Probing the dynamics of polyatomic multichannel elementary reactions by crossed molecular beam experiments with soft electron-ionization mass spectrometric detection
Piergiorgio
Casavecchia
*,
Francesca
Leonori
,
Nadia
Balucani
,
Raffaele
Petrucci
,
Giovanni
Capozza
and
Enrico
Segoloni
Dipartimento di Chimica, Università degli Studi di Perugia, 06123, Perugia, Italy. E-mail: piero@dyn.unipg.it
First published on 12th November 2008
Abstract
In this Perspective we highlight developments in the field of chemical reaction dynamics. Focus is on the advances recently made in the investigation of the dynamics of elementary multichannel radical–molecule and radical–radical reactions, as they have become possible using an improved crossed molecular beam scattering apparatus with universal electron-ionization mass spectrometric detection and time-of-flight analysis. These improvements consist in the implementation of (a) soft ionization detection by tunable low-energy electrons which has permitted us to reduce interfering signals originating from dissociative ionization processes, usually representing a major complication, (b) different beam crossing-angle set-ups which have permitted us to extend the range of collision energies over which a reaction can be studied, from very low (a few kJ mol−1, as of interest in astrochemistry or planetary atmospheric chemistry) to quite high energies (several tens of kJ mol−1, as of interest in high temperature combustion systems), and (c) continuous supersonic sources for producing a wide variety of atomic and molecular radical reactant beams. Exploiting these new features it has become possible to tackle the dynamics of a variety of polyatomic multichannel reactions, such as those occurring in many environments ranging from combustion and plasmas to terrestrial/planetary atmospheres and interstellar clouds. By measuring product angular and velocity distributions, after having suppressed or mitigated, when needed, the problem of dissociative ionization of interfering species (reactants, products, background gases) by soft ionization detection, essentially all primary reaction products can be identified, the dynamics of each reaction channel characterized, and the branching ratios determined as a function of collision energy. In general this information, besides being of fundamental relevance, is required for a predictive description of the chemistry of these environments via computer models. Examples are taken from recent on-going work (partly published) on the reactions of atomic oxygen with acetylene, ethylene and allyl radical, of great importance in combustion. A reaction of relevance in interstellar chemistry, as that of atomic carbon with acetylene, is also discussed briefly. Comparison with theoretical results is made wherever possible, both at the level of electronic structure calculations of the potential energy surfaces and dynamical computations. Recent complementary CMB work as well as kinetic work exploiting soft photo-ionization with synchrotron radiation are noted. The examples illustrated in this article demonstrate that the type of dynamical results now obtainable on polyatomic multichannel radical–molecule and radical–radical reactions might well complement reaction kinetics experiments and hence contribute to bridging the gap between microscopic reaction dynamics and thermal reaction kinetics, enhancing significantly our basic knowledge of chemical reactivity and understanding of the elementary reactions which occur in real-world environments.
 Piergiorgio Casavecchia Piergiorgio Casavecchia | Piergiorgio Casavecchia received his degree in chemistry from the University of Perugia, Italy then worked as a postdoctorate in Berkeley, California with Yuan T. Lee. He began his research at the University of Perugia by building a high-resolution “universal” crossed molecular beam apparatus. In 1990 he moved to reactive scattering researching the dynamics of elementary reactions. He is a member of the editorial board of Phys. Chem. Chem. Phys. and the international advisory board of J. Phys. Chem. and Chem. Phys. He received the 2008 Polanyi Medal at the 20th International Symposium on Gas Kinetics. |
1. Introduction
Understanding how chemical reactions occur at the microscopic level is a central goal of chemistry. The field of gas-phase reaction dynamics, a branch of physical chemistry/chemical physics, is concerned with this issue and has been having an increasingly strong impact in many other areas of chemistry.1–6 Reaction dynamics was born theoretically following the advent of quantum mechanics, bloomed experimentally and also theoretically in the 1960–1970s, reached its maturity in the 1980–1990s, and is still continuing to attain new highs at the beginning of the new millennium.1–6 It should be noted that pivotal to the strong progress in the field has always been the strong synergic interplay between experiment and theory. Experimentally, studies of reaction dynamics are carried out under single-collision conditions, which can be best obtained using molecular beams or pump–probe laser spectroscopic techniques. Molecular beam techniques, often coupled to laser methods, permit us to explore the individual reactive collision event under well controlled initial conditions and hence to derive the most detailed experimental observable for an elementary gas-phase reaction, that is the state-to-state differential cross section (DCS). For simple 3-atom systems this quantity can currently be calculated by rigorous quantum mechanical methods on accurate ab-initio potential energy surfaces (PESs), allowing for very detailed comparisons between experiment and theory.7–25Although the field has expanded continuously over the past 50 years, much of the focus has been on the detailed understanding of simple prototype reactions from both the experimental and theoretical points of view. Especially during the past 10–15 years there have been very significant advances in our fundamental understanding of chemical reactivity due to strong progress in experiment and computational capabilities. This is especially true for simple three-atom,5–33 some four-atom34–36 and also some poly-atom17,37–39 elementary gas-phase reactions which involve only one product channel, such as X + H2(CH4) → HX + H(CH3) (X = H, F, Cl, OH, CN, O(1D), N(2D), C(1D), S(1D)).
The recent progress on simple reactions is arguably marking the “end of the beginning” for the field of reaction dynamics. Now that simple systems are well understood (including the role of the breakdown of the Born–Oppenheimer approximation in three-atom reactions23,25) attention is being turned to polyatomic elementary reactions involving a radical and a closed-shell molecule, or, a more challenging task, two radicals. These are, indeed, the reactions most relevant in areas of practical interest—from combustion environments and plasmas to terrestrial/planetary atmospheres and interstellar clouds. In these environments reactions commonly encompass polyatomic molecules or radicals as reactants/products, and often have a wealth of energetically allowed competing product channels. To characterize the chemical evolution of these media it is essential to determine for the relevant reactions as a function of temperature (or collision energy) (a) the nature of the primary products, (b) the mechanism of each reaction channel and (c) the relative yield of the competing channels (branching ratios). This information is particularly important for the theoretical modeling of these chemical systems. However, that goal is usually very difficult to achieve experimentally, not only by classical kinetics experiments, but even by microscopic dynamical investigations, especially when products include polyatomic radicals/molecules. In fact, despite the progress in sensitive laser-based spectroscopic techniques for product detection in crossed-beam experiments, such as the H-Rydberg-atom-tagging time-of-flight (TOF) spectroscopy method,7,21,36 the Doppler-selected REMPI technique,18,35 and the velocity-map ion-imaging techniques37–40 (which have fostered an exciting progress in reaction dynamics studies of simple reactions at the quantum state-resolved level), these techniques are not “universal”. In addition, for their application the reaction products need to be known and to have a well characterized spectroscopy, while often the nature itself of the products is not known. Undoubtedly the most suitable experimental technique for tackling the above challenge remains the crossed molecular beam (CMB) scattering technique with velocity analysis, based on electron-ionization (EI) mass-spectrometric (MS) detection.41,42 In fact, electron-ionization affords a “universal” detection method, because every species can be ionized at the electron energies normally used in MS instruments; however, since reactive scattering signals are typically low, hard ionization (i.e., using 60–200 eV electrons) has always been used for maximizing signal intensities (EI cross sections of most molecules and radicals exhibit a maximum at around 60–70 eV). The advantages of the CMB method with respect to the more common flow-reactor kinetic experiments using MS detection is that the reaction of interest is studied under single-collision conditions, and no complications are caused by secondary and wall collisions. In addition, the reactive differential cross section, a basic quantity characterizing the chemical reaction, is also obtained from measurements of product angular and velocity distributions, and this allows one to unambiguously assign the signals to the different reaction products.42 Because of its advantages, the MS detection technique has been extensively used in CMB investigations of the dynamics of fundamental bimolecular reactions during the past 40 years.5,43–45 By providing information on the reaction dynamics (primary products, branching ratios, microscopic reaction mechanisms, product energy partitioning) and in turn on the underlying potential energy surfaces governing the transformation from reactants to products, CMB studies can complement the kinetic data and furnish a more complete characterization of the elementary reactions which are responsible for the macroscopic observations. Nevertheless, when applied to the study of polyatomic multichannel reactions the CMB method has been often hampered by the problem of “dissociative ionization” of the reaction products and/or reagents and/or background gases, which may make very difficult or even preclude the detection of the primary products.42
This Perspective article focuses on the investigation of polyatomic multichannel reaction dynamics and provides an overview of the recent progress that has permitted us, in many cases, to achieve the above goal, as well as to extend dynamical investigation by the CMB technique to radical + radical reactions, a category of reactions of great relevance in many environments, but whose dynamics have been scarcely investigated up to date because of experimental difficulties (see section 3.3). The main issue of this contribution is to illustrate the advantages of using soft ionization in CMB experiments to reduce the dissociative ionization of reactants, products and background gases, which is perhaps the main reason why until very recently detailed studies of multichannel polyatomic reactions in crossed-beams have been limited.
The dissociative ionization of products implies that they ionize not only to a mass-to-charge ratio (m/z) equal to the mass of the neutral, but also to that of many “daughter” ions. This phenomenon might represent a serious complication in CMB-MS experiments, as for instance when the signal at a certain m/z originates from more than one product. This is a common situation for reactions producing organic radicals/molecules because of their tendency to fragment in the ionizer and only in favorable cases it is possible to distinguish whether an ion at a given m/z originates from different neutral products by exploiting energy and momentum conservation.42 Especially troublesome is the case in which interference for product detection comes from elastically scattered reactants, because reactant beams are typically very intense and elastic cross sections very large.
To overcome these problems Nobel laureate Y. T. Lee and coworkers have introduced during the mid-1990s soft (i.e., non-dissociative) ionization, which was implemented by replacing the EI ionizer with a bright tunable VUV radiation beam from a third generation synchrotron.46,47 By using quasicontinuous (500 MHz) VUV radiation, tunable in a broad energy range (5–30 eV) and by tuning the photon energy below the threshold for dissociative ionization of interfering species, one can often suppress interfering signals that would complicate or prohibit the detection of reaction products. Operating below energies that induce the product dissociative ionization, the products are exclusively detected at the parent m/z ratio. In addition, because of the high energy resolution (ΔE/E ≤ 0.05), product isomeric species which have slightly different ionization energies can be selectively ionized and distinguished. SoftPI by VUV synchrotron radiation has been particularly successful in a large variety of photodissociation studies;48–52 for instance, the 11 photo-fragments (including H and H2) in the photodissociation of propene at 157 nm have been successfully detected.53 Although there were only a few examples of reactive scattering studies exploiting soft PI,54–56 recent work57,58 on the multichannel reactions O(3P,1D) + SiH4 and O(3P,1D) + C2H4 using an apparatus with improved detection efficiency, in which TOF measurements of product parent ions below the dissociative ionization thresholds were performed, shows the great potential of this method. Notably, the capability of identifying isomeric product species from their PI thresholds has recently been exploited in kinetic experiments on low-pressure flames.59,60 In the same vein, single photon PI by a 157 nm F2 laser (7.9 eV radiation) has been used in CMB reactive scattering studies of transition metals, exploiting the low ionization potentials (<7.9 eV) of these metals and their compounds.44,61,621
In our laboratory we have recently proposed an attractive alternative to softPI, namely soft EI using a hot-filament ionizer with tunable electron energy (see section 2). This approach, which is well known in discharge-flow mass-spectrometric kinetics, has never been applied in CMB reactive scattering experiments prior to our recent work, because EI cross sections drop drastically toward threshold. However, we have successfully demonstrated the feasibility of soft EI on an improved CMB instrument.63–65
Besides the advantages of the soft ionization approach, this article addresses also an aspect of the long-standing gap between microscopic dynamics and macroscopic kinetics studies of elementary reactions. In fact, the collision energies that can be readily obtained in CMB experiments are usually significantly higher than those (~4 kJ mol−1) corresponding to typical thermal kinetic conditions (300 K). In order to bridge this gap, we have implemented a variable beam crossing-angle set-up, so that we can cross reactant beams not only at 90° (the classic configuration), but also at angles both smaller (45°) and larger (135°). Because the collision energy depends also on the beam crossing angle (see section 2), this has permitted us to widen significantly the range of collision energies that can be attained in a CMB experiment with MS detection.31,63,66,67 Collision energies as low as a few kJ mol−1 have become possible, which overlaps with the conditions of room temperature kinetics and allow us to also approach the actual conditions prevailing in several practical environments, such as the atmosphere of the Earth and also of Titan, up to the interstellar medium.66,67
The content of the article can be schematized as follows. After some general experimental considerations, we emphasize the power of soft EI detection and variable beam crossing angle set-ups in CMB experiments, and the new insight that can be gained from these experiments. Examples include the reactions of C(3P) with acetylene, O(3P) with acetylene and ethylene, and O(3P) with allyl radical. Synergic contributions from theory, both at the level of electronic structure calculations of the PESs and of dynamical computations are discussed, and comparison with theoretical results made wherever possible. Recent complementary CMB work as well as kinetic work exploiting soft PI with synchrotron radiation are noted. We will conclude with an outlook towards the prospects of a truly “universal” product detection in chemical reactions and the synergies between experimental and theoretical studies of gas phase polyatomic multichannel reactions.
