrations are 100% unless otherwise stated. Note the unusual case of Re [NiFe]-MBH, where H2 production is inhibited much less by CO than by H2.](/image/article/2009/CS/b801144n/b801144n-f6.gif) | ||
| Fig. 6 Experiments to compare the inhibition of H2 production by H2 and CO across a range of hydrogenases. All experiments were performed at –0.45 V vs.SHE at an electrode rotation rate of 3000 rpm. Experiments on Re [NiFe]-MBH and Dg [NiFe]-H were conducted at pH 5.5, 30 °C, whilst those on Dd [FeFe]-H were conducted at pH 6.0, 10 °C. All gas concentrations are 100% unless otherwise stated. Note the unusual case of Re [NiFe]-MBH, where H2 production is inhibited much less by CO than by H2. | ||
Inhibition by CO
Carbon monoxide is usually a potent inhibitor of hydrogenases, for both H2oxidation and H2 production. Interesting exceptions are the [NiFe]-MBH enzymes from Ralstonia species for which CO is an extremely weak (essentially ineffective) inhibitor of H2oxidation24 and very much weaker than H2 as an inhibitor of H2 production.23Fig. 6 also compares CO and H2 as inhibitors of H2 production by Re [NiFe]-MBH,23Dg [NiFe]-H and Dd [FeFe]-H. These experiments show the effects of switching between inert and inhibitor gases. For Dd [FeFe]-H and Dg [NiFe]-H, switching from N2 to CO immediately results in almost complete loss of H+ reduction current, then upon switching back to N2 there is a short lag period before the current begins to recover. In contrast, Re [NiFe]-MBH shows very much weaker inhibition by CO and recovery begins immediately upon switching the gas flow back to N2. When N2 is switched to 100% H2, the greatest effect is seen for Re [NiFe]-MBH (immediate and almost complete inhibition, then a lag period upon switching back to N2) and the smallest effect is seen for Dd [FeFe]-H. The rate of re-activation of CO-inhibited Dd [FeFe]-H is light sensitive and depends on potential.22
The observation that H2 and CO have such contrasting affinities for the same type of active site in different hydrogenases suggests that the active site in each case is sterically constrained so much as to be able to distinguish between the favoured binding geometries (sideways-on vs. end-on) of these ligands.
Inhibition by O2—the question of ‘O2 tolerance’
To enable hydrogenases or small molecular analogues to be used as H2 cycling catalysts in technological applications, they must be capable of operating in the presence of O2, even if this means just transient exposure to low levels. However, all hydrogenases that have so far been studied are inactivated or permanently damaged by O2, in many cases even at trace levels. Hydrogen-cycling organisms were on Earth long before O2 appeared in the atmosphere: as O2 levels began to build up organisms had to adapt to survive and one way to do this was to protect the active sites of hydrogenases against irreversible damage by O2. The ability of hydrogenases to function in the presence of O2 is known as ‘O2 tolerance’ and it is now an established property of some [NiFe]-hydrogenases.In contrast, it is widely reported25 that [FeFe]-hydrogenases (which are usually isolated from facultative anaerobic organisms) cannot sustain H2-oxidation activity in the presence of even traces of O2. Unlike CO, O2 induces substantial changes to the active site, causing oxidation of metal ions and sulfur, and attack by reactive products of O2reduction. Only for some [NiFe]-hydrogenases are the products of O2 attack well established, namely Ni-B and Ni-A (also known as ‘Unready’ or Niu*), which contain, respectively, a OH– ligand or the bound product (a peroxide or oxygenated cysteine-S atom) of incomplete reduction of O2 (see Scheme 2).26–28
![Representations of the products of aerobic inactivation of [NiFe]-hydrogenases. Depending on the availability of electrons, O2 is either reduced completely or trapped as a reactive oxygen species.](/image/article/2009/CS/b801144n/b801144n-s2.gif) | ||
| Scheme 2 Representations of the products of aerobic inactivation of [NiFe]-hydrogenases. Depending on the availability of electrons, O2 is either reduced completely or trapped as a reactive oxygen species. | ||
For [FeFe]-hydrogenases, the products of O2 reaction have not yet been characterised. There are major efforts to engineer greater O2 tolerance into hydrogenases, the main aim being to develop biosystems that can sustain high H2 production by photosynthesis or fermentation when O2 is present. The main focus so far has been on selectively restricting the access of O2 to the active site, the assumption being that H2 diffuses through the enzyme using tight gas channels.30 Although we agree that a diffusion filter may be operative, we believe that other factors are also important in enabling a hydrogenase to be O2-tolerant. The experiments shown in Fig. 7 illustrate the wide spectrum of O2 tolerance among hydrogenases.13,22,28,29 In each case, a small aliquot of O2-saturated solution is injected whilst H2oxidation is monitored as the potential is scanned in the positive direction. The O2-tolerant enzyme Re [NiFe]-MBH is only partially inhibited by O2 although the drop in current is rapid. At higher potential the hydrogenase undergoes anaerobic inactivation, then almost full re-activation occurs on the return scan. The switch potential is about +150 mV, the same as observed for anaerobic inactivation alone. Addition of O2 to Av [NiFe]-MBH causes rapid and complete inactivation, and (slow) re-activation occurs only when the potential on the return scan is taken below –0.1 V at which point in time all O2 has also been removed. The Db [NiFeSe]-H is also completely inhibited by a small amount of O2 but subsequently recovers activity (rapidly) below a potential of –0.3 V.29 The [FeFe]-hydrogenase reacts more slowly with O2, but shows only a tiny current when a more reducing potential is applied.22 Interestingly, Baffert et al. have reported that the initial reaction of O2 with Ca [FeFe]-H is reversible31 and we return to this later. For the [NiFe]-hydrogenases, there is a correlation between the switch potential and O2 tolerance in H2oxidation, because under aerobic conditions, each enzyme must remain inactive at potentials more positive than this value.