2. Experimental
The principles of CMB reactive scattering experiments with MS detection and TOF analysis have been discussed at length in reviews5 and book chapters.42,44,63 Briefly, in the typical arrangement of a CMB apparatus (see Fig. 1, lhs), two beams of radicals and molecules with narrow angular and velocity spread are crossed at 90° in a high-vacuum chamber. The product angular and TOF distributions are recorded after well defined single collisional events take place. The detector is usually an electron-impact ionizer, operated under hard ionization conditions (60–200 eV electrons), followed by a quadrupole mass filter and an ion counting device; the whole detector unit can be rotated in the collision plane around the axis passing through the collision center. The quantities which are measured are the product intensity as a function of the scattering angle, which we call the laboratory angular distribution, N(Θ), and the product intensity as a function of the scattering angle Θ and arrival time t, which we call the time-of-flight spectrum, N(Θ, t). The measurements are carried out in the laboratory (LAB) system of coordinates, but for the physical interpretation of the scattering data it is necessary to perform a coordinate transformation and move to the center-of-mass (CM) reference frame. It can be easily demonstrated42 that, for each reaction channel, the relation between LAB and CM product flux is given by ILAB(Θ,v) = ICM(θ,u)v2/u2, where Θ and v are the LAB scattering angle and velocity, respectively, while θ and u are the corresponding CM quantities. Since the EI mass spectrometric detector measures the number density of products, N(Θ), rather than the flux, the actual relation between the LAB density and the CM flux is given by NLAB(Θ,v) = ICM(θ,u)v/u2. Analysis of the laboratory data is usually performed by forward convoluting tentative CM distributions over the experimental conditions. In other words, the CM angular and velocity distributions are assumed, averaged and transformed to the LAB frame for comparison with the experimental distributions and the procedure is repeated until a satisfactory fit of the experimental distributions is obtained. The CMdifferential cross section ICM(θ, u) is commonly factorized into the product of the velocity (or translational energy) distribution, P(u) (or P(E′T)), and the angular distribution, T(θ): ICM(θ, E′T) = T(θ) P(E′T). In some cases the coupling between the T(θ) and P(E′T) functions needs to be accounted for. The T(θ) and P(E′T) functions contain all the information about the reaction dynamics. When multiple reaction channels contribute to the signal at a given m/z ratio, a more complex situation arises. In these cases a weighted total CM differential cross section reflecting the various possible contributions is used in the data analysis of the LAB distributions for a specific m/z ratio, that is: ICM(θ, E′T) = Σiwi × [T(θ) × P(E′T)]i with the parameter wi representing the relative contribution of the integral reactive cross section of the ith channel.31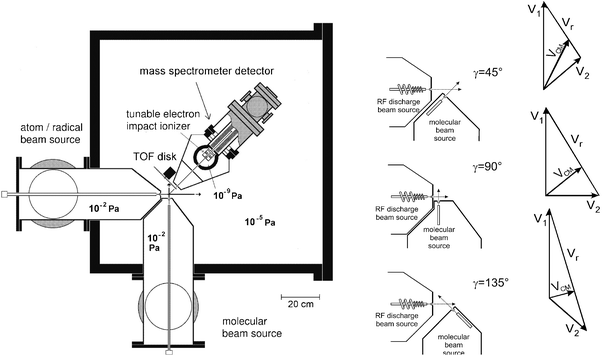 | ||
| Fig. 1 Schematic view of the Perugia crossed molecular beam apparatus (lhs). The three different crossing beam set-ups are also shown on the rhs together with the corresponding (schematic) velocity vector (“Newton”) diagrams. Here the reactant velocities, v1 and v2, the center of mass velocity, vCM, and the relative velocity vr are indicated. Note how vr increases and vCM decreases in passing from γ = 45° to γ = 135°. | ||
2.1 Soft electron-ionization (EI) detection
Soft EI by electrons with low, tunable energy (7–100 eV)63–65 represents a simple and low-cost alternative to the use of softPI by synchrotron radiation46,47 to fight the problem of dissociative ionization. Although not affording the same degree of selectivity as VUV synchrotron radiation (the FWHM electron energy spread from a hot tungsten filament is about 0.7 eV68 and EI cross sections in general do not exhibit sharp rises), the soft EI approach offers in many cases similar advantages, because often the main aim is simply to suppress interferences from dissociative ionization processes and to improve the signal-to-noise (S/N) ratio. In addition, soft EI provides the possibility of determining branching ratios more readily than soft PI, because absolute EI cross sections are often known or can be reliably estimated from additivity rules based on atomic polarizabilities.50,69,70To understand why soft EI has become possible in CMB experiments, we recall that for a given process the reactive scattering signal is proportional, among other factors, to the density of the two reactant beams at the collision region and to the detection efficiency.42 The detection efficiency decreases with decreasing electron energy, but what matters is the S/N ratio and this may actually increase by using soft EI, partly because the fragmentation of the parent ion decreases with decreasing electron energy (see section 3.1) and partly because the background can decrease dramatically. However, the key is the beam densities at the collision region. By changing the source chamber geometries which permitted us to move the two nozzles of the reactants closer to the collision region by about a factor two, an overall gain of about a factor 25 in the signal intensity was achieved and this has permitted us to compensate for the signal loss due to the lower ionization cross section at the reduced energy of the ionizing electrons.
Besides making possible the detailed investigation of polyatomic multichannel reaction dynamics, the new capability gained by using a variable-energy EI source in the MS detector is also permitting us to measure the EI efficiency curves of reactants and products down to their ionization thresholds. That is, we have developed the capability of estimating the ionization energy of transient (radical) species by “synthesizing” them in CMB reactive scattering experiments. To this end, one important aspect is to know the absolute electron energy scale. This has been calibrated by using beams of pure rare gases (He, Ne, Ar), atomic species (C, O, S), and molecular species (O2, C2H2,C3H6) to cover an energy range from about 9 to 25 eV (see section 3.1).
2.2 CMB experiments with variable beam crossing angle
In many practical cases it is useful to study the reaction dynamics as a function of collision energy, Ec. In particular, it is of considerable interest to explore collision energies corresponding to typical kinetic measurements at room temperature. It may also be useful to attain even lower Ecs, as of interest in planetary atmospheres or the interstellar medium, or to explore very high Ecs, corresponding to high temperature combustion and plasma systems. CMB experiments with a fixed 90° crossing angle configuration had difficulties to meet these criteria, because there are limits to the lowest and highest velocities that a species can attain in a supersonic beam. In fact, the relative collision energy in a CMB experiment is given by Ec = 1/2 µvr2, where µ is the reduced mass of the system and vr is the relative velocity. In general, vr2 = (v12 + v22 – 2 v1v2 cos γ), where v1 and v2 are the two reactant beam velocities in the LAB frame and γ the crossing angle of the two beams. Traditionally, CMB instruments with a rotatable MS detector have always featured a beam crossing angle of 90° (see, for instance, Fig. 1 (lhs)); in this case vr2 = v12 + v22. In order to change Ec one needs to change vr and this, in apparatuses with γ = 90°, is achieved by changing the velocity of one or both beams. Unfortunately, as already mentioned there are limits to the maximum and minimum beam velocity that one can attain; also, an increase in beam velocities leads to a reduction in angular and velocity resolution, because the CM velocity, vCM, increases as well and consequently the Newton circle within which the product is scattered moves away from the LAB velocity origin, with the consequence that the product is scattered in the LAB frame over a narrower angular range and is faster. Furthermore, varying the beam velocity often implies varying also the velocity spread, the density, and the internal quantum state distribution of the species of interest.Clearly, it is desirable to vary the collision energy while keeping the same beam characteristics (i.e., internal temperature and speed ratio); this can be achieved by varying the intersection angle of the two beams, as commonly done in pulsed CMB apparatuses featuring pulsed laser product detection (see ref. 11, 71–75). The new beam crossing-angle set-ups in our CMB apparatus with the two reactant beams crossing at γ = 45°, 90° or 135° (see Fig. 1—rhs) allow us to vary the collision energy over a much wider range than previously possible (A continuously variable crossing angle would have required, for geometrical reasons, an increase in the nozzle-collision centre distance with its consequent loss of beam intensities and therefore was not pursued). In addition, by using the γ = 135° geometry we can increase the collision energy (i.e., vr increases with respect to γ = 90°, see Fig. 1—rhs) and at the same time, increase both angular and velocity resolution. In this case, in fact, the Newton circle within which the products are energetically confined becomes closer to the LAB velocity origin (i.e., vCM becomes smaller) than in the γ = 90° case (see Fig. 1—rhs). Conversely, the γ = 45° arrangement is intended for reaching very low Ecs, because the relative velocity, vr decreases significantly with respect to the γ = 90° set-up (see Fig. 1—rhs) and this is of interest when studying reactions of relevance in astrochemistry, such as those of C(3P) with unsaturated hydrocarbons. For instance, by using the γ = 45°, 90° and 135° configurations we have recently been able to explore in detail the dynamics of the C(3P) + C2H2 reaction from 3.6 kJ mol−1 up to about 50 kJ mol−1.66,67 Here we now discuss briefly the results on the branching ratios as a function of collision energy (temperature) for this important reaction67 which is of interest both in interstellar chemistry and combustion.
| C(3P) + C2H2(X1Σg+) → l–C3H(X2Π1/2) + H(2S1/2) ΔH°0 = −1.67 kJ mol−1 | (1a) |
| → c–C3H (X2B2) + H(2S1/2) ΔH°0 = −8.8 kJ mol−1 | (1b) |
| → C3(X1Σg+) + H2(X1Σg+) ΔH°0 = −105.9 kJ mol−1 | (1c) |
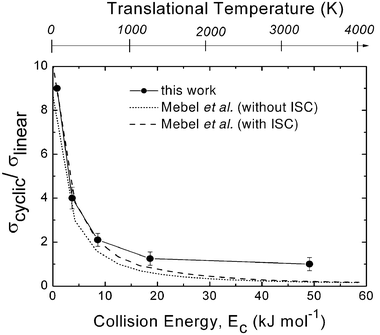 | ||
| Fig. 2 Ratio of cross sections for cyclic and linear-C3H formation, σcyclic/σlinear, as a function of collision (translational) energy, Ec, as derived from crossed molecular beam studies of the C(3P) + C2H2 reaction. The corresponding translational temperature scale is indicated on the top abscissa. The solid line joining the data points is drawn to guide the eye only. The ratio obtained from statistical calculations on ab initio potential surfaces by Mebel et al.80 is reported with dotted line (on the triplet PES only) and with dashed line (including ISC) for comparison. Reproduced with permission from J. Phys. Chem. A, 2008, 112, 1363–1379. Copyright 2008 American Chemical Society. | ||
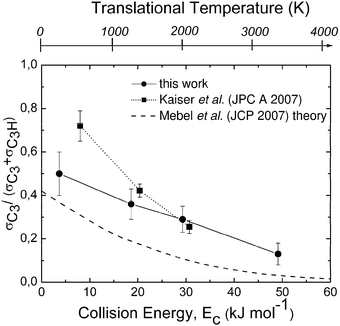 | ||
| Fig. 3 Branching ratio σC3/(σC3 + σC3H) as a function of collision (translational) energy, Ec, as derived from crossed molecular beam studies of the C(3P) + C2H2 reaction. The corresponding translational temperature scale is indicated on the top abscissa. The solid line joining the data points (solid circles) is drawn to guide the eye only. The branching ratio obtained by Mebel et al.80 from statistical calculations on ab initio potential surfaces including ISC is reported with a dashed line for comparison. The results of Guo et al.82 obtained at three collision energies are also shown as solid squares; again, the dotted line joining the experimental points is drawn to guide the eye only. Reproduced with permission from J. Phys. Chem. A, 2008, 112, 1363–1379. Copyright 2008 American Chemical Society. | ||
We conclude this section by emphasizing that variable beam crossing angle set-ups are crucial for exploring the reaction dynamics in a wide range of collision energies, as the example discussed here clearly witnesses.
2.3 Supersonic sources of radical beams
A prerequisite for studying in crossed beams radical–molecule and radical–radical reactions is the capability of generating intense, continuous (or pulsed) supersonic beams of the radical reactant (the generation of intense supersonic beams of stable molecules is straightforward).5 In our laboratory a quite general technique has been used for a number of years; this is based on a high-pressure, high-power radio-frequency (RF) discharge beam source,85 originally developed for generating O(3P,1D) beams,86 and that we have adapted for producing supersonic beams of a wide variety of atomic and molecular radicals (O,87N,88–91C,66,67,74,75,91,92Cl,8,19,85,93 S,94OH,34,87,95–97 CN,91 C298) starting from dilute mixtures (1–5%) of suitable precursor molecules (O2, N2, CO2, Cl2, SO2, H2O, CO2/N2, CO) seeded in a rare gas carrier. Recently, continuous supersonic beams of hydrocarbon radicals (CH3, C2H5, C3H5) were also produced by means of a continuous flash pyrolysis beam source (see section 3.3), which is based on that developed by Chen and coworkers99 for the production of pulsed radical beams. By crossing a radical beam produced by the RF discharge source with another one produced by the flash-pyrolysis source it has become possible to tackle reactive scattering between two radicals (see section 3.3). The detailed characteristics of the beams of the various species can be found in the relevant references.3. Polyatomic multichannel reactions: examples
We now discuss a few examples illustrating how soft EI detection permits us to approach universal product detection in CMB experiments, and thus to identify all primary reaction products of multichannel polyatomic reactions, determine their branching ratios, and characterize the dynamics of each channel. The advantages obtained using crossing beam angles ≠ 90° are also highlighted. Focus is on some reactions of atomic oxygen with unsaturated hydrocarbons and hydrocarbon radicals.3.1 Reaction O(3P) + C2H2
The O(3P) + C2H2 reaction is one of the most important reactions in fuel rich hydrocarbon flames, because acetylene is easily formed during the combustion of methane, larger aliphatic hydrocarbons, and aromatics.100,101 The reaction has three energetically and spin-allowed competing channels:| O(3P) + C2H2 → H + HCCO(X2A′′) ΔH°0 = –81.6 kJ mol−1 | (2a) |
| → CH2(3B1) + CO ΔH°0 = –197.1 kJ mol−1 | (2b) |
| → H2 + CCO(X3Σ−g) ΔH°0 = –93.7 kJ mol−1 | (2c) |
| Reference | 112 | 103 | 102 | 111 | 106 | 107 | 108 | 64 | 113 |
|---|---|---|---|---|---|---|---|---|---|
| Year | 1986 | 1986 | 1987 | 1989 | 1990 | 1992 | 1994 | 2004 | 2006 |
| H + HCCO (%) | 70 ± 10 | 62 ± 23 | 64 ± 15 | 42 ± 10 | 80 ± 15 | — | 83 ± 8 | 79 ± 5 | 80 |
| CH2(X3B1) + CO (%) | — | — | — | 58 ± 10 | — | 17 ± 8 | — | 21 ± 5 | 20 |
Recently, by exploiting (i) an increased instrumental sensitivity, (ii) an improved resolution for measuring product angular and TOF distributions, and (iii) the new ability to detect cleanly the CH2 radical using soft EI, it has been possible to obtain improved CMB results,64 which have corroborated the branching ratios obtained from the kinetic studies and have deepened significantly our understanding of this important combustion reaction, as summarized here.
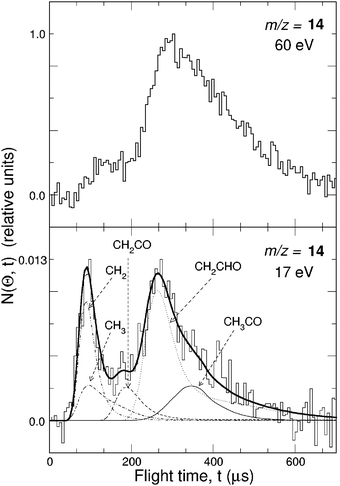
While detection of HCCO from channel(2a) is straightforward in CMB experiments, there are complications arising when one attempts to detect the CO product, because of the inherently high background at m/z = 28 in every UHV chamber and the dissociative ionization to CO+ of the HCCO product. Detection of CH2 (m/z = 14) is therefore desirable, but also problematic when employing hard EI, because of the relatively high inherent background at this mass due to dissociative ionization of residual CH4 to CH2+ and N2 to N+, and of the interference from the dissociative ionization to 13CH+ of the main HCCO product, and especially of the elastically scattered, intense C2H2 beam to 13CH+ and to also CH2+. Although 13C represents only a very small fraction of the total carbon in HCCO and C2H2, the 13CH+ signal from the above processes is comparable to the CH2 reactive scattering signal from reaction (2b) when using 60–200 eV electrons. For all these reasons, detection of CH2 from reaction (2b) was not attempted in the early CMB studies. This, however, became easy by using soft EI.64 Because the appearance energy (A.E.) of N+ from N2 is A.E. = 24.3 eV, of 13CH+ and CH2+ from C2H2 A.E. = 20.9 eV and 19.7 eV,116 respectively, and of 13CH+ from HCCO A.E. ~14 eV, by using 17 eV electron energy we were able to detect CH2 cleanly without essentially any interference from the above dissociative ionization processes, as shown in Fig. 4-top. This figure compares the TOF spectra at m/z = 14 recorded at the LAB angle of 22° using 60 and 17 eV electrons; as can be seen, at 60 eV the spectrum exhibits, in addition to a fast feature due to reactive signal from channel(2b), a slow broad peak due to dissociative ionization of HCCO and elastically/inelastically scattered C2H2. By using an electron energy of 17 eV, interference from dissociative ionization is completely suppressed, leaving only the signal from reaction channel(2b). This has permitted us to measure the angular distribution of the sole CH2 product in the usual manner, i.e., by modulating the acetylene beam for background subtraction.64 It should be noted that at lower electron energy the fragmentation of the HCCO product is strongly reduced (see Fig. 4—bottom), and therefore one “gains back” in the relative intensity of the parent ion when operating at low electron energies. Fig. 5(a) shows the angular distributions of the HCCO and CH2 products at Ec = 39.7 kJ mol−1, obtained by crossing the reactant beams at γ = 90°. Experiments were also performed with γ = 135°; in this case, Ec = 52.7 kJ mol−1.117TOF spectra were measured at numerous LAB angles for HCCO and CH2; two typical spectra are shown in Fig. 5(b).
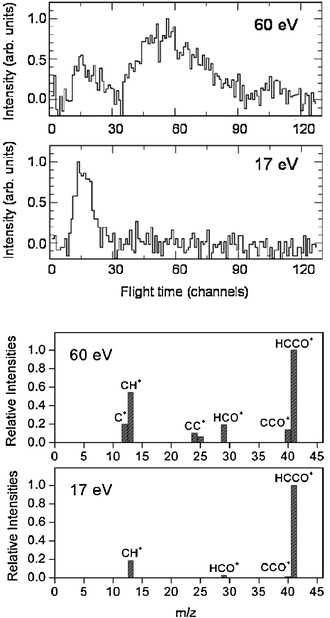 | ||
| Fig. 4 Top: Comparison of m/z = 14 TOF spectra (5 µs/channel) at the lab angle of 30° for the reaction O(3P) + C2H2 (Ec = 39.7 kJ mol−1) using an electron energy of 60 eV and 17 eV. Bottom: Comparison of relative fragmentation patterns of the HCCO product at 60 eV and 17 eV. | ||
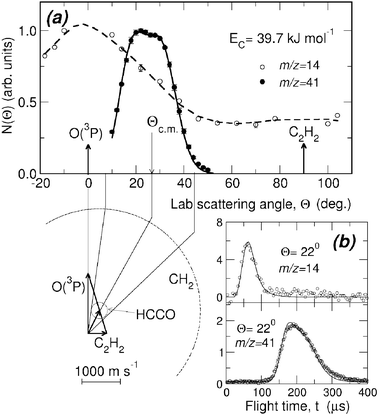 | ||
| Fig. 5 (a) HCCO and CH2 product lab angular distributions from the O(3P) + C2H2 reaction at Ec = 39.7 kJ mol−1. Solid and dashed lines are best-fit curves obtained from the best-fit product angular and translational energy distributions. The Newton diagram of the experiment is also shown; there the circles delimit the maximum velocity that the indicated products can attain assuming that all the available energy is channeled into translation. (b) TOF spectra of m/z = 14 and m/z = 41 products at the lab angle of 22°. The CH2 and HCCO spectra are recorded at 5 and 2 µs/channel, respectively, using the single-shot TOF method; solid lines are best-fit curves obtained from the best-fit CM functions. Reproduced with permission from J. Chem. Phys., 2004, 120, 4557–4560. Copyright 2004 American Chemical Society. | ||
As can be seen the heavy HCCO product left by the very light H counterpart, is confined in a narrow angular range around the CM (Fig. 5(a)) and is quite slow in the lab frame (Fig. 5(b)), while the lighter CH2 product, left by the heavy CO counter-fragment, has a very broad angular distribution (Fig. 5(a)) and is very fast in the lab frame (Fig. 5(b)) because of linear momentum conservation and, to a minor extent, also because of a larger reaction exoergicity. When suppression of dissociative ionization is not as complete as in this case, it may still be possible to separate the contributions from two or more competing channels, provided that they are comparable in size and are fully or at least partially resolved in the TOF spectra (see section 3.2).
The N(Θ) and N(Θ, t) at m/z = 41 (HCCO+) and 40 (CCO+) were found to be superimposable, indicating that the m/z = 40 signal is all coming from the dissociative ionization of HCCO, and not from the dynamically and energetically different H2 elimination channel. This suggests that the H2 elimination pathway (2c) is closed at these Ecs, most likely because it is characterized by a very high exit potential barrier.
From LAB product angular and TOF distributions at m/z = 41 and 14, CM product angular and translational energy distributions were derived for both channels (2a) and (2b).64,117 The HCCO CM angular distribution has nearly constant intensity over the entire angular range, with slightly less intensity in the backward direction (with respect to the O-atom beam direction); the slight asymmetry (which increases with increasing Ec) indicates that the reaction proceeds through a long-lived complex that just starts to osculate.117 The CH2CM angular distribution is also forward biased and this indicates that pathway (2b) is also occurring through an osculating complex,118i.e., a complex whose lifetime is comparable to its rotational period. According to accurate ab initio electronic structure calculations, the mechanism sees the initial electrophilic attack of the O atom to the triple bond of the C2H2 molecule with formation of a triplet diradical adduct (HCCHO) that, under single collision conditions can undergo competitively CH bond cleavage to HCCO + H and isomerization to triplet ketene (H2CCO) followed by CC bond rupture to triplet-CH2 + CO.112,113,106 The fraction of total available energy channeled into translation is 0.40 for channel(2a) and 0.42 for channel(2b), which indicates the presence of an exit potential barrier for both processes, as confirmed by electronic structure calculations of the PES.113 Interestingly, the P(E′T) distribution of the CH2 + CO channel was found to peak at a high energy value (ca. 70 kJ mol−1), and this is consistent with a dissociation process characterized by a high energy barrier in the exit channel, as in triplet-CH2CO → CH2(3B1) + CO. In contrast, it is known119 that the process singlet-CH2CO → CH2(1A1) + CO occurs without an exit potential barrier, and it should exhibit a P(E′T) distribution peaking near zero. This indicates that singlet CH2 is not formed in this reaction and therefore no ISC between triplet and singlet PESs occurs to any appreciable extent, in contrast to theoretical suggestions.120 This conclusion corroborates that obtained in previous kinetic work.107
Once the CM angular and translational energy distributions for the two competing channels (2a) and (2b) were derived, it was possible to determine the branching ratios. To estimate the branching ratios from CMB results it is required to know: (i) the absolute beam intensities; (ii) the size of the collision volume; and (iii) the detection efficiency.111 These quantities are not easy to determine accurately in a CMB experiment; however, since the first two are constant and the third can be reasonably estimated, we can easily determine relative cross sections.111 We obtained64,117 the following branching ratio: σ(2b)/[σ(2a) + σ(2b)] = 0.19 ± 0.04 that implies σ(2a)/[σ(2a)+σ(2b)] = 0.81 ± 0.04 at both Ecs investigated. The derived values are in excellent agreement with the accurate kinetic determinations of Peeters et al.107,108 (see above and Table 1). It is gratifying to note that these branching ratios are also corroborated by very recent statistical predictions based on both the ground and first excited ab initio triplet PESs.113
In conclusion, CMB dynamic experiments confirm that the dominant channels of the reaction between O(3P) and acetylene are HCCO + H (80%) and triplet-CH2 + CO (20%), and indicate that the H2 elimination channel(2c) is not occurring to any appreciable extent and ISC (leading to 1CH2 + CO) is not playing a significant role. We can say that dynamics, kinetics and theory have finally converged for the important O(3P) + C2H2combustion reaction.
A final point to mention about this reaction is that the capability of tuning the electron energy has permitted us to also obtain information on the ionization energy (I.E.) of the ketyl radical product. The HCCO radical is a particularly important intermediate in combustion, atmospheric chemistry, and interstellar clouds, and many of its physical-chemical properties (see, for instance, ref. 121), as well as its reaction kinetics,107,114 have been recently investigated. However, no direct experimental nor theoretical information is available about its ionization potential. By exploiting soft EI we have measured the EI efficiency curve (i.e., the ionization cross section as a function of electron energy) of the HCCO product from the O(3P) + C2H2 reaction by detecting the m/z = 41 signal intensity at the CM angle as a function of electron energy. This is shown in Fig. 6 together with the EI efficiency curves of acetylene (I.E. = 11.41 eV)116 and allene (I.E. = 9.69 eV)116 produced in supersonic beams of the pure species, where they are expected to be vibrationally and also rotationally cold, and which are here used for calibrating the absolute electron energy scale. The corresponding ionization threshold of HCCO was determined to be 9.8 ± 0.3 eV from the simple straight-line extrapolation method (the same used to calibrate the energy scale by using the two stable hydrocarbon species which have a known I.E.). It should be noted that the probed HCCO radical is formed by the O(3P) + C2H2 reaction and therefore contains a certain amount of internal energy. From the experimentally determined P(E′T) distribution we derive an average ro-vibrational energy content of 0.8 eV, with the maximum value, on the basis of energy conservation, being 1.3 eV. Consequently, our approximate I.E., which is a vertical ionization energy, is expected to be significantly red-shifted (by a maximum of 1.3 eV) with respect to that of internally cold HCCO (not available in the literature). High-level CCSD(T) calculations give a vertical (adiabatic) I.E. for HCCO of 11.0 (10.7) eV.117 Considering the expected red-shift for “hot” HCCO of about 1.3 eV, there appears to be a good agreement between experiment and theory. This type of information can be obtained for a variety of radical products “synthesized” via a bimolecular reaction in a CMB experiment. Very recently we have also measured the EI efficiency curve of the related HCCS (thioketyl) radical produced from the reaction S(1D) + C2H2 at Ec = 35.6 kJ mol−1.122 In the next section we also report an estimate of the ionization energy of “hot” CH2CHO (vinoxy) radical.