 | ||
| Fig. 7 Inactivation and subsequent re-activation of various hydrogenases after reaction with O2. Each O2 injection involved introduction of 0.2 mL of O2-saturated buffer into a 2 mL cell solution, resulting in ca. 90 µM O2 in the cell solution. The constant H2 flow rate and electrode rotation rate ensure that the O2 is flushed out after injection, and after 300 s no O2 remains in solution. All experiments were conducted at pH 6, 30 °C, 1 bar H2, at an electrode rotation rate of 2500 rpm. Panels 1, 2 and 4 are reprinted, with permission, from ref. 13 (copyright 2005 American Chemical Society), and panel 3 is reprinted, with permission, from ref. 29 (copyright 2008 American Chemical Society). | ||
Although not an O2-tolerant hydrogenase, Av [NiFe]-MBH has been extensively studied, first by EPR and IR spectroscopy, then by protein film voltammetry.13,28,32–34 Reaction with O2 produces the two inactive Ni(III) species Ni-A and Ni-B, in amounts that vary with the conditions under which O2 reacts with the active site. Under 100% H2 and applying a more negative electrode potential, most of the product is Ni-B, whereas when O2 is introduced under N2 and a more positive electrode potential, Ni-A predominates. This is easily seen in experiments (Fig. 8) that record the progress of reductive re-activation after a potential step. Re-activation is biphasic—the fast stage corresponding to reactivation of Ni-B whereas the slow phase corresponds to Ni-A. The effect of potential on the ratio of Ni-A to Ni-B produced by reaction with O2 suggests that Ni-A results from incomplete reduction of O2 in the active site and the formation of a ‘reactive oxygen species’ (ROS), whereas Ni-B results from complete reduction of O2. We have also noted, for all [NiFe]-hydrogenases, that reaction with O2 is much faster than anaerobic inactivation, showing that under O2, Ni-B forms by a different route, i.e. direct attack by O2.
![Experiments to investigate the formation and re-activation of the Unready (Ni-A) state of Av [NiFe]-MBH. Panel A shows the time-course of re-activation at −158 mV following injection of O2 under the conditions indicated for each trace. Note here that following exposure to O2 at low potential, the fast phase corresponding to Ready (Ni-B) predominates, whereas following exposure to O2 at high potential, the slow phase corresponding to Ni-A predominates. Panels B and C show the dependence of reactivation of Ni-A on electrode potential. All experiments were conducted at 45 °C, at an electrode rotation rate of 2500 rpm, and at 1 bar H2 and pH 6 unless otherwise stated. Panel A is reprinted, with permission, from ref. 28 (copyright 2004 American Chemical Society), and Panels B and C are reprinted, with permission, from ref. 27 (copyright 2005 American Chemical Society)](/image/article/2009/CS/b801144n/b801144n-f8.gif) | ||
| Fig. 8 Experiments to investigate the formation and re-activation of the Unready (Ni-A) state of Av [NiFe]-MBH. Panel A shows the time-course of re-activation at −158 mV following injection of O2 under the conditions indicated for each trace. Note here that following exposure to O2 at low potential, the fast phase corresponding to Ready (Ni-B) predominates, whereas following exposure to O2 at high potential, the slow phase corresponding to Ni-A predominates. Panels B and C show the dependence of reactivation of Ni-A on electrode potential. All experiments were conducted at 45 °C, at an electrode rotation rate of 2500 rpm, and at 1 bar H2 and pH 6 unless otherwise stated. Panel A is reprinted, with permission, from ref. 28 (copyright 2004 American Chemical Society), and Panels B and C are reprinted, with permission, from ref. 27 (copyright 2005 American Chemical Society) | ||
Based upon X-ray structure analysis, Ni-A has been proposed to be a peroxo species,26 which is consistent with the more oxidising conditions required for its formation. The electrochemistry has been studied in some detail: the kinetics of re-activation are first order and potential-dependent, reaching a limiting rate of 0.0025 s–1 (half-life 280 s) at 45 °C. The mechanism is of the ‘EC’ type, with the ‘C’ step rate-determining but not final: the last stage it seems is achieved by the binding of H2 or, surprisingly, CO.27 For Av [NiFe]-MBH, Ni-A and Ni-B appear to re-activate at very similar potentials. Evidently a HO– and a (putative) HOO– confer very similar influences on the Ni(III)/Ni(II) reduction potential. In contrast, experiments on the Db [NiFeSe]-H, which has not been well characterised in any oxidised inactive form, show that the product of reaction with O2 re-activates at a much lower potential than the species that is formed upon anaerobic inactivation.29
The slow rate of reactivation of Ni-A is sufficient to prevent most [NiFe]-hydrogenases from oxidising H2 in the presence of even trace O2. The [NiFe]-MBH enzymes from Ralstonia sp. are unusual in that they have the ability to oxidise H2 even in the presence of atmospheric levels of O2. Under conditions of 20% O2/80% H2, significant H2oxidation activity is retained, although the extent of O2 inhibition is strongly potential dependent. Significantly, EPR studies show that Ni-A is not formed when these enzymes react with O2, but signals are observed that correspond to Ni-B, the rapidly re-activating ‘resting’ state.35 The reason why these enzymes are O2 tolerant therefore is due, at least in part, to the fact that Ni-A is not formed, and in part to the high potential at which the inactive state, presumably Ni-B, is rapidly re-activated. One way to view the [NiFe]-hydrogenases is that O2 is a substrate that competes with H2, but causes reverse electron flow and produces a more complicated choice of reaction intermediates and rates of interconversion. This idea, that [NiFe]-hydrogenases are also oxidases (catalysing O2reduction albeit at rates that are sometimes very slow) is represented in Scheme 3.