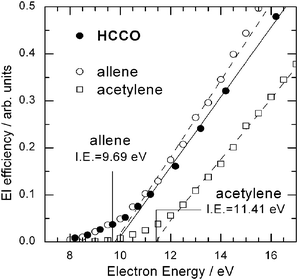 | ||
| Fig. 6 Electron ionization efficiency as a function of electron energy of the HCCO radical product (m/z = 41) from the O(3P) + C2H2 reaction at Ec = 39.7 kJ mol−1, measured at the CM angle, compared to those of allene and acetylene contained in a supersonic beam of the pure species. The I.E. of allene and acetylene is indicated. The estimated ionization threshold of the “hot” HCCO reaction product is 9.8 ± 0.3 eV (see text). | ||
3.2 Reaction O(3P) + C2H4
Analogously to O(3P) + C2H2, the O(3P) + C2H4 reaction plays a central role, in addition to the combustion of ethylene itself, in the overall mechanism for hydrocarbon combustion.123 It exhibits six competing energetically allowed pathways:| O(3P) + C2H4 → H + CH2CHO ΔH°0 = −71 kJ mol−1 | (3a) |
| → H + CH3CO ΔH°0 = −114 kJ mol−1 | (3b) |
| → H2 + CH2CO ΔH°0 = −356 kJ mol−1 | (3c) |
| → CH3 + HCO ΔH°0 = −113 kJ mol−1 | (3d) |
| → CH2 + HCHO ΔH°0 = −29 kJ mol−1 | (3e) |
| → CH4 + CO ΔH°0 = −488 kJ mol−1 | (3f) |
Since the 1950s many research groups have investigated reaction (3) by employing a variety of experimental techniques in different pressure and temperature regimes, but have identified only some of the possible products.123–125 While the overall rate constant has been well established (k298 = 7.5 × 10−13 cm3 molecule−1 s−1),126 the question of the identity of the primary reaction products and their relative importance has been a subject of considerable controversy over the years (see Table 2, where the history of the branching ratios is summarized).
| Product | CH2CHO | CH3CO | CH2CO | CH3 | CH2 |
|---|---|---|---|---|---|
| a See ref. 65 and 70. b Ref. 124. c Ref. 125. d Ref. 70. e Ref. 65. f Ref. 127. g Ref. 128. h Ref. 129. | |||||
| Reference | Vinoxy | Acetyl | Ketene | Methyl | Methylene |
| Cvetanovica (1955) 300 K | <0.10 | — | 0.04 | — | — |
| Avramenkoa (1963) | — | — | — | Large | — |
| Gutmana (1972) | <<0.10 | — | Large | Large | — |
| Blumenberga (1977) | — | — | Small | — | — |
| Hunzingera (1981) | 0.36 ± 0.04 | — | — | 0.52/0.58 | — |
| Kaufmana (1983) | 0.79 ± 0.14 | — | — | — | — |
| Klemma (1986) 300–769 K | 0.27/0.35 ± 0.05 | — | — | — | — |
| Endob (1986) 290 K | 0.4 ± 0.1 | — | — | 0.5 ± 0.1 | 0.1 ± 0.05 |
| Tempsc (1988) 290 K | 0.50 ± 0.10 | — | 0 | 0.44 ± 0.15 | 0.06 ± 0.03 |
| Schmoltnerd (1989) 25 kJ mol−1 | 0.29 ± 0.11 | — | — | 0.71 ± 0.26 | — |
| (0.38 ± 0.05)h | |||||
| Casavecchiae (2005) 54 kJ mol−1 | 0.27 ± 0.06 | 0.01 ± 0.005 | 0.13 ± 0.03 | 0.43 ± 0.11 | 0.16/0.08 |
| Peetersf (2005) 54 kJ mol−1 | 0.271 | 0.033 | 0.033 | 0.444 | 0.17 |
| Schatzg (2008) 54 kJ mol−1 | 0.22 | — | 0/0.21 | 0.55 | 0.03 |
Again, CMB investigations were undertaken in the early 1980s to assist kinetic work in the elucidation of the reaction mechanism and dynamics. Two early CMB studies115,130 at Ec ≈ 25 kJ mol−1 confirmed the occurrence of channel(3a), the easiest to detect for kinematics reasons. Channels (3c, d, e, f) are much more difficult to detect due to unfavorable kinematics and to the fact that the expected ion signals from products of channel(3c) at m/z = 42, of channel(3d) at m/z = 15 or 29, and of channel(3e) at m/z = 14 or 30, coincide with significant background peaks and/or with peaks coming from dissociative ionization of the most intense signal corresponding to CH2CHO formation and of the elastically scattered C2H4 reagent. Channel(3f) should be negligible; in any case, it is very hard to tackle it by the CMB technique because one of the two products (CH4) has the same mass as one of the two reagents and the other (CO) corresponds to the highest background mass (after H2) in any UHV chamber. Only the problem connected with channel(3d) was partly overcome in one of the previous CMB studies70 by using a beam of isotopically labeled 18O, which permitted the detection of the HCO product of channel(3d) at m/z = 31 (HC18O+) and 30 (C18O+), and allowed an estimation of the branching ratio between channel(3a) and (3d), σ(3a)/[σ(3a) + σ(3d)], of 0.71 ± 0.26, a value which is somewhat larger than kinetic estimates, which gave values ranging from 0.44 to 0.55 (see Table 2). A recent re-evaluation by Butler and coworkers129 of this branching ratio, originally estimated by Schmoltner et al.,70 yielded a value of 0.38 ± 0.05 which is in better agreement with the most recent kinetic result of 0.44 ± 0.15125 (see Table 2).
The development of soft ionization detection has permitted us to shed new light on the dynamics of this important reaction. From a detailed series of angular and velocity distribution measurements at m/z = 42, 15, and 14 in CMB experiments at Ec = 54.0 kJ mol−1 utilizing universal soft EI detection we have been able to unambiguously detect products from the five reaction pathways (3a–e), determine their branching ratio and characterize their dynamics.65,131 Here we summarize some of these results, which well exemplify the power of soft EI. A very recent CMB study58 of this reaction at Ec = 26.8 kJ mol−1 with soft PI detection by VUV synchrotron light has provided some additional information and is also noted here.
In our study, reactive signals were observed at m/z = 43, 42, 29, 15, and 14. At the CM angle the signal intensity at m/z = 43, corresponding to the vinoxy parent ion CH2CHO+ from channel(3a) (and possibly also to the acetyl parent ion CH3CO+ from channel(3b)), is only 2.7% of that at m/z = 42 which is the largest signal and corresponds to the parent ion of ketene (channel(3c)) and to the daughter ion of vinoxy (channel(3a)); this fraction is very small, but higher than the expected 13C isotope fraction of m/z = 42, and therefore indicates that some m/z = 43 parent ion is actually formed. From measurements of the EI efficiency curves at various m/z ratios (m/z = 15, 42, and 43) at the CM angle (see Fig. 7, top), and from the shape of the angular and TOF distributions at m/z = 42 it was concluded that the m/z = 42 signal mostly comes from dissociative ionization of CH2CHO (vinoxy), indicating that CH2CHO+ is not very stable and fragments heavily to m/z = 42, especially at electron energies higher than 12.5 eV, as the middle panel of Fig. 7, where the ratio (m/z = 43)/(m/z = 42) is plotted as a function of electron energy, shows. Consequently, all measurements of product angular and TOF distributions for channels (3a) (and possibly (3b)) were carried out at the daughter ion m/z = 42; however, the signal from channel(3c) is also expected to contribute at this mass (see below). From the EI efficiency curve of m/z = 43 (see Fig. 7-bottom), an ionization threshold for the hot vinoxy radical of 9.5 ± 0.7 eV was estimated, which should be significantly red-shifted with respect to the adiabatic I.E., because the detected vinoxy is “synthesized” in our CMB experiment with an internal energy of ~75 kJ mol−1 (~0.8 eV) (the maximum internal energy is 1.3 eV). We note that an ionization threshold of 9.3 ± 0.1 eV was very recently determined from the PI efficiency curve with synchrotron radiation for hot vinoxy produced by the same reaction studied in pulsed CMB at Ec = 26.8 kJ mol−1.58 In that work58 an adiabatic I.E. of 10.24 eV was also computed for vinoxy and the significant red-shift attributed to the internal energy content (estimated to be ~0.56 eV) of the nascent vinoxy radical. We note that there is a good agreement between the onset of ionization of “hot” vinoxy obtained by EI and PI, the latter technique affording smaller uncertainties because of the higher ionizing energy resolution Again, before these EI and PI efficiency curve measurements there existed only an indirect suggestion that the ionization energy of the CH2CHO radical was 10.85 eV, obtained from an early conventional fast-flow mass-spectrometric kinetic study of the reaction of nitrogen atoms with acetaldehyde.132
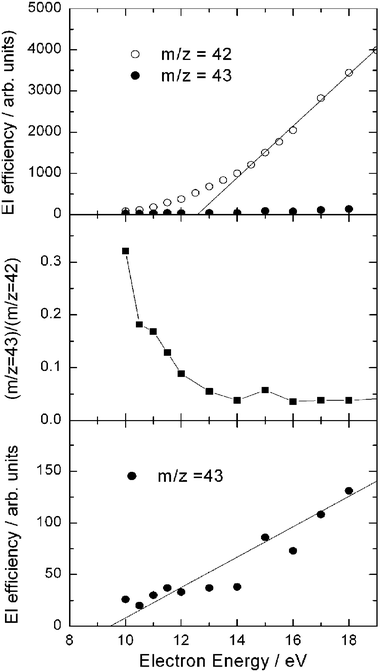 | ||
| Fig. 7 Top: electron ionization efficiency as a function of electron energy of m/z = 42 and 43 products from the O(3P) + C2H4 reaction at EC = 54.0 kJ mol−1, measured at the CM angle. Middle: ratio of m/z = 43 and m/z = 42 signals as a function of electron energy. Bottom: electron ionization efficiency as a function of electron energy of m/z = 43 (vinoxy radical product) from the O(3P) + C2H4 reaction at EC = 54.0 kJ mol−1, measured at the CM angle (note that here the vertical scale has been amplified by a factor 25 with respect to the top panel) The estimated ionization threshold of the “hot” H2CCHO reaction product is 9.5 ± 0.7 eV (see text). | ||
Product angular distributions were recorded only at m/z = 42 and m/z = 15, the two strongest signals. Fig. 8 shows the LAB angular distribution at these two masses using 17 eV electron energy, with the Newton diagram of the experiment at Ec = 54.0 kJ mol−1 (the crossing beam configuration with γ = 135° was used for higher angular and especially TOF resolution). Two channels (3a and 3d) contribute to the signal at m/z = 15, while three channels (3a, 3b, and 3c) contribute to that at m/z = 42; the relative contributions are disentangled through TOF measurements at selected LAB angles. As an example, we show in Fig. 9 the TOF spectrum for Θ = 34° at m/z = 42 (top-left) and that at m/z = 15 (top-right). We were also able to detect the momentum-matched HCO counter-fragment of CH3 (see Fig. 9-bottom-right), but because of the contribution from 13C in the C2H4 beam (which cannot be suppressed even by soft EI or PI), a correction for elastically scattered C2H4 to the data at m/z = 29 was needed to derive the TOF spectrum shown in Fig. 9, which exhibits a slow peak due to dissociative ionization of vinoxy (perhaps some acetyl also contributes to it) and a fast peak due to HCO from channel(3d). In the study at Ec = 26.8 kJ mol−1 with synchrotron radiation,58HCO was detected cleanly at 9.8 eV and 10.3 eV, avoiding interference from C2H4 which has an I.E. = 10.51 eV.116Fig. 10 (bottom) shows instead the TOF spectrum recorded at m/z = 14, again with an electron energy of 17 eV at the same LAB angle of Θ = 34°. As derived from the data analysis the TOF spectra at m/z = 14, 15 and 42 carry the fingerprints of all five product channels (3a–e). Specifically, the m/z = 15 TOF spectrum (see Fig. 9—top-right) exhibits a fast peak which is unambiguously due to the methyl radical from the CH3 + HCO channel (reaction 3d), and a slower, more intense peak, due to dissociative ionization in the ionizer of the vinoxy radical, corresponding to the CH2CHO + H channel (reaction 3a). This has been recently confirmed by soft PI measurements at 10.3, 11.9 and 13.8 eV in the study of the O + C2H4 reaction at Ec = 26.8 kJ mol−1.58 At m/z = 42 the TOF spectrum (see Fig. 9-top-left) exhibits: (i) a dominant peak, analogous to the main peak observed at m/z = 15, which is due to dissociative ionization of vinoxy (reaction 3a); (ii) a fast peak, which appears as a shoulder on the main peak and is unambiguously attributed, on the basis of energy and linear momentum conservation, to the ketene product from the channel CH2CO + H2 (reaction 3c); (iii) a small component, peaked at the CM velocity, which is attributed to formation of the acetyl radical from the channel CH3CO + H (reaction 3b) with a very small recoil energy. From the comparison of the m/z = 15 and 42 spectra we infer that at 17eV CH3CO (m/z = 43) fragments very little to m/z = 15.
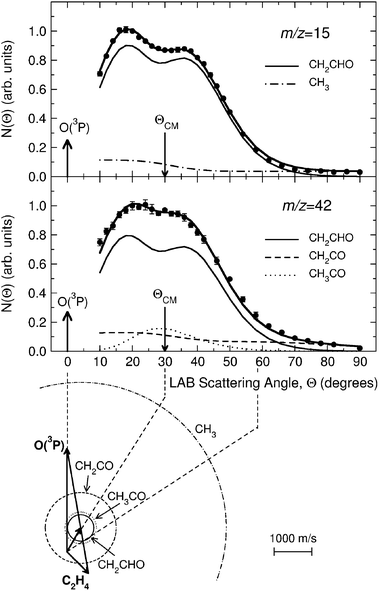 | ||
| Fig. 8 LAB angular distribution at m/z = 15 (top) and m/z = 42 (bottom) from the reaction O(3P) + C2H4 at Ec = 54.0 kJ mol−1, obtained by using an electron energy of 17 eV, together with the Newton diagram of the experiment. Error bars are indicated when visible outside the experimental dots. The circles in the Newton diagram delimit the maximum speed that the indicated products can attain on the basis of energy and linear momentum conservation if all the available energy goes into product translation. The heavy solid line is the total angular distribution calculated from the best-fit product CM translational energy and angular distributions, the separate contributions from the CH2CHO and CH3 products from channels (3a) and (3d) are shown with light solid and dashed-dotted lines, respectively (top), and from the CH2CHO, CH2CO and CH3CO products from channels (3a), (3c) and (3b) are shown with light solid, dashed, and dotted lines, respectively (bottom). Top graph: reproduced with permission from J. Phys. Chem. A, 2005, 109, 3527–3530. Copyright 2005 American Chemical Society. | ||
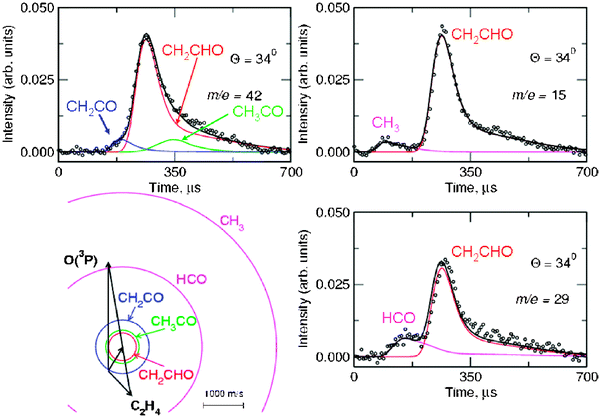 | ||
| Fig. 9 TOF spectra at Θ = 34° for the O(3P) + C2H4 reaction at Ec = 54.0 kJ mol−1 recorded at m/z = 42 (top-left)), m/z = 15 (top-right) and m/z = 29 (bottom-right) using an electron energy of 17 eV. Open circles are experimental points; heavy solid lines are the total TOF distributions calculated from the best-fit product CM translational energy and angular distributions for the contributing channels. The various contributions (depicted with different color lines) are marked with the formula of the corresponding product. Note that three product channels contribute to the signal detected at m/z = 42, two product channels to the signal detected at m/z = 15, and two product channels to the signal detected at m/z = 29. The circles in the Newton diagram delimit the maximum speed that the indicated products (same colour coding as circles) can attain on the basis of energy and linear momentum conservation if all the available energy goes into product translation. | ||
 | ||
| Fig. 10 TOF spectra at Θ = 34° for the O(3P) + C2H4 reaction at Ec = 54.0 kJ mol−1 recorded at m/z = 14 at 60 eV (top) and 17 eV (bottom). Top: m/z = 14 signal is coming from dissociative ionization of elastically scattered C2H4 reagent and to a negligible extent (~1%) from the reaction. Bottom: m/z = 14 signal is coming exclusively from reaction (note the relative scale with respect to the signal at 60 eV in the top panel); heavy solid line is the total TOF distribution of reactively scattered signals calculated from the best-fit product CM translational energy and angular distributions for the indicated contributing product channels. Note that five product channels contribute to the signal detected at m/z = 14 at 17 eV (see text). | ||
It should be noted that the m/z = 42 angular and TOF distributions can equally well be measured using hard (60 eV) EI, simply in this case the fast shoulder in the TOF spectra due to ketene formation is somewhat less intense than at 17 eV, because ketene fragments relatively less at 17 eV than at 60 eV. In contrast, for m/z = 15 it was necessary to perform measurements at 17 eV to suppress a pronounced elastic contribution (both in the angular and TOF distributions) from dissociative ionization of C2H4 (the appearance energies, A.E., of CH3+ and 13CH2+ from C2H4 are 17 and 18 eV, respectively).116
The m/z = 14 TOF spectrum at 17 eV (Fig. 10—bottom) exhibits, in addition to contributions from fragmentation of the ketene, vinoxy and acetyl products, very clearly also a fast peak which can only correspond, on the basis of energy and linear momentum conservation, to methylene formation from the channel CH2 + HCHO (formaldehyde) (3e). A small contribution to this fast peak also comes from fragmentation of the CH3 radical product from channel(3d), but this is small, considering that the appearance energy of CH2+ from CH3 dissociative ionization is A.E. = 15.1 eV.116 This was readily estimated from the known EI cross section of CH3 leading to CH3+ and CH2+ + H. It is worth noting that the detection of methylene from reaction channel(3e) was only possible because of the use of soft EI. In fact, by using an electron energy of 17 eV it was possible to remove completely the elastic contribution (which at 60 eV is about two orders of magnitude larger than the reactive signal; see Fig. 10—top) due to C2H4 scattered from the various components (O, O2, He) of the atomic oxygen beam, because the A.E. of CH2+ from C2H4 is 18 eV.116 We did not attempt to detect the counter-fragment HCHO (formaldehyde) at m/z = 30 because of some elastic interference present in the O beam even at 17 eV. However, m/z = 30 was cleanly detected by soft PI at 11.9 eV in the synchrotron work (see Fig. 4 in ref. 58).
From LAB angular and velocity distributions, product angular and translational energy distributions in the CM system were derived, and the branching ratios between the various competing channels were estimated.65,131 The angular distributions of vinoxy, ketene, and methyl products exhibit intensity over all of the CM angular range, with slightly more intensity in the forward direction (with respect to the O-atom direction). This indicates that the reaction is proceeding through an osculating complex mechanism.118 The T(θ) of the H-elimination channel, in addition, exhibits a peculiar form with a pronounced sideways scattering that can be rationalized in terms of the partitioning of the total angular momentum of the decomposing C2H4O complex (analogously to what was observed for the related F + C2H4 reaction133). The average fraction of energy in translation is 48% for channel(3a) and reflects a sizeable exit potential barrier. This is confirmed by the ab initio calculations of the PES.127 The molecular products of channel(3c) feature a P(E′T) peaking at about 165 kJ mol−1 (about 46% of the total available energy), and this reflects a large exit potential barrier, again in agreement with theoretical calculations of the PES.127 In contrast, the CH3 + HCO channel exhibits a P(E′T) peaking at a very low energy and this reflects a very high internal (ro-vibrational) excitation of the two molecular radical products (of these, HCO is expected to significantly decompose to H + CO because of the weak H–C bond before reaching the detector).
From our work65,131 it was found that formation of CH3 + HCO is the major channel (43%), followed by CH2CHO + H (27%); it was also firmly established, for the first time, that formation of molecular products, CH2CO + H2, is a sizeable channel in the O(3P) + ethylene reaction, accounting for about 13% of the yield, contrasting the conclusions of the most recent and reliable kinetic studies124,125 (see Table 2 where the present branching ratios are reported together with estimation from previous kinetic and dynamic studies, and also from theory). For the first time, it was also shown that a small fraction (1%) of acetyl radicals is formed, and finally, we have observed unambiguously, under truly single collision conditions, formation of methylene + formaldehyde at the level of 16%, corroborating two of the most recent kinetic investigations.124,125 It should be noted that the fraction of CH3CO may be somewhat underestimated in our study, because highly internally excited acetyl radicals rapidly decompose to CH3 + CO before reaching the detector, due to a relatively low (75 ± 12 kJ mol−1) dissociation barrier.
The observation of channels (3b), (3c), and (3d), which account for about 2/3 of the overall reaction yield, can only be rationalized assuming that ISC between the triplet and singlet PESs is occurring very efficiently. This is supported by recent theoretical work127 using various quantum mechanical methods and statistical rate theory on the PESs for both the triplet and singlet electronic states for the O(3P) + C2H4 reaction. It should be noted that formation of HCO + CH3 on the triplet PES is prevented, at least at low Ecs, by a high isomerization barrier from the initial diradical adduct to triplet acetaldehyde; this barrier is calculated to be ~8 kcal mol−1 above the reactant asymptote. Calculated product branching ratios on the ab initio PESs assuming a ratio for total triplet and total singlet yields of about 45% versus 55% were found127 to be in excellent agreement with the branching ratio results derived from our CMB study (see Table 2), with the exception that the statistical theory underestimates the yield of the molecular channel leading to CH2CO + H2; however, this is not surprising considering that this channel is found to be highly non-statistical from our experiments, with a P(E′T) peaking at very high values and reflecting a high potential barrier in the exit channel. The statistical calculations predict also a few percent of channel(3f) at our Ec, but this is very hard to probe experimentally (even with soft PI). Very recently, dynamical calculations by the QCT method with surface hopping, with the inclusion of non-adiabatic couplings between the triplet and singlet PESs of C2H4O, have been reported by Schatz and coworkers.128 While this work contributes significantly to our understanding of the complex non-adiabatic dynamics of this important reactive system, only semiquantitative agreement is found with our results (see Table 2). One of the main complications is perhaps an accurate estimate of the non-adiabatic singlet–triplet spin–orbit coupling elements which determine the extent of ISC.
In the recent pulsed CMB study at Ec = 26.8 kJ mol−1 using tunable VUV synchrotron radiation for ionization, TOF spectra of all carbon-containing products from the four exit (3a,c,d,e) channels were measured, but the branching ratios were not estimated.58 For completeness, we note that in that work58 some signals were attributed to excited O(1D) atoms also present in the beam in the amount of 4% and producing 3-body channels (as CH2O + H + H), which represents a considerable complication. We did not have evidence of this, despite the presence of a comparable concentration of O(1D) also in our atomic oxygen beam.
3.3 Radical–radical reactions
Elementary reactions between two radicals play an important role in numerous gaseous environments – such as flames and plasma-assisted processes – and in the chemical evolution of terrestrial and planetary atmospheres and the interstellar medium. Kinetic studies have particularly focused on radical–radical reactions that affect combustion chemistry,126 such as those involving polyatomic hydrocarbon radicals. For these reactions, several channels are usually open and their branching ratios might be difficult to estimate.Scarce dynamical studies exists on radical–radical reactions. Pioneering work on radical–radical reaction dynamics was carried out by Dagdigian et al.134 by LIF spectroscopy. Choi et al. carried out a similar study of the reactions O(3P) + propargyl,135O(3P) + allyl,136,137 and O(3P) + t-butyl,138 employing crossed jets and detecting the OH product ro-vibrational distributions and H-atom Doppler profile by LIF. Again, CMB experiments with “universal” MS detection are desirable to look at the possible channels of radical–radical reactions. However, despite the potentiality of the method, very little has been done up to now. This is mainly because of the difficulty in generating two beams of unstable species with a density high enough to perform a reactive scattering experiment. The only prior CMB study performed with MS detection on a radical–radical reaction was carried out in the 1990s on the reaction C(3P) + propargyl, and used pulsed beams;139 in that study from a very limited number of TOF spectra the product CM angular and translational energy distributions were derived. Since then no other attempts were made to measure differential cross sections of radical–radical reactions until very recently, when, using our improved CMB instrument and exploiting the novel capability of producing intense continuous supersonic beams of hydrocarbon radicals by a flash pyrolytic beam source, we have undertaken the investigation of atomic radical + hydrocarbon radical reactions. The first reaction we have looked at is that between atomic oxygen and the allyl radical.140 Preliminary data have also been obtained for the O(3P) + CH3 (methyl) reaction.
A quite different picture of the O(3P) + C3H5 reaction has emerged from a recent paper reporting ab initio electronic structure calculations of the stationary points of the relevant PES and RRKM estimates on the importance of the accessible reaction channels.142 According to the ab initio calculations of the lowest doublet C3H5O PES, nine reaction channels are open, as they are connected to the reactants by one or more adiabatic pathways:
| O(3P) + C3H5 → CH2CHCHO(acrolein) + H ΔH°0 = −279 kJ mol−1 | (4a) |
| CH2(CO)CH2(cyclopropanone) + H ΔH°0 = −186 kJ mol−1 | (4b) |
| H2CO + C2H3 ΔH°0 = −228 kJ mol−1 | (4c) |
| C2H4 + HCO ΔH°0 = −324 kJ mol−1 | (4d) |
| CH2CO(ketene) + CH3 ΔH°0 = −321 kJ mol−1 | (4e) |
| C2H5 + CO ΔH°0 = −410 kJ mol−1 | (4f) |
| CH2CCH2 + OH ΔH°0 = −190 kJ mol−1 | (4g) |
| CH3CCH + OH ΔH°0 = −196 kJ mol−1 | (4h) |
| CH2(CH)CH2(cyclopropene) + OH ΔH°0 = −104 kJ mol−1 | (4i) |
By using the CMB technique with soft EI MS detection we were able to characterize the dynamics of the H-elimination channel and explore some C–C bond breaking channels.140 Useful complementary information is provided by new experiments by Butler and co-workers143 in which the O + allyl reaction pathways are investigated from the OCH2CHCH2 radical intermediate obtained by UV photodissociation of a suitable precursor (epichlorohydrin).
The LAB distributions recorded at m/z = 56 (C3H4O+, the acrolein parent peak) and 55 (the daughter ion C3H3O+) at Ec = 73.0 kJ mol−1 were found to be superimposable, thus indicating that the only detected product in this range of masses is C3H4O and that it partly fragments to C3H3O+ in the ionizer. In order to have the best S/N ratio, all of the final measurements were carried out at m/z = 55. Note that for this set of measurements, the ionizing electron energy was 60 eV, as no significant improvement in the S/N was obtained by soft EI. The product LAB angular distribution together with the most probable Newton diagram (showing the kinematics of the process) is reported in Fig. 11 with open circles. The dashed line superimposed on the experimental results is the total calculated curve when using the best-fit CM functions for the H-channel (see ref. 140).