![Schematic catalytic cycles for competing H2oxidation (left) and aerobic inactivation (right) of [NiFe]-hydrogenases.](/image/article/2009/CS/b801144n/b801144n-s3.gif) | ||
| Scheme 3 Schematic catalytic cycles for competing H2oxidation (left) and aerobic inactivation (right) of [NiFe]-hydrogenases. | ||
A model for the O2 tolerance of certain [NiFe]-hydrogenases can be proposed, based upon consideration of three distinct stages of the disruption caused by O2 to the normal catalytic cycle:
Access. How quickly does O2 reach the active site?
Reaction. What happens when O2 reaches the active site?
Recovery. How quickly can the active site recover?
‘Access’ refers to the so-called ‘gas-channel effect’ in which access of H2 and other gases to the active site is controlled by the size of channels in the protein.19,30,36,37 These channels are likely to be dynamic and are not necessarily directly revealed in static crystal structures.30 However, limiting access of O2 to the active site is insufficient itself to confer O2 tolerance, as some O2 will invariably reach the active site.
‘Reaction’ involves two sequential steps. First; the binding of O2 to the active site, which may be in direct competition with H2. Second; reduction of O2, either by four electrons to give Ni-B and, eventually, two H2O molecules, or by fewer than four electrons, which will result in a reactive oxygen species and formation of Ni-A. Thus O2 tolerance could be conferred by the availability of an additional electron source close to the active site. This raises an interesting point—an enzyme attached to an electrode (and by extension a biological membrane loaded with reduced quinol) ought to be better able to avoid ROS formation than a free enzyme molecule in solution.
‘Recovery’ involves the thermodynamics and kinetics of reductive activation. A high reduction potential for the inactive state (i.e. minimal driving force for reactivation) is advantageous and an inherently fast reduction process is essential in order for hydrogen oxidation to persist under aerobic conditions.
In summary, for O2 tolerant H2oxidation, access of O2 must be slow, the reaction of O2 must be highly selective for the formation of an easily-recovered state, and recovery from this state should be fast. Although a hydrogenase can thus survive O2 reaction by acting as an oxidase, the extent of this reverse electron flow is never sufficient to prevent the biological action of the enzyme.
A similar model could be useful for [FeFe]-hydrogenases. However, the products of O2 attack have yet to be characterised and most studies to date suggest that the formation of the oxidised inactive states is irreversible. The irreversible reaction of O2 at the active site implies that limiting its access is crucial as this is the only available mechanism for restricting the rate at which O2 can react. This draws attention again to the suggestion by Baffert et al., that for Ca [FeFe]-H the initial attack of O2 on the active site is reversible, but the O2-adduct undergoes further irreversible reaction.31
Inhibition by H2S
Hydrogen sulfide is another potentially potent inhibitor and was the subject of earlier controversy regarding the active site structure of Dv [NiFe]-MBH.38 Initial crystallographic studies on this enzyme by Ogata and colleagues showed that a sulfur atom rather than an oxygen atom was coordinated to Ni in the bridging position. Additionally, H2S liberation was detected during the re-activation of as-purified enzyme.39,40 A question we addressed41 was how easy it would be to introduce a sulfur ligand into the active site then remove it by reduction. Since H2S is produced by sulfate-reducing bacteria the conditions under which it inhibits are particularly relevant.Fig. 9 shows experiments to investigate the reaction of Dv [NiFe]-MBH with sulfide and the mechanism by which the inactive product is re-activated. Most experiments employed injection of a solution of Na2S dissolved in buffer at pH 6: the pK for H2S/HS– is 6.8 so the predominant species is H2S. Injecting the sulfide solution when the potential is more positive than +100 mV causes immediate inactivation; but if sulfide is injected at a more negative potential, no inactivation is observed until the potential has climbed above 0 V. Activity is recovered on the return scan, at a potential that is some 130 mV higher than that observed for recovery from O2 under the same conditions. Reaction of the hydrogenase with sulfide is thus reversible, but the inactive species that is formed is stable only under quite oxidising conditions (at a positive potential). It is certain that these conditions must have determined the state of the active site when crystals of the sulfide-form of Dv [NiFe]-MBH were grown. However, since sulfide is not bound at less oxidising potentials, it is unlikely that the enzyme is inhibited in vivo, even when large amounts of H2S are being produced.