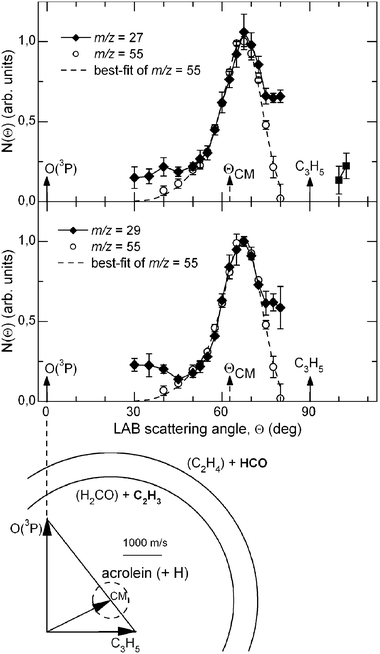 | ||
| Fig. 11 LAB angular distributions for the reactions O(3P,1D) + C3H5 at Ec = 73.0 kJ mol−1 of (top) the C3H4O product at m/z = 55 (open circles) and C2H3 product at m/z = 27 (diamonds), (bottom) the C3H4O product at m/z = 55 (open circles) and the HCO product at m/z = 29 (diamonds), measured at the electron energy of 17 eV. The best-fit total LAB angular distribution of the acrolein product from the H-elimination channel is indicated with dashed line. (see text and ref. 140). For the C–C bond breaking channels the data points are joined by a line to simply guide the eye. The velocity vector diagram showing the kinematics of the experiment is also shown; there the circles delimit the maximum speed achievable by the acrolein, C2H3 (vinyl) and HCO (formyl) products formed in channels (4a), (4c) and (4d). (Phys. Chem. Chem. Phys., 2007, 9, 1307–1311)—reproduced by permission of the PCCP owner societies. | ||
The peculiar shape of the CM angular distribution (exhibiting some sideways scattering) was qualitatively interpreted in the light of the recent ab initio calculations of the C3H5O PES142 and connected to the geometry of the involved decomposing transition states. As for the product energy release, the fraction (0.58) of energy released as product translational energy in our CMB study at Ec = 73.0 kJ mol−1 is much larger than that determined in the previous CMB study at Ec = 26.8 kJ mol−1, where H atoms were detected by VUV-LIF Lyman-α spectroscopy.136 A direct comparison cannot be made, because the Ecs of the two experiments differ significantly. However, in that study a surprisingly small fraction of energy in product translation was derived, being only 0.054. It should be noted that exit barriers of 19.7 and 30.1 kJ mol−1 with respect to products have been calculated by the same authors for the reaction proceeding through INT1 and INT4, respectively,142 and that these barriers should channel some of the available energy into product translational energy. At this stage it is unclear what the origin of the discrepancy between our results140 and the previous work is.136
According to the ab initio calculations and RRKM estimates,142 there are at least two C–C bond breaking channels wich are competitive with acrolein formation: channel (c) which is the result of a C(2)–C(3) bond cleavage of INT1, CH2CH-CH2O, and channel (d) which is the result of the C(2)-C(3) bond cleavage of INT4, CH2CH2-CHO. In particular, formation of C2H4 and HCO is predicted by RRKM calculations to be by far the dominant pathway originating from INT4 and, therefore, from one of the three addition intermediates. With the aim of verifying whether one or both of these channels are open, we have explored the relevant range of masses to examine the possible formation of these products. We have been able to observe reactive signals at m/z = 27 and 29 and we have measured LAB angular distributions for both masses (see Fig. 11) and have taken TOF spectra for the fragment at m/z = 27 (see Fig. 12). In order to do that, we took full advantage of the soft EI technique, as both distributions could be measured only by reducing the background signal coming from the dissociative ionization of species contained in the allyl beam by employing a 17 eV electron energy (see Fig. 13, where the detector mass spectra during experiment at the CM angle are reported for 60 and 17 eV; note here the dramatic simplification of the mass spectrum at the lower electron energy because of the reduction of dissociative ionization processes). As can be seen from Fig. 11 showing the LAB angular distributions measured at 17 eV, even at this low electron energy there is a significant contribution arising from the dissociative ionization of acrolein. However, some product intensity is visible at LAB angles which are outside the angular range amenable to the acrolein+H products. As can be seen from Fig. 12 the m/z = 27 spectrum shows, in addition to a main, slow peak coming from dissociative ionization of acrolein (and therefore similar to that recorded at m/z = 55), a fast shoulder which is expected to originate mainly from C2H3 (vinyl) from the H2CO + C2H3channel(4c) and, to a lower extent at this relatively low electron energy, from dissociative ionization of C2H4 product from the C2H4 + HCO channel(4d). The fact that the shapes of the m/z = 27 and 29 angular distributions are significantly different (the m/z = 29 distribution appears more forward peaked than that of m/z = 27, see Fig. 11) implies that the two signals are originating from two dynamically different channels. The m/z = 29 signal is expected to mostly originate from HCO (channel 4d) and to a minor extent from dissociative ionization of H2CO (channel 4c). At the moment, the experimental results are not sensitive enough to allow us to extract the relevant CM functions and to establish in detail the reaction mechanisms; further experiments are planned to address this point. Although not conclusive, the scattering results provide evidence that at the collision energy of the experiment, at least two C–C bond breaking channels are also open, those leading to C2H4 + HCO and H2CO + C2H3. According to the calculated PESs, the latter channel can occur in competition with the acrolein + H channelvia INT1, while the former is formed from INT4 once reached from INT1 viaisomerization. The more backward–forward symmetric m/z = 27 angular distribution correlates well with the less exoergic C2H3 + H2CO channel, while the more forward biased m/z = 29 angular distribution correlates well with the much more exoergic C2H4 + HCO channel. From the extent of the reactive intensities between signals coming from dissociative ionization of acrolein and that from C–C bond breaking channels we anticipate that the cross sections for the latter processes are comparable (likely larger) with respect to that of the H-elimination channel, at least at the Ec of the experiment.
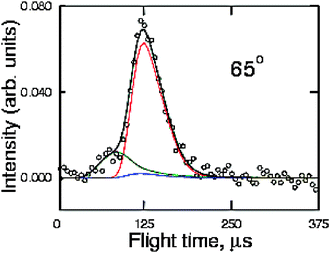 | ||
| Fig. 12 TOF distribution of the m/z = 27 product from the reactions O(3P,1D) + C3H5 at Ec = 73.0 kJ mol−1 measured at a LAB angle of 65 using 17 eV electron energy. The main peak and the faster shoulder are attributed to dissociative ionization of the acrolein product and to the parent ion of the vinyl radical product (channel 4c), respectively. | ||
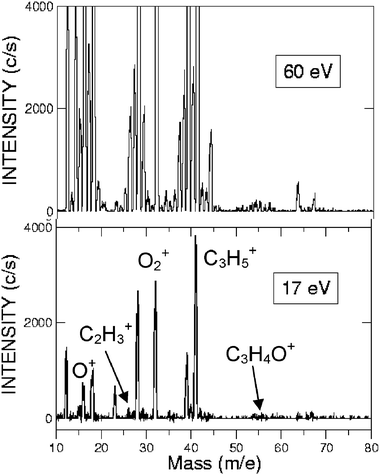 | ||
| Fig. 13 Detector mass spectra at 60 eV (top) and 17 eV (bottom) recorded at the CM angle during the CMB experiment on the reactions O(3P,1D) + C3H5 at Ec = 73.0 kJ mol−1. Note the dramatic suppression of background mainly due to a strongly reduced fragmentation when using soft ionization at 17 eV; here some mass peaks are identified with the parent ion of the two reagents and the identified products. | ||
Very recent work by Butler and coworkers143 uses photodissociation (at 193 nm) of epicholorohydrin to produce the cyclic-OCH2CHCH2 radicals with an internal energy content ranging from 17 to 70 kcal mol−1 with a maximum at 38 kcal mol−1. This radical has enough internal energy to undergo a facile ring opening and isomerize to the straight chain OCH2CHCH2 radical intermediate, which can dissociate to one or more product channels of the O + allyl bimolecular reaction. Photo-ionization by VUV synchrotron light in a crossed laser-beam experiment is then used to detect the velocity distribution of the possible products. The data (TOF spectra at m/z = 56, 30, 29, 28, and 27) identify three dominant product channels as C3H4O (acrolein) + H, C2H4 + HCO and H2CO (formaldehyde) + C2H3 (vinyl). The branching ratio of the acrolein channel was estimated to be 0.18 while those of the C–C bond breaking channels were not derived. Notably, the most exoergic C2H5 + CO channel was not observed to contribute significantly, while a small signal from CH2CO (ketene) was also detected. Unfortunately, a direct comparison between Butler’s results and our CMB results is not possible because the internal energy content of the OCH2CHCH2 radical intermediate in our CMB experiment is above the O + allyl reactant asymptote, while in the case of Butler’s study is significantly below.
In conclusion, it appears that also in polyatomic radical–radical reactions all (or nearly all) the energetically allowed channels actually occur, as was observed for the O + alkene reactions. In the case of the O + allyl reaction it is now clear that H elimination, H abstraction, and C–C bond breaking are all occurring at some extent. This calls for experiments aimed at examining a variety of radical–radical reactions using the universal CMB technique with soft EI (or PI), possibly complemented by theoretical studies of the relevant PESs as well as by studies on the dissociation of radical intermediates.
4. Conclusion and outlook
We have reviewed some significant advances made in our laboratory over the last few years in the field of reaction dynamics, with particular attention to the investigation of polyatomic reactions exhibiting multiple channels. We have emphasized that the implementation of soft EI for product detection in CMB experiments has been central for progress in this area, because the use of low-energy tunable electrons for product ionization has permitted us to reduce interfering signals originating from the dissociative ionization of products, reactants and background gases, which usually represents a major complication. In addition, the implementation of a variable beam crossing angle set-up in a universal CMB instrument has permitted us to extend the range of collision energies over which a given reaction can be studied. By exploiting these new features in an improved CMB instrument and the capability of generating continuous supersonic beams of a variety of radicals, we have elucidated the dynamics of important reactions, such as those of O(3P) and C(3P) with acetylene and ethylene, which are, besides the fundamental interest, of significant relevance in combustion chemistry and astrochemistry. Furthermore, we have demonstrated that the experimental investigation of polyatomic multichannel reactions involving an atomic and a hydrocarbon radical is now feasible in CMB experiments with MS detection.In the near future we plan to use the same experimental approach to tackle detailed dynamical studies of some other reactions of practical relevance. Besides more reactions involving O and C atoms, we envisage the investigation of reactions of N and S atoms and of molecular radicals, such as OH, CN and C2 or polyatomic radicals, such as hydrocarbon radicals. In studies of reactions of molecular radical produced by a discharge or thermal dissociation it is important to know the internal energy content of the produced radicals. For this reason we have built a new experimental set-up to characterize the supersonic radical beams by LIF measurements98 and soon also by REMPI (when applicable). Some reactions involving several of the above atomic and molecular radical species have already been investigated in our laboratory, by observing mainly the H-elimination channel, but the novel capability of exploring all possible product channels is an incentive to re-investigate them. Contributions in this area are also expected from numerous other laboratories where work on polyatomic radical reactions is being pursued using pulsed CMB techniques with EI-MS detection,144VUV PI by synchrotron radiation MS detection,57,58 and ion-imaging detection,145 laser photo-initiated methods in co-expanded reagents with REMPI detection,146,147 as well as standard kinetic techniques empowered with VUV tunable synchrotron radiation detection.60,148
Another class of reactions that is very interesting to continue to study are those involving two open-shell reactants. Following the success in the study of the O(3P) + C3H5 reaction, we plan to study other similar reactions of relevance in combustion such as those of O(3P) atoms with CH3 (methyl), C2H5 (ethyl), and C3H3 (propargyl). Interesting reactions are also those of N atoms with alkyl radicals (CH3 and C2H5), of particular relevance in the chemistry of the atmosphere of Saturn’s moon Titan, which has been highlighted by the recent Cassini–Huygens mission. Finally, especially interesting are those radical–radical reactions, such as N + OH, C + OH, O + OH and S + OH, which are simple enough to be within the capabilities of current theoretical treatments, both at the level of accurate electronic structure calculations of the PES and of dynamical calculations by exact quantum and/or quasi-classical trajectory methods.149
Looking to the future, one may think to attempt state-resolved experiments on these polyatomic systems to gain further information on the dynamics. Valuable contributions are expected from CMB experiments with laser spectroscopic detection, possibly coupled to ion-imaging techniques.39,40 Unfortunately, there are fundamental limitations to obtaining quantum-state specific scattering data for polyatomic reactions, especially radical–radical reactions, with the exception perhaps of reaction channels leading to products that can be probed very efficiently by REMPI, such as HCl146 and CH339 or an atom.150 In the realm of polyatomic multichannel radical–molecule and radical–radical reactions proceeding via the addition/elimination mechanism, very promising is the new approach proposed by Butler and co-workers,143 where a particular isomeric form of an unstable radical intermediate along a bimolecular reaction coordinate is generated under collision-less conditions and investigate the branching between the ensuing product channels of the energized radical as a function of its internal energy. These experiments allow probing key portions of the reaction dynamics and also of the PES, such as isomerization and dissociation barrier heights. The use of VUV PI with tunable synchrotron radiation has revealed itself to be extremely useful to detect selectively the possible products and hence to probe the barriers encountered by the radical intermediates as they proceed towards one of the product channels of the bimolecular reaction. In principle the methodology can probe the dynamics resulting from each of several possible radical intermediates, provided that they can be formed from a controlled photodissociation of suitable precursor molecules.
Future CMB experiments with beam crossing angles down to 25–30° will permit us to reach collision energies ≤1 kJ mol−1, which is of particular interest for the chemistry of cold planetary atmospheres and the interstellar medium. These experiments will be a valuable supplement to the successful contribution of new kinetic measurements by the CRESU technique151 down to very low temperatures, as well as of pulsed CMB experiments with LIF/REMPI detection at the collision center down to very low collision energies.66,73–75 Significant complementary contributions are also expected to come from CMB experiments with tunable VUV synchrotron radiation detection. Overall, CMB studies with either soft EI or PI will complement strongly the new kinetic experiments exploiting VUV PI detection, which have demonstrated selective isomer product detection capability.60 In this regard, photoionization spectroscopy and reliable absolute PI efficiency measurements are required60 to proceed in parallel with flame and kinetics measurements, for instance, as well as with reactive scattering and photodissociation investigations,50,143 if reliable branching ratios are to be derived.
Crucial synergic contributions to our progress in the understanding of the kinetics and dynamics of polyatomic multichannel reactions is expected from ab-initio electronic structure calculations of the stationary points of the relevant reaction PESs. One particularly appealing aspect is the prospect of developing a full-dimensional PESs for some of these complex systems on which to carry out QCT dynamical calculations.79 Unfortunately, for polyatomic reactions it is not possible to perform rigorous quantum dynamical calculations on accurate multidimensional PESs. Direct dynamics QCT calculations “on-the-fly” are perhaps the most promising approach to treat theoretically the dynamics of polyatomic reactions. Such calculations are starting to be performed for reactions such as F + CH4 → HF + CH3152 and Cl + RH → HCl + R.146 Extensions of this approach to the multichannel reactions discussed in this article have also started to appear128 and more are to be expected in the near future. Advances in experiment and theory will expectedly make reactions such as those of O(3P) and C(3P) with C2H2 and C2H4 prototypical for detailed studies using the latest methods of reaction dynamics and related computational techniques.
CMB studies of polyatomic reactions such as those described in this Perspective article, together with synergic related theoretical work and complementary experimental dynamical studies, are expected to contribute to bridge the gap between crossed-beam dynamics and thermal kinetics research, and therefore to deepen our understanding of chemical reactivity by widening detailed investigations to a large variety of real-world reactions.
Acknowledgements
We acknowledge financial support from the Italian “Ministero Istruzione, Università e Ricerca (MIUR)” (FIRB 2001 and several PRIN projects, 2001-2003-2005-2007). Partial support from the EC Research Training Network “REACTION DYNAMICS” (contract HPRN-CT-1999-00007), Marie Curie Research Training Network “MOLECULAR UNIVERSE” (contract MCRTN-CT-2004-512302) and Coordination Action “EuroPlanet” (Contract 001637) is also gratefully acknowledged. Special thanks go to Emeritus Professor G. G. Volpi for continuous inspiration and support over the years. We also thank Astrid Bergeat, Kevin Hickson, and Domenico Stranges for their valuable contribution to some aspects of the work discussed here. Finally, this article is a contribution from a member (PC) of the Physical Chemistry Division of the Italian Chemical Society (SCI) in occasion of the 100th anniversary of SCI, to be celebrated at the 2009 SCI national meeting in Sorrento.References
- (a) R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics and Chemical Reactivity, New York, Oxford University Press, 1987 Search PubMed; (b) R. D. Levine, Molecular Reaction Dynamics, Cambridge, Cambridge University Press, 2005 Search PubMed.
- Y. T. Lee, Nobel Prize lecture, “Molecular Beam Studies of Elementary Chemical Processes” reprinted in Science, 1987, 236, 793, and in Angew. Chem., Int. Ed. Engl., 1987, 26, 939.
- D. R. Herschbach, Nobel Prize lecture, “Molecular Dynamics of Elementary Chemical Reactions” reprinted in Angew. Chem., Int. Ed. Engl., 1987, 26, 1223.
- J. C. Polanyi, Nobel Prize lecture, “Some Concepts in Reaction Dynamics” reprinted in Science, 1987, 236, 680, and in Angew. Chem., Int. Ed. Engl., 1987, 26, 952.
- P. Casavecchia, Rep. Prog. Phys., 2000, 63, 355 CrossRef CAS , and references therein.
- Modern Trends in Chemical Reaction Dynamics: Experiment and Theory (Part I & II), ed. X. Yang and K. Liu, Adv. Ser. Phys. Chem. vol. 14, World Scientific, Singapore, 2004 Search PubMed.
- L. Schnieder, K. Seekamp-Rahn, J. Borkowski, E. Wrede, K. H. Welge, F. J. Aoiz, L. Banares, M. J. D’Mello, V. J. Herrero, V. Saez Rabanos and R. E. Wyatt, Science, 1995, 269, 207 CrossRef CAS.
- M. Alagia, N. Balucani, L. Cartechini, P. Casavecchia, E. H. van Kleef, G. G. Volpi, F. J. Aoiz, L. Bañares, D. W. Schwenke, T. C. Allison, S. L. Mielke and D. G. Truhlar, Science, 1996, 273, 1519 CAS.
- Skouteris, D. E. Manolopoulos, W. Bian, H.-J. Werner, L.-H. Lai and K. Liu, Science, 1999, 286, 1713 CrossRef CAS.
- T. Skodje, D. Skouteris, D. E. Manolopoulos, S.-H. Lee, F. Dong and K. Liu, J. Chem. Phys., 2000, 112, 4536 CrossRef CAS.
- K. Liu, Annu. Rev. Phys. Chem., 2001, 52, 139 CrossRef CAS , and references therein.
- K. Liu, R. T. Skodje and D. E. Manolopoulos, PhysChemComm, 2002, 5, 27 RSC.
- A. Harich, D. Dai, C. C. Wang, X. Yang, S. D. Chao and R. T. Skodjie, Nature, 2002, 419, 281 CrossRef CAS.
- S. C. Althorpe, F. Fernandez-Alonso, B. D. Bean, J. D. Ayers, A. E. Pomerantz, R. N. Zare and E. Wrede, Nature, 2002, 416, 67 CrossRef CAS.
- F. Fernandez-Alonso and R. N. Zare, Annu. Rev. Phys. Chem., 2002, 53, 67 CrossRef CAS.
- D. Dai, C. C. Wang, S. A. Harich, X. Wang, X. Yang, S. D. Chao and R. T. Skodje, Science, 2003, 300, 1730 CrossRef CAS.
- S. C. Althorpe and D. C. Clary, Annu. Rev. Phys. Chem., 2003, 54, 493 CrossRef CAS.
- K. Liu, in Modern Trends in Chemical Reaction Dynamics: Experiment and Theory (Part II), ed. X. Yang and K. Liu, Adv. Ser. Phys. Chem. vol. 14, World Scientific, Singapore, 2004, ch. 1 Search PubMed.
- N. Balucani, D. Skouteris, G. Capozza, E. Segoloni, P. Casavecchia, M. H. Alexander, G. Capecchi and H.-J. Werner, Phys. Chem. Chem. Phys., 2004, 6, 5007 RSC.
- R. T. Skodje and X. Yang, Int. Rev. Phys. Chem., 2004, 23, 253 CrossRef CAS.
- X. Yang, Int. Rev. Phys. Chem., 2005, 24, 37 CrossRef CAS.
- M. Qiu, Z. Ren, L. Che, D. Dai, S. A. Harich, X. Wang, X. Yang, M. Gustafsson, R. T. Skodje, C. Xu, D. Xie, Z. Sun and D. H. Zhang, Science, 2006, 311, 1440 CrossRef.
- L. Che, Z. Ren, X. Wang, W. Dong, D. Dai, X. Wang, D. H. Zhang, X. Yang, L. Sheng, G. Li, H.-J. Werner, F. Lique and M. H. Alexander, Science, 2007, 317, 1061 CrossRef CAS.
- X. Wang, W. Dong, M. Qiu, Z. Ren, L. Che, D. Dai, X. Wang, X. Yang, Z. Sun, B. Fu, S.-Y. Lee, X. Xu and D. H. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6227 CrossRef CAS.
- X. Wang, W. Dong, C. Xiao, L. Che, Z. Ren, D. Dai, X. Wang, P. Casavecchia, X. Yang, B. Jiang, D. Xie, Z. Sun, S.-Y. Lee, D. H. Zhang, H.-J. Werner and M. H. Alexander, Science, 2008, 322, 573 CrossRef CAS.
- N. Balucani, M. Alagia, L. Cartechini, P. Casavecchia, G. G. Volpi, L. A. Pederson and G. C. Schatz, J. Phys. Chem. A, 2001, 105, 2414 CrossRef CAS.
- N. Balucani, L. Cartechini, G. Capozza, E. Segoloni, P. Casavecchia, G. G. Volpi, F. J. Aoiz, L. Bañares, P. Honvault and J.-M. Launay, Phys. Rev. Lett., 2002, 89, 013201 CrossRef.
- N. Balucani, G. Capozza, L. Cartechini, A. Bergeat, R. Bobbenkamp, P. Casavecchia, F. J. Aoiz, L. Bañares, P. Honvault, B. Bussery-Honvault and J.-M. Launay, Phys. Chem. Chem. Phys., 2004, 6, 4957 RSC.
- N. Balucani, G. Capozza, E. Segoloni, A. Russo, R. Bobbenkamp, P. Casavecchia, T. Gonzalez-Lezana, E. J. Rackham, L. Bañares and F. J. Aoiz, J. Chem. Phys., 2005, 122, 234309 CrossRef.
- N. Balucani, P. Casavecchia, L. Bañares, F. J. Aoiz, T. Gonzalez-Lezana, P. Honvault and J.-M. Launay, J. Phys. Chem. A, 2006, 110, 817 CrossRef CAS.
- N. Balucani, G. Capozza, F. Leonori, E. Segoloni and P. Casavecchia, Int. Rev. Phys. Chem., 2006, 25, 109 CrossRef CAS.
- L. Banares, F. J. Aoiz and J.-M. Launay, J. Phys. Chem. A, 2004, 108, 1616 CrossRef CAS.
- F. J. Aoiz, L. Banares and V. J. Herrero, J. Phys. Chem. A, 2006, 110, 12546 CrossRef CAS.
- M. Alagia, N. Balucani, P. Casavecchia, D. Stranges, G. G. Volpi, D. C. Clary, A. Kliesch and H.-J. Werner, Chem. Phys., 1996, 207, 389 CrossRef CAS.
- D.-C. Che and K. Liu, Chem. Phys., 1996, 207, 367 CrossRef CAS.
- B. R. Strazisar, C. Lin and H. F. Davis, Science, 2000, 290, 958 CrossRef CAS.
- J. J. Lin, J. Zhou, W. Shiu and K. Liu, Science, 2003, 300, 966 CrossRef CAS.
- S. Yan, Y.-T. Wu, B. Zhang, X.-F. Yue and K. Liu, Science, 2007, 316, 1723 CrossRef CAS.
- K. Liu, Phys. Chem. Chem. Phys., 2007, 9, 17 RSC , and references therein.
- M. N. R. Ashfold, N. H. Nahler, A. J. Orr-Ewing, O. P. J. Vieuxmaire, R. L. Toomes, T. N. Kitsopoulos, I. A. Garcia, D. A. Chestakov, S.-M. Wu and D. H. Parker, Phys. Chem. Chem. Phys., 2006, 8, 26 RSC , and references therein.
- Y. T. Lee, J. D. McDonald, P. R. LeBreton and D. R. Herschbach, Rev. Sci. Instrum., 1969, 40, 1402 CrossRef CAS.
- Y. T. Lee, in Atomic and Molecular Beam Methods, ed. G. Scoles, Oxford University Press, New York, 1987, vol. 1, pp. 553–568 Search PubMed.
- R. I. Kaiser, Chem. Rev., 2002, 102, 1309 CrossRef CAS.
- J. J. Schroden and H. F. Davis, in Modern Trends in Chemical Reaction Dynamics: Experiment and Theory (Part II), Adv. Ser. Phys. Chem. vol. 14, ed. X. Yang and K. Liu, World Scientific, Singapore, 2004, ch. 5 Search PubMed.
- X. Yang, Phys. Chem. Chem. Phys., 2006, 8, 205 RSC , and references therein.
- X. Yang, J. Lin, Y. T. Lee, D. A. Blank, A. G. Suits and A. M. Wodtke, Rev. Sci. Instrum., 1997, 68, 3317 CrossRef CAS.
- C. C. Wang, J. Shu, J. J. Lin, Y. T. Lee and X. Yang, J. Chem. Phys., 2002, 117, 153 CrossRef CAS.
- D. A. Blank, W. Sun, A. G. Suits, Y. T. Lee, S. W. North and G. E. Hall, J. Chem. Phys., 1998, 108, 5414 CrossRef CAS.
- W. Sun, K. Yokoyama, J. C. Robinson, A. G. Suits and D. M. Neumark, J. Chem. Phys., 1999, 110, 4363 CrossRef CAS.
- J. C. Robinson, S. A. Harris, W. Sun, N. E. Sveum and D. M. Neumark, J. Am. Chem. Soc., 2002, 124, 10211 CrossRef CAS.
- J. J. Lin, Y. Chen, Y. Y. Lee, Y. T. Lee and X. Yang, Chem. Phys. Lett., 2002, 361, 374 CrossRef CAS.
- L. R. McCunn, K.-C. Lau, M. J. Krisch, L. Butler, J.-W. Tsung and J. J. Lin, J. Phys. Chem. A, 2006, 110, 1625 CrossRef CAS.
- S.-H. Lee, Y.-Y. Lee, Y. T. Lee and X. Yang, J. Chem. Phys., 2003, 119, 827 CrossRef CAS.
- D. A. Blank, N. Hemmi, A. G. Suits and Y. T. Lee, Chem. Phys., 1998, 231, 261 CrossRef CAS.
- N. Hemmi and A. G. Suits, J. Chem. Phys., 1998, 109, 5338 CrossRef CAS.
- H. F. Davis, J. Shu, D. Peterka and M. Ahmed, J. Chem. Phys., 2004, 121, 6254 CrossRef.
- C. Chaudhuri, I.-C. Lu, J. J. Lin and S.-H. Lee, Chem. Phys. Lett., 2007, 444, 237 CrossRef CAS.
- S.-H. Lee, W.-J. Huang and W.-K. Chen, Chem. Phys. Lett., 2007, 446, 276 CrossRef CAS.
- F. Goulay, D. L. Osborn, C. A. Taatjes, P. Zou, G. Meloni and S. R. Leone, Phys. Chem. Chem. Phys., 2007, 9, 4291 RSC.
- C. A. Taatjes, N. Hansen, D. L. Osborn, K. Kohse-Höinghaus, T. A. Cool and P. R. Westmoreland, Phys. Chem. Chem. Phys., 2008, 10, 20 RSC.
- P. A. Willis, H. U. Stauffer, R. Z. Hinrichs and H. F. Davis, Rev. Sci. Instrum., 1999, 70, 2606 CrossRef CAS.
- R. Z. Hinrichs, J. J. Schroden and H. F. Davis, J. Am. Chem. Soc., 2003, 125, 860 CrossRef CAS , and references therein.
- P. Casavecchia, G. Capozza and E. Segoloni, in Modern Trends in Chemical Reaction Dynamics Experiment and Theory (Part II), ed. X. Yang and K. Liu, Adv. Ser. Phys. Chem. vol. 14, World Scientific, Singapore, 2004, ch. 7 Search PubMed.
- G. Capozza, E. Segoloni, F. Leonori, G. G. Volpi and P. Casavecchia, J. Chem. Phys., 2004, 120, 4557 CrossRef CAS.
- P. Casavecchia, G. Capozza, E. Segoloni, F. Leonori, N. Balucani and G. G. Volpi, J. Phys. Chem. A, 2005, 109, 3527 CrossRef CAS.