![Voltammetric experiments to investigate the reaction of sulfide with a [NiFe]-hydrogenase (Dv [NiFe]-MBH). All experiments were conducted at pH 6, 45 °C, electrode rotation rate of 2500 rpm, under 1 bar H2. The voltammograms in panel A were recorded at 3 mV s–1 and the voltammograms in Panel B were recorded at 1 mV s–1. Figure reproduced with permission from ref. 41 (copyright 2006 American Chemical Society).](/image/article/2009/CS/b801144n/b801144n-f9.gif) | ||
| Fig. 9 Voltammetric experiments to investigate the reaction of sulfide with a [NiFe]-hydrogenase (Dv [NiFe]-MBH). All experiments were conducted at pH 6, 45 °C, electrode rotation rate of 2500 rpm, under 1 bar H2. The voltammograms in panel A were recorded at 3 mV s–1 and the voltammograms in Panel B were recorded at 1 mV s–1. Figure reproduced with permission from ref. 41 (copyright 2006 American Chemical Society). | ||
As the pH is raised, the inactivation becomes less rapid and the switch potential for reactivation shifts to lower values. As a higher pH lowers the amount of H2S available to attack the active site, the results suggest that the attacking species is H2S rather than HS−, although HS− is likely to be the ligand actually coordinated. Another interesting observation is that active hydrogenase reacts with sulfide, but the inactive ‘resting states’ Ni-B and Ni-A do not. This supports the proposal that the site of reaction is the active site rather than one of the FeS clusters. Ogata and colleagues reported that Ni-A is formed in particularly high yield if the enzyme is first exposed to sulfide before reacting with O2.40 This ‘new route’ to Ni-A was thus tested electrochemically. The green trace in Fig. 9B shows the reactivation of hydrogenase in which the sulfide adduct was subsequently treated with O2 while holding the electrode potential at a positive value; from its much lower re-activation potential, the sulfide adduct is no longer present. Chronoamperometric studies showed that the product behaves like ‘pure’ Ni-A (i.e., there is a single phase having the same rate of reactivation as Ni-A generated from serial O2 injections, cf.Fig. 8) rather than the mixtures of Ni-A and Ni-B that are always formed upon direct reaction with O2. One possibility is that the sulfide entity is quite easily released from a state that is otherwise very electron-poor, and we have seen above, with Av [NiFe]-MBH, how more oxidising conditions favour formation of Ni-A over Ni-B.
Similar experiments have shown that both Dg [NiFe]-H and Df [NiFe]-H, also both from sulfate-reducing bacteria, form sulfide adducts, and Av [NiFe]-MBH also reacts with sulfide but to a much lesser extent than the Desulfovibrio species. In addition to the [NiFe]-containing hydrogenases, we have observed sulfide inhibition for Db [NiFeSe]-H and Dd [FeFe]-H. However, the O2-tolerant Re [NiFe]-MBH shows no reaction with sulfide over a wide range of conditions. As with CO, this lack of inhibition is informative, but only once structural information becomes available will an explanation be found.
H2 production in the presence of O2
Concepts behind electrochemical measurements of H2 production in the presence of O2
Microbes produce H2 not only by anaerobic fermentation, allowing its generation from organic waste, but also in photosynthetic processes that allow it to be generated from sunlight. Microalgae and cyanobacteria are attracting considerable interest regarding their possible exploitation in ‘H2 farms’;25,42 however, they carry out oxygenic photosynthesis which poses a problem because the O2 released by Photosystem II must be prevented from inactivating the hydrogenase. Hydrogenases with the ability to catalyse H2 production under aerobic conditions have thus become the ‘Holy Grail’ of the effort to photosynthesise H2. We have already discussed O2 tolerance in the context of H2oxidation, and we pointed out that aside from ‘designing’ enzymes to physically restrict access of O2 to the active site, a chemical tolerance to O2 can also be conferred upon the active site.Several studies have been carried out to evaluate the ability of hydrogenases to carry out sustained H2 production in the presence of O2.23,29,31,32 This type of experiment poses a problem for conventional methods because O2 reacts rapidly with (and consumes) the low-potential electron donors that provide the electrons for H2 production; but this need not be a problem when the hydrogenase is attached to an electrode. Our experiments begin by forming a film of hydrogenase and setting the electrode potential at a value below that for the reversible 2H+/H2 couple: we are thus examining zone 1 of the voltammograms shown in Fig. 3. A steady-state reduction current is observed which corresponds to H2 production. When O2 is introduced, two things occur regardless of whether or not the hydrogenase is inactivated; first, there should be an increase in reduction current due to O2reduction at the graphite electrode; second, this consumes some of the O2 and may produce partially-reduced oxygen species. The problem of monitoring H2 production when O2 is present is solved by introducing a gaseous inhibitor of hydrogenase and measuring the changes in reduction current as it first attacks and is then flushed from the cell. The actual O2 concentration experienced by the hydrogenase at any particular electrode potential is estimated by carrying out control experiments on O2reduction at a bare PGE rotating disc electrode.
Hydrogen production in air by the ‘O2-tolerant’ [NiFe]-MBH enzymes from aerobes (Ralstonia)
Although the [NiFe]-MBH enzymes from Ralstonia (Re and Rm) are very poor at catalysing H2 production (see Fig. 3 and 5), part of this is due to product inhibition and these hydrogenases can produce H2 provided it is continually removed. With this fact in mind, the high O2 tolerance of these hydrogenases suggested them as candidates for investigations of H2 production in the presence of O2. An experiment demonstrating H2 production when air is flushed through the cell is shown in Fig. 10.23 Because CO is such a poor inhibitor of Rm [NiFe]-MBH, we used H2 as the inhibitor (to quantify its own production). The experiment commences with N2 flowing through the headspace of the cell. The current due to H+ reduction at –0.45 V drops immediately when 10% H2 is introduced into the cell then recovers as the inhibitor is removed from the headspace. When 21% O2 is flushed through the headspace, there is a large increase in current resulting from O2reduction at the electrode. From control experiments we estimated that under these reducing conditions approximately 14% O2 survives at the electrode surface to be ‘sensed’ by the enzyme. However, when H2 is introduced into the headgas (i.e., the composition becomes 21% O2, 10% H2, 69% N2), a further decrease in current is recorded. This decrease is similar in magnitude to that prior to O2 introduction. This result shows that hydrogenase-catalysed H2-production must have contributed to the total current prior to introduction of the inhibitor. When the headgas is restored to 21% O2 in N2, the increase in reduction current signifies recovery of enzyme-catalysed H2 production. When O2 is flushed out, the current corresponding solely to enzymatic H2 production is re-established. Finally, a repeat of the initial inhibition phase is performed to confirm the activity of the enzyme. The surprising significance of these results is that for Rm [NiFe]-MBH, O2 behaves as a much weaker inhibitor than H2.![H2 production by Rm [NiFe]-MBH in the presence of 21% O2. Experimental conditions: pH 5.5, potential –0.45 V vs.SHE, electrode rotation rate 3000 rpm, 40 °C, N2 is the carrier gas throughout the experiment and the concentrations of H2 and O2 indicated are introduced into the head gas. Figure reproduced with permission from ref. 23 (copyright 2008 American Chemical Society).](/image/article/2009/CS/b801144n/b801144n-f10.gif) | ||
| Fig. 10 H2 production by Rm [NiFe]-MBH in the presence of 21% O2. Experimental conditions: pH 5.5, potential –0.45 V vs.SHE, electrode rotation rate 3000 rpm, 40 °C, N2 is the carrier gas throughout the experiment and the concentrations of H2 and O2 indicated are introduced into the head gas. Figure reproduced with permission from ref. 23 (copyright 2008 American Chemical Society). | ||
Hydrogen production by a [NiFeSe]-hydrogenase in the presence of low-level O2
The Db [NiFeSe]-H is a much better H2 producer than the [NiFe]-hydrogenases we have investigated.29 We saw in Fig. 7 that this enzyme cannot be an O2-tolerant H2 oxidiser because although it recovers from O2 attack, it only does this rapidly at a potential that is almost as negative as the 2H+/H2 couple. But the rapidity of this re-activation, determined in potential-step experiments, suggested that the hydrogenase could instead be a H2-producer in the presence of O2. The experiment confirming this proposal is shown in Fig. 11.29 Note that when O2 is introduced into the cell the reduction current decreases rapidly as the enzyme is immediately inhibited before levelling off as the O2 equilibrates with the cell solution. When O2 is removed from the cell, the current initially decreases slightly (due to decreased O2-reduction at the electrode, circled) but then increases as hydrogenase-catalysed H2 production recovers.![H2 production in the presence of O2 by Db [NiFeSe]-H. Experimental conditions: electrode rotation 3000 rpm, –0.45 V vs.SHE, pH 6.0, 30 °C, N2 is the carrier gas throughout the experiment and the concentrations of CO and O2 indicated are introduced into the head gas. Figure reproduced, with permission, from ref. 29 (copyright 2008 American Chemical Society).](/image/article/2009/CS/b801144n/b801144n-f11.gif) | ||
| Fig. 11 H2 production in the presence of O2 by Db [NiFeSe]-H. Experimental conditions: electrode rotation 3000 rpm, –0.45 V vs.SHE, pH 6.0, 30 °C, N2 is the carrier gas throughout the experiment and the concentrations of CO and O2 indicated are introduced into the head gas. Figure reproduced, with permission, from ref. 29 (copyright 2008 American Chemical Society). | ||
Demonstrations of possible future applications of hydrogenases
Comparing hydrogenases with platinum
It is very timely to compare the electrocatalytic activities of hydrogenases with the activity of a Pt electrocatalyst. So far, in simple experiments, and based on estimates of enzyme coverage, the H2 production rate by Ca [FeFe]-H (HydA)43 and the H2oxidation rates of Av [NiFe]-MBH44 and Db [NiFeSe]-H45 all seem to compare quite favourably with Pt at modest temperatures and neutral pH. However, it matters whether our comparisons relate to practical or fundamental considerations. The H2 pressures provided to a normal fuel cell are much higher than the levels used in experiments on hydrogenases, and very much higher than the enzymes would experience in an organism. It is very likely that evolutionary pressure on H2 oxidising enzymes favoured a high affinity for H2 (a low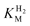 ) and ability to deal with inhibitors such as CO and O2, which would be more important than simply having a high turnover frequency (kcat). The H2 oxidising hydrogenases might then be superior to Pt only when compared at low H2 pressures (too low to be useful in normal fuel cells) and where a membrane is undesirable (e.g. in miniaturised fuel cells). Another practical disadvantage is that the ‘footprint’ of an enzyme molecule on an electrode is orders of magnitude larger than the tiny cluster of Pt atoms on which H2 is produced or oxidised. However, at the fundamental level, the activity per active site of enzyme could be much higher than for the minimum catalytic Pt unit. Generally speaking, enzymes are restricted to operate within a narrow range of temperature and pH and they are usually considered rather unstable. Against this, neither are Pt fuel cell electrodes stable at the atomic level (high Pt loadings are needed to counteract loss from the support) and Pt is particularly susceptible to inactivation by H2S and CO. In contrast the activity of hydrogenases is finely tuned by the ligand and supramolecular environment around the active site and inhibition by these gases (and O2) can be prevented or minimised.
) and ability to deal with inhibitors such as CO and O2, which would be more important than simply having a high turnover frequency (kcat). The H2 oxidising hydrogenases might then be superior to Pt only when compared at low H2 pressures (too low to be useful in normal fuel cells) and where a membrane is undesirable (e.g. in miniaturised fuel cells). Another practical disadvantage is that the ‘footprint’ of an enzyme molecule on an electrode is orders of magnitude larger than the tiny cluster of Pt atoms on which H2 is produced or oxidised. However, at the fundamental level, the activity per active site of enzyme could be much higher than for the minimum catalytic Pt unit. Generally speaking, enzymes are restricted to operate within a narrow range of temperature and pH and they are usually considered rather unstable. Against this, neither are Pt fuel cell electrodes stable at the atomic level (high Pt loadings are needed to counteract loss from the support) and Pt is particularly susceptible to inactivation by H2S and CO. In contrast the activity of hydrogenases is finely tuned by the ligand and supramolecular environment around the active site and inhibition by these gases (and O2) can be prevented or minimised.