- M. Costes, N. Daugey, C. Naulin, A. Bergeat, F. Leonori, E. Segoloni, R. Petrucci, N. Balucani and P. Casavecchia, Faraday Discuss., 2006, 133, 157 RSC.
- F. Leonori, R. Petrucci, E. Segoloni, A. Bergeat, K. M. Hickson, N. Balucani and P. Casavecchia, J. Phys. Chem. A, 2008, 112, 1363 CrossRef CAS.
- R. C. Wetzel, F. A. Baiocchi, T. R. Hayes and R. S. Freunf, Phys. Rev. A, 1987, 35, 559 CrossRef CAS.
- W. L. Fitch and A. D. Sauter, Anal. Chem., 1983, 55, 832 CrossRef CAS.
- A. M. Schmoltner, P. M. Chu, R. J. Brudzynski and Y. T. Lee, J. Chem. Phys., 1989, 91, 6926 CrossRef CAS.
- R. Gentry, in Atomic and Molecular Beam Methods, ed. G. Scoles, Oxford University Press, New York, 1987, vol. 1 Search PubMed.
- K. Liu, Int. Rev. Phys. Chem., 2001, 20, 189 CAS , and references therein.
- C. Naulin and M. Costes, Chem. Phys. Lett., 1999, 310, 231 CrossRef CAS.
- L. Cartechini, A. Bergeat, G. Capozza, P. Casavecchia, G. G. Volpi, W. D. Geppert, C. Naulin and M. Costes, J. Chem. Phys., 2002, 116, 5603 CrossRef CAS.
- D. Geppert, C. Naulin, M. Costes, G. Capozza, L. Cartechini, P. Casavecchia and G. G. Volpi, J. Chem. Phys., 2003, 119, 10607 CrossRef CAS.
- E. Buonomo and D. C. Clary, J. Phys. Chem. A, 2001, 105, 2694 CrossRef CAS.
- T. Takayanagi, Chem. Phys., 2005, 312, 61 CrossRef CAS.
- T. Takayanagi, J. Phys. Chem. A, 2006, 110, 361 CrossRef CAS.
- W. K. Park, J. Park, S. C. Park, B. J. Braams, C. Chen and J. M. Bowman, J. Chem. Phys., 2006, 125, 081101 CrossRef.
- A. M. Mebel, V. V. Kislov and M. Hayashi, J. Chem. Phys., 2007, 126, 204310 CrossRef CAS.
- A. Bergeat and J.-C. Loison, Phys. Chem. Chem. Phys., 2001, 3, 2038 RSC.
- Y. Guo, A. M. Mebel, X. Gu and R. I. Kaiser, J. Phys. Chem. A, 2007, 111, 2980 CrossRef.
- E. Herbst, Annu. Rev. Phys. Chem., 1995, 46, 27 CrossRef CAS.
- E. Herbst, J. Phys. Chem. A, 2005, 109, 4017 CrossRef CAS.
- M. Alagia, V. Aquilanti, D. Ascenzi, N. Balucani, D. Cappelletti, L. Cartechini, P. Casavecchia, F. Pirani, G. Sanchini and G. G. Volpi, Isr. J. Chem., 1997, 37, 329 CAS.
- S. J. Sibener, R. J. Buss, C. Y. Ng and Y. T. Lee, Rev. Sci. Instrum., 1980, 51, 167 CrossRef CAS.
- M. Alagia, N. Balucani, P. Casavecchia, D. Stranges and G. G. Volpi, J. Chem. Soc., Faraday Trans., 1995, 91, 575 RSC.
- P. Casavecchia, N. Balucani, M. Alagia, L. Cartechini and G. G. Volpi, Acc. Chem. Res., 1999, 32, 503 CrossRef CAS.
- N. Balucani, M. Alagia, L. Cartechini, P. Casavecchia, G. G. Volpi, K. Sato, T. Takayanagi and Y. Kurosaki, J. Am. Chem. Soc., 2000, 122, 4443 CrossRef CAS.
- N. Balucani, L. Cartechini, M. Alagia, P. Casavecchia and G. G. Volpi, J. Phys. Chem. A, 2000, 104, 5655 CrossRef CAS.
- P. Casavecchia, N. Balucani, L. Cartechini, G. Capozza, A. Bergeat and G. G. Volpi, Faraday Discuss., 2001, 119, 27 Search PubMed.
- A. Bergeat, L. Cartechini, N. Balucani, G. Capozza, L. Phillips, P. Casavecchia, G. G. Volpi, L. Bonnet and J. C. Rayez, Chem. Phys. Lett., 2000, 327, 197 CrossRef CAS.
- N. Balucani, D. Skouteris, L. Cartechini, G. Capozza, E. Segoloni, P. Casavecchia, M. H. Alexander, G. Capecchi and H.-J. Werner, Phys. Rev. Lett., 2003, 91, 013201 CrossRef CAS.
- P. Casavecchia et al., in preparation.
- M. Alagia, N. Balucani, P. Casavecchia, D. Stranges and G. G. Volpi, J. Chem. Phys., 1993, 98, 2459 CrossRef CAS.
- M. Alagia, N. Balucani, P. Casavecchia, D. Stranges and G. G. Volpi, J. Chem. Phys., 1993, 98, 8341 CrossRef CAS.
- P. Casavecchia, N. Balucani and G. G. Volpi, Advanced Series in Physical Chemistry—vol. 6: The Chemical Dynamics and Kinetics of Small Radicals, ed. K. Liu and A. Wagner, World Scientific, Singapore, 1995, ch. 9, pp. 365–437 Search PubMed.
- F. Leonori, R. Petrucci, K. M. Hickson, E. Segoloni, S. Le Picard, N. Balucani, P. Foggi and P. Casavecchia, Planet. Space Sci., 2008, 56, 1658 CrossRef CAS.
- D. W. Kohn, H. Clauberg and P. Chen, Rev. Sci. Instrum., 1992, 63, 4003 CrossRef CAS.
- J. Peeters, J. Bull. Soc. Chim. Belg., 1997, 106, 357 Search PubMed , and references therein.
- D. J. Hucknall, Chemistry of Hydrocarbon Combustion, Chapman Hall, New York, 1985 Search PubMed.
- K. Mahmud and A. Fontijn, J. Phys. Chem., 1987, 91, 1918 CrossRef CAS , and references therein.
- J. Peeters, M. Schaekers and C. Vinckier, J. Phys. Chem., 1986, 90, 6552 CrossRef CAS.
- P. Frank, K. A. Bhaskaran, T. H. Just, 21th Symp. (Int.) Combustion, 1986, p. 885.
- J. Peeters, S. Vanhaelemeersch, J. Van Hoeymissen, R. Borms and D. J. Vermeylen, J. Phys. Chem., 1989, 93, 3892 CrossRef CAS.
- J. V. Michael and A. F. Wagner, J. Phys. Chem. A, 1990, 94, 2453 Search PubMed.
- W. Boullart and J. Peeters, J. Phys. Chem., 1992, 96, 9810 CrossRef CAS.
- J. Peeters, W. Boullart and I. Langhans, Int. J. Chem. Kinet., 1994, 26, 869 CAS.
- X. Huang, G. Xing and R. Bershon, J. Chem. Phys., 1994, 101, 5818 CrossRef CAS.
- V. Chikan and S. R. Leone, J. Phys. Chem. A, 2005, 109, 2525 CrossRef CAS.
- A. M. Schmoltner, P. M. Chu and Y. T. Lee, J. Chem. Phys., 1989, 91, 5365 CrossRef CAS.
- L. B. Harding and A. F. Wagner, J. Phys. Chem., 1986, 90, 2974 CrossRef CAS.
- T. L. Nguyen, L. Vereecken and J. Peeters, J. Phys. Chem. A, 2006, 110, 6696 CrossRef CAS.
- S. A. Carl, L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2007, 9, 4071 RSC.
- A. R. Clemo, G. L. Duncan and R. Grice, J. Chem. Soc., Faraday Trans. 2, 1982, 78, 1231 RSC.
- NIST Chemistry WebBook, 2002.
- F. Leonori, G. Capozza, E. Segoloni, N. Balucani, G. G. Volpi, M. Rosi, P. Casavecchia, to be published.
- G. A. Fisk, J. D. McDonald and D. R. Herschbach, Discuss. Faraday Soc., 1967, 44, 228 Search PubMed.
- C. C. Hayden, D. M. Neumark, K. Shobatake, R. K. Sparks and Y. T. Lee, J. Chem. Phys., 1982, 76, 3607 CrossRef CAS.
- D. R. Yarkony, J. Phys. Chem. A, 1998, 102, 5305 CrossRef CAS.
- D. L Osborn, D. H. Mordaunt, H. Choi, R. T. Bise, D. M. Neumark and C. M. Rohlfing, J. Chem. Phys., 1997, 106, 10087 CrossRef CAS.
- F. Leonori, R. Petrucci, N. Balucani, M. Rosi, P. Casavecchia, to be published.
- R. J. Cvetanović and D. L. Singleton, Rev. Chem. Int., 1984, 5, 183 Search PubMed , and references therein.
- Y. Endo, S. Tsuchiya, C. Yamada, E. Hirota and S. Koda, J. Chem. Phys., 1986, 85, 4446 CrossRef CAS.
- U. Bley, P. Dransfeld, B. Himme, M. Koch, F. Temps and H. Gg. Wagner, Twenty-Second Symposium (International) on Combustion, The Combustion Institute, Pittsburgh, 1988, p. 997 Search PubMed.
- D. L. Baulch, C. T. Bowman, C. J. Cobos, R. A. Cox, Th. Just, J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 2005, 34, 757 CrossRef CAS.
- T. L. Nguyen, L. Vereecken, H. J. Hou, M. T. Nguyen and J. Peeters, J. Phys. Chem. A, 2005, 109, 7489 CrossRef CAS.
- W. Hu, G. Lendvay, B. Maiti and G. C. Schatz, J. Phys. Chem. A, 2008, 112, 2093 CrossRef CAS.
- M. L. Morton, D. E. Szpunar and L. J. Butler, J. Chem. Phys., 2001, 115, 204 CrossRef CAS.
- R. J. Buss, R. J. Baseman, G. He and Y. T. Lee, J. Photochem., 1981, 17, 389 CrossRef CAS.
- G. Capozza, E. Segoloni, F. Leonori, N. Balucani, G. G. Volpi, and P. Casavecchia, to be published.
- R. M. Lambert, M. I. Christie, R. C. Golesworthy and J. N. Linnet, Proc. R. Soc. London, Ser. A, 1968, 302, 167 CrossRef.
- G. N. Robinson, R. E. Continetti and Y. T. Lee, J. Chem. Phys., 1990, 92, 275 CrossRef CAS.
- D. Patel-Misra, D. G. Sauter and P. J. Dagdigian, J. Chem. Phys., 1991, 95, 955 CrossRef.
- H. Lee, S. K. Joo, L. K. Kwon and J. H. Choi, J. Chem. Phys., 2004, 120, 2215 CrossRef CAS.
- S.-K. Joo, L.-K. Kwon, H. Lee and J.-H. Choi, J. Chem. Phys., 2004, 120, 7976 CrossRef CAS.
- H.-C. Kwon, J.-H. Park, H. Lee, H.-K. Kim, Y.-S. Choi and J.-H. Choi, J. Chem. Phys., 2002, 116, 2675 CrossRef CAS.
- M. J. Nam, S. E. Youn and J. H. Choi, J. Chem. Phys., 2006, 124, 104307 CrossRef.
- R. I. Kaiser, W. Sun, A. G. Suits and Y. T. Lee, J. Chem. Phys., 1997, 107, 8713 CrossRef CAS.
- F. Leonori, N. Balucani, G. Capozza, E. Segoloni, D. Stranges and P. Casavecchia, Phys. Chem. Chem. Phys., 2007, 9, 1307 RSC.
- I. R. Slagle, J. R. Bernhardt and D. Gutman, J. Phys. Chem., 1990, 94, 3652 CrossRef CAS.
- J. H. Park, H. Lee and J.-H. Choi, J. Chem. Phys., 2003, 119, 8966 CrossRef CAS.
- B. L. FitzPatrick, K.-C. Lau, L. J. Butler, S.-H. Lee and J. Jr.-M. Lin, J. Chem. Phys., 2008, 129, 084301 CrossRef.
- F. Zhang, X. Gu, Y. Guo and R. I. Kaiser, J. Phys. Chem. A, 2008, 112, 3284 CrossRef CAS , and references therein.
- C. Huang, W. Li, A. D. Estillore and A. G. Suits, J. Chem. Phys., 2008, 129, 074301 CrossRef , and references therein.
- C. Murray, J. K. Pearce, S. Rudic, B. Retail and A. J. Orr-Ewing, J. Phys. Chem. A, 2005, 109, 11093 CrossRef CAS.
- B. Retail, R. A. Rose, J. K. Pearce, S. J. Greaves and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2008, 10, 1675 RSC , and references therein.
- T. M. Selby, G. Meloni, F. Goulay, S. R. Leone, A. Fahr, C. A. Taatjes and D. L. Osborn, J. Phys. Chem. A, 2008, 112, 9376.
- S. Y. Lin, H. Guo, P. Honvault, C. X. Xu and D. Q. Xie, J. Chem. Phys., 2008, 128, 014303 CrossRef.
- B. Retail, S. J. Greaves, J. K. Pearce, R. A. Rose and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2007, 9, 3261 RSC.
- H. Sabbah, L. Biennier, I. R. Sims, Y. Georgievskii, S. J. Klippenstein and I. W. M. Smith, Science, 2007, 317, 5834 , and references therein.
- J. F. Castillo, F. J. Aoiz, L. Bañares, E. Martinez-Nuñez, A. Fernández-Ramos and S. Vazquez, J. Phys. Chem. A, 2005, 109, 8459 CrossRef CAS.
| This journal is © the Owner Societies 2009 |