A hydrogen fuel cell running on a dilute mixture of H2 in air
The high H2 affinity and O2-tolerance of Rm [NiFe]-MBH46 was demonstrated through its use as the anode catalyst in a membraneless fuel cell, generating power from an atmosphere of 3% H2 in air.47,48 As shown schematically in Fig. 12A, the hydrogenase-modified anode was placed in a pH 5 solution close to a cathode modified with laccase from Trametes versicolor. Laccase is a ‘blue’ multi-copper enzyme which catalyses four-electron reduction of O2 to H2O at a potential within 100–200 mV of the thermodynamically reversible value. The power produced was sufficient to allow a digital watch to run for over 24 hours (Fig. 12B), and a maximum power output of ca. 5 µW cm–2 was recorded (Fig. 12C).![H2oxidation in air and power generation in a membraneless H2/O2fuel cell. Panel A: Schematic diagram summarising the reactions taking place in the fuel cell: desirable reactions are shown in blue whilst undesirable reactions are shown in red. Panel B: Using the fuel cell to generate useful power. Panel C: Power vs. applied resistance curves show that power is not produced when the O2-tolerant Rm [NiFe]-MBH was replaced with the O2-sensitive Av [NiFe]-MBH, or when either enzyme was omitted. Experimental conditions were pH 5, 25 °C. Panel D: Cyclic voltammograms of Rm [NiFe]-MBH under conditions of 1% H2 in N2 (dotted) and 1% H2 in air (bold); the response of a blank electrode in 1% H2 in air is also shown (dashed). All pH 5.5, 30 °C, scan rate 2 mV s−1. Panels A and B are reprinted with permission from ref. 47, reproduced by permission of the Royal Society of Chemistry, and Panel D is reprinted with permission from ref. 46 (copyright 2008 American Chemical Society).](/image/article/2009/CS/b801144n/b801144n-f12.gif) | ||
| Fig. 12 H2oxidation in air and power generation in a membraneless H2/O2fuel cell. Panel A: Schematic diagram summarising the reactions taking place in the fuel cell: desirable reactions are shown in blue whilst undesirable reactions are shown in red. Panel B: Using the fuel cell to generate useful power. Panel C: Power vs. applied resistance curves show that power is not produced when the O2-tolerant Rm [NiFe]-MBH was replaced with the O2-sensitive Av [NiFe]-MBH, or when either enzyme was omitted. Experimental conditions were pH 5, 25 °C. Panel D: Cyclic voltammograms of Rm [NiFe]-MBH under conditions of 1% H2 in N2 (dotted) and 1% H2 in air (bold); the response of a blank electrode in 1% H2 in air is also shown (dashed). All pH 5.5, 30 °C, scan rate 2 mV s−1. Panels A and B are reprinted with permission from ref. 47, reproduced by permission of the Royal Society of Chemistry, and Panel D is reprinted with permission from ref. 46 (copyright 2008 American Chemical Society). | ||
Although the absolute amount of power generated from this setup was small, the experiment stands as a proof of concept, demonstrating what is possible under the most disadvantageous conditions with a sufficiently selective catalyst. For practical purposes, the device is perhaps better employed as a H2 sensor. Additionally, these experiments are valuable as an alternative to ‘conventional' electrochemical experiments in probing the reactions of hydrogenases, providing probably the closest way in which we can explore how certain enzymes would behave in vivo.
At low loads, the power generated by the fuel cell drops sharply (Fig. 12C), reflecting the high-potential inactivation process at the hydrogenase, which could be ‘re-activated' only by application of open-circuit conditions for a short period, presumably in order to reduce the hydrogenase through enforcement of a low electrode potential. Fig. 12D shows a voltammetric experiment carried out in an atmosphere containing low-level H2 in air, and demonstrates the narrow potential region in which a net oxidation current can be observed.46 A similar effect would presumably also occur in vivo if the cell redox potential was to become too positive.
Coupling H2oxidation to catalysis by a second enzyme on conducting particles
Pyrolytic graphite is easily ground into particles, which produces some interesting possibilities. We are developing the concept of attaching complementary pairs of redox enzymes to particles of graphite and other conducting materials, in order to create selective heterogeneous redox catalysts.49 Electrons required by one enzyme (the electron acceptor enzyme) are supplied, via conduction, from the reaction catalysed by the electron-donor enzyme. Hydrogenases are the obvious candidates as electron-donor enzymes, making it possible to carry out enzymatic reductions simply by placing a suspension of particles under a H2 atmosphere. The particles can be easily separated by centrifugation and stored between use. The concept is illustrated in Fig. 13. | ||
| Fig. 13 A schematic representation of catalytic electron flow through a conducting particle to which are attached a hydrogenase and another enzyme capable of catalysing a reduction process using electrons produced through H2oxidation. | ||
As a proof of concept, we carried out experiments using Av [NiFe]-MBH as the electron-donor enzyme and the respiratory nitrate reductase from E. coli (NarGHI) as electron-acceptor enzyme. The reaction product, nitrite, was easy to assay using the colorimetric Griess reagent and quantitative conversions were observed.49
Photochemical H2 production
With the goal of achieving photocatalytic H2 production, we have investigated the electrochemistry of hydrogenases adsorbed on TiO2-ITO electrodes (ITO = indium-doped tin oxide). Experiments with Db [NiFeSe]-H which is both a good H2 producer and reasonably O2 tolerant in H2 production29 (see above) showed that it displayed excellent electrocatalytic voltammetry with very promising stability.11This allowed us to develop a rational basis for the interpretation of earlier reports of photo-H2 production by hydrogenases, including highly O2-sensitive enzymes, adsorbed on TiO2 upon UV band-gap irradiation.50 To achieve visible light-driven water reduction, we adsorbed Db [NiFeSe]-H on TiO2nanoparticles to which we had attached the photosensitizer [RuII(bipy)2(4,4′-(PO3H2)2-bipy)]Br2 (RuP; bipy = 2,2′-bipyridine). We then suspended the particles in a buffer solution containing EDTA/Tris as sacrificial electron donor (Fig. 14). Visible light irradiation (λ > 420 nm) excites the RuP photo-sensitizer, which injects electrons into the conduction band of TiO2 and on to the hydrogenase, resulting in efficient and catalyticH+ reduction. The O2-tolerance of Db [NiFeSe]-H for H2 production may allow this to be extended to include a water oxidation catalyst thereby leading to photochemical water splitting, resembling artificial photosynthesis.
![(A) Schematic representation of visible light-driven H2 production with Db [NiFeSe]-H attached on ruthenium-dye sensitized TiO2nanoparticles, in the presence of a sacrificial electron donor D. (B) Time-dependent H2 evolution from bio-photocatalytic experiments (λ > 420 nm) with this system at 25 °C in buffered neutral water (squares) measured by gas chromatography. The turnover numbers (TON) are defined as mol H2 produced per mol of hydrogenase (shown in thousands) and a control experiment (no dye) is also shown (circles). Chem. Commun., DOI: 10.1039/b817371k. Reproduced by permission of the Royal Society of Chemistry.](/image/article/2009/CS/b801144n/b801144n-f14.gif) | ||
| Fig. 14 (A) Schematic representation of visible light-driven H2 production with Db [NiFeSe]-H attached on ruthenium-dye sensitized TiO2nanoparticles, in the presence of a sacrificial electron donor D. (B) Time-dependent H2 evolution from bio-photocatalytic experiments (λ > 420 nm) with this system at 25 °C in buffered neutral water (squares) measured by gas chromatography. The turnover numbers (TON) are defined as mol H2 produced per mol of hydrogenase (shown in thousands) and a control experiment (no dye) is also shown (circles). Chem. Commun., DOI: 10.1039/b817371k. Reproduced by permission of the Royal Society of Chemistry. | ||
Outlook
Protein film voltammetry is revealing a wealth of important information on hydrogenases, and complementing structural and spectroscopic studies on this class of enzyme. Electrochemical methods provide a sharp tool with which to navigate an extremely complex subject. The breadth of experiments now being conducted suggests that ‘Protein Film Electrochemistry’ (PFE) might be a more suitable term.In order to make further progress in determining the mechanisms of O2 tolerance it is essential to obtain structural and spectroscopic data for the relevant states of O2-tolerant hydrogenases. The tiny sample requirements and high resolution of kinetic and thermodynamic factors make PFV studies very valuable for comparing different enzymes and pinpointing even the subtlest of differences that should eventually be traceable at the atomic level.
A significant observation, when comparing hydrogenases with model complexes that have been prepared so far, is that hydrogenases, like platinum, catalyse reversible H2 cycling without any overpotential. This is even true of those hydrogenases from Ralstonia, that are strongly inhibited by H2—take away the H2 and they will produce H2 with minimal overpotential. This fact sets an important benchmark for the development of alternative catalysts to platinum for fuel cells. The intrinsic activity of the hydrogenase active site is very high and is comparable with that of Pt. Despite disadvantages such as the low practical power density (due to the voluminous nature of enzymes) it is possible that hydrogenases could be used in niche applications, including sensors.
Could an isolated active site work without the protein ‘casing’? It would be highly desirable to have molecular catalysts that were almost as small as the active sites, because enzymes have limited stability, are expensive to isolate, and take up so much space on an electrode that we would need hundreds of monolayers to achieve acceptable current densities. The difficulty of synthesising small model analogues that have all their component atoms ‘in the right place’ cannot be overstated. The coordination spheres within either the [NiFe] and [FeFe] classes probably differ little between enzymes that otherwise display wide variations in O2 tolerance and catalytic bias for H2 production vs.oxidation; so the protein environment plays a very important role in tuning the activity and deterring inhibitors.
Assuming that in the right context hydrogenases will be applied in new electrochemical technologies, it will be necessary to establish reliable procedures to achieve strong and robust attachment to their electrode or other conducting support. Achieving this goal would be useful also in particle technologies such as the coupling of hydrogenases to other enzymes or in developments along the lines of the photo H2 production we described previously.
Biological production of H2 on a large scale brings certain practical requirements that need to be addressed at the molecular as well as the microbial level. The hydrogenases should be good H2 producers and for photosynthetic H2 production the enzymes must be able to sustain activity in the presence of O2. The electrochemical studies are providing important new data and generating clearer insight as to how this might be achieved.
Acknowledgements
Research on the electrocatalytic properties of hydrogenases carried out in the Armstrong group has been supported by grants from the BBSRC (43/E16711 and BB/D52222X/1) and EPSRC (Supergen and quota awards). K. A. V. is a Royal Society Research Fellow.References
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS.
- R. J. Maier, Biochem. Soc. Trans., 2005, 33, 83–85 CrossRef CAS.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS.
- C. Léger, A. K. Jones, W. Roseboom, S. P. J. Albracht and F. A. Armstrong, Biochemistry, 2002, 41, 15736–15746 CrossRef CAS.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS.
- K. A. Vincent and F. A. Armstrong, Inorg. Chem., 2005, 44, 798–809 CrossRef CAS.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef CAS.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef CAS.
- F. J. M. Hoeben, I. Heller, S. P. J. Albracht, C. Dekker, S. G. Lemay and H. A. Heering, Langmuir, 2008, 24, 5925–5931 CrossRef CAS.
- O. Rüdiger, J. M. Abad, E. C. Hatchikian, V. M. Fernandez and A. L. De Lacey, J. Am. Chem. Soc., 2005, 127, 16008–16009 CrossRef.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun. 10.1039/b817371k.
- A. K. Jones, S. E. Lamle, H. R. Pershad, K. A. Vincent, S. P. J. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2003, 125, 8505–8514 CrossRef CAS.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179–18189 CrossRef CAS.
- H. R. Pershad, J. L. C. Duff, H. A. Heering, E. C. Duin, S. P. J. Albracht and F. A. Armstrong, Biochemistry, 1999, 38, 8992–8999 CrossRef CAS.
- C. Léger, A. K. Jones, S. P. J. Albracht and F. A. Armstrong, J. Phys. Chem. B, 2002, 106, 13058–13063 CrossRef CAS.
- S. Dementin, V. Belle, P. Bertrand, B. Guigliarelli, G. Adryanczyk-Perrier, A. L. De Lacey, V. M. Fernandez, M. Rousset and C. Léger, J. Am. Chem. Soc., 2006, 128, 5209–5218 CrossRef CAS.
- C. Léger, S. Dementin, P. Bertrand, M. Rousset and B. Guigliarelli, J. Am. Chem. Soc., 2004, 126, 12162–12172 CrossRef CAS.
- M. Ludwig, J. A. Cracknell, O. Lenz, K. A. Vincent and F. A. Armstrong, J. Biol. Chem. DOI:10.1074/jbc.M803676200.
- F. Leroux, S. Dementin, B. Burlat, L. Cournac, A. Volbeda, S. Champ, L. Martin, B. Guigliarelli, P. Bertrand, J. C. Fontecilla-Camps, M. Rousset and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11188–11193 CrossRef CAS.
- F. A. Armstrong and S. P. J. Albracht, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 937–954 CrossRef CAS ; discussion 1035–1040.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS.
- A. Parkin, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2006, 128, 16808–16815 CrossRef CAS.
- G. Goldet, A. F. Wait, J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 11106–11113 CrossRef CAS.
- K. A. Vincent, J. A. Cracknell, O. Lenz, I. Zebger, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16951–16954 CrossRef CAS.
- M. L. Ghirardi, M. C. Posewitz, P.-C. Maness, A. Dubini, J. Yu and M. Seibert, Annu. Rev. Plant Biol., 2007, 58, 71–91 Search PubMed.
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. J. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg. Chem., 2005, 10, 591–591 CrossRef CAS.
- S. E. Lamle, S. P. J. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 6595–6604 CrossRef CAS.
- S. E. Lamle, S. P. J. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2004, 126, 14899–14909 CrossRef CAS.
- A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS.
- J. Cohen, K. Kim, P. Kimg, M. Seibert and K. Schulten, Structure, 2005, 13, 1321–1329 CrossRef CAS.
- C. Baffert, M. Demuez, L. Cournac, B. Burlat, B. Guigliarelli, P. Bertrand, L. Girbal and C. Léger, Angew. Chem., Int. Ed., 2008, 47, 2052–2054 CrossRef CAS.
- B. Bleijlevens, F. A. Broekhuizen, A. L. Lacey, W. Roseboom, V. M. Fernandez and S. P. J. Albracht, J. Biol. Inorg. Chem., 2004, 9, 743–752 CAS.
- B. Bleijlevens, B. W. Faber and S. P. J. Albracht, J. Biol. Inorg. Chem., 2001, 6, 763–769 CrossRef CAS.
- S. J. George, S. Kurkin, R. N. F. Thorneley and S. P. J. Albracht, Biochemistry, 2004, 43, 6808–6819 CrossRef CAS.
- F. Lendzian, personal communication.
- Y. Montet, P. Amara, A. Volbeda, X. Vernede, E. C. Hatchikian, M. J. Field, M. Frey and J. C. Fontecilla-Camps, Nat. Struct. Biol., 1997, 4, 523–526 CrossRef CAS.
- J. Cohen, K. Kim, M. Posewitz, M. L. Ghirardi, K. Schulten, M. Seibert and P. King, Biochem. Soc. Trans., 2005, 33, 80–82 CrossRef CAS.
- Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549–556 CrossRef CAS.
- M. Higuchi and T. Yagi, Biochem. Biophys. Res. Commun., 1999, 255, 295–299 CrossRef CAS.
- H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635–1642 CrossRef CAS.
- K. A. Vincent, N. A. Belsey, W. Lubitz and F. A. Armstrong, J. Am. Chem. Soc., 2006, 128, 7448–7449 CrossRef CAS.
- A. Melis and T. Happe, Plant Physiol., 2001, 127, 740–748 CrossRef CAS.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS.
- A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC.
- A. A. Karyakin, S. V. Morozov, E. E. Karyakina, N. A. Zorin, V. V. Perelygin and S. Cosnier, Biochem. Soc. Trans., 2005, 33, 73–75 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 424–425 CrossRef CAS.
- K. A. Vincent, J. A. Cracknell, J. R. Clark, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, Chem. Commun., 2006, 5033–5035 RSC.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS.
- K. A. Vincent, X. Li, C. F. Blanford, N. A. Belsey, J. H. Weiner and F. A. Armstrong, Nat. Chem. Biol., 2007, 3, 761–762 CrossRef CAS.
- P. Cuendet, K. K. Rao, M. Grätzel and D. O. Hall, Biochimie, 1986, 68, 217–221 CrossRef CAS.
Footnote |
| † Part of the renewable energy theme issue. |
| This journal is © The Royal Society of Chemistry 2009 |