What can be learned from single molecule spectroscopy? Applications to sol–gel-derived silica materials
Fangmao
Ye
a,
Maryanne M.
Collinson
*b and
Daniel A.
Higgins
*a
aDepartment of Chemistry, Kansas State University, KS, 66506, Manhattan, USA. E-mail: higgins@ksu.edu
bDepartment of Chemistry, Virginia Commonwealth University, VA, 23229, Richmond, USA. E-mail: mmcollinson@vcu.edu
First published on 12th November 2008
Abstract
Single molecule spectroscopic methods are now being widely employed to probe the nanometer scale properties of sol–gel-derived silica materials. This article reviews a subset of the recent literature in this area and provides salient examples of the new information that can be obtained. The materials covered include inorganic and organically-modified silica, along with surfactant-templated mesoporous materials. Studies of molecule–matrix interactions based on ionic, hydrogen bonding and hydrophobic interactions are reviewed, highlighting the impacts of these interactions on mass transport phenomena. Quantitative investigations of molecular diffusion by single molecule tracking and fluorescence correlation spectroscopy are also covered, focusing on the characterization of anisotropic and hindered diffusion in mesoporous systems. Single molecule polarity studies are described and the new information that can be obtained from the resulting inhomogeneous distributions is discussed. Likewise, single molecule studies of silica acidity properties are reviewed, including observation of nanoscale buffering phenomena due to the chemistry of surface silanols. Finally, related single nanoparticle studies of macroporous silicas are also discussed.
 Fangmao Ye Fangmao Ye | Fangmao Ye received his BS degree in polymer materials & engineering from Hefei University of Technology in 2001 and got his MS degree in polymer science from The University of Science and Technology of China in 2004. He joined the department of chemistry at Kansas State University as a PhD candidate in 2004. His current research interests are in single molecule diffusion and adsorption in sol–gel derived silica films. |
 Maryanne M. Collinson Maryanne M. Collinson | Dr Maryanne M. Collinson received her BS in chemistry and forensic science from the University of Central Florida and a PhD under the direction of Edmond F. Bowden from North Carolina State University. After her PhD, Maryanne was a postdoctoral research associate at the University of North Carolina at Chapel Hill where she worked under the direction of R. Mark Wightman. Dr Collinson started her academic career at Kansas State University and is currently a Professor in Chemistry at Virginia Commonwealth University. Her research interests include electrochemical, microscopic and spectroscopic characterization of microporous and mesoporous materials and their applications in analytical science. |
 Daniel A. Higgins Daniel A. Higgins | Daniel A. Higgins received a BA in chemistry from St. Olaf College in 1988. He received a PhD in chemistry under the direction of Robert M. Corn from the University of Wisconsin-Madison in 1993. He subsequently worked as a postdoctoral fellow under Prof. Paul F. Barbara at the University of Minnesota. He joined the faculty at Kansas State University in 1996, where he currently serves as professor of chemistry. His research interests are in the optical microscopic and spectroscopic characterization of nano- and meso-structured thin film materials. |
1. Introduction
Sol–gel-derived materials find numerous applications in a variety of fields, having been used in sensors,1–3catalysts,4,5 separations media,6–8 ion-exchange coatings,9,10 nonlinear optics11 and solid-state electrochemical devices.12,13 Key advantages of the sol–gel process are its economy and the relative simplicity of materials preparation and modification. Typically, silica materials are prepared via acid- or base-catalyzed hydrolysis and condensation of alkoxysilanes such as tetramethoxysilane (TMOS)14,15 in homogeneous mixtures of alcohol and water. The mild polymerization conditions (the reaction is often run at room temperature) make it possible to encapsulate many different reagents (enzymes, receptors, catalysts, dyes, cellsetc.)1–3,16–19 within the silica matrix while retaining these reagents’ desired functional characteristics after gelation.Silica materials can also be prepared, in whole or in part, from organoalkoxysilanes, which can act as network modifiers.20–28 The resulting materials have been termed organically modified silicas (ORMOSILs).20 The preparation of ORMOSILs allows for further control over the physical properties and chemical composition of the final materials.21–28 Sol–gel-prepared silica materials can also be modified by reaction with organosilanes, using well-known post-synthesis procedures.29
While relatively simple to make, sol–gel materials are microscopically complex and thus require sophisticated tools to unravel this complexity. To date, many different methods have been used to follow the hydrolysis and condensation processes in sols prepared from both alkoxysilanes and organoalkoxysilanes, and to study the physical and chemical properties of the resulting gels.14 The vast majority of these can be classified as bulk techniques. They include NMR,30X-ray scattering,31 gas adsorption,32 Raman33,34 and FTIR35 spectroscopies, electrochemical methods,27,36UV-Vis absorption37,38 and fluorescence spectroscopies.17,38–40 Such bulk studies have provided many useful insights into the average chemical and physical properties of sol–gel materials. However, almost all such materials are heterogeneous, exhibiting variations on nanometer (and longer) length scales in terms of their acidity, polarity, porosity, surface charge and other properties. Importantly, such variations may not be random, leading to certain classes of heterogeneity (i.e. due to phase separation, domain formation, etc.) that may be masked, overlooked or otherwise misinterpreted when observed by bulk methods alone.
A number of research groups have recently begun to use single molecule spectroscopic methods as a means to better understand the impacts of nanoscale heterogeneity on the properties of sol–gel-derived silica materials. Single molecule fluorescence experiments probe many of the same phenomena probed by bulk methods, while also providing detailed new information about individual nanoscale environments and materials microheterogeneity.41–46 In such studies, the silica materials are investigated by doping them with very low (i.e. nanomolar) concentrations of fluorescent dyes. The dye molecules are then spatially isolated from each other using sample scanning, beam scanning, or wide-field optical microscopy methods and their emission characteristics probed.44 Since individual molecules and/or individual nanoscale environments are interrogated one at a time in single molecule experiments, detailed new information on the variations in materials properties is obtained. By judicious choice of the probe molecule, materials properties such as surface charge, surface silanol density, matrix polarity and matrix acidity can be investigated. By recording the translational or rotational motions of charged or neutral dyes of different sizes, molecular mobility within these materials can also be explored.
This review is focused specifically on the use of single molecule fluorescence microscopy and spectroscopy as means to probe sol–gel-derived silica materials. The purpose is to provide representative examples of the experiments that have been performed to date and the important new knowledge that has been gained. It is not meant as a comprehensive review of silica-based materials studies. Neither is it meant as a review of all possible single molecule methods, or even all single molecule studies of silica materials. Pioneering studies of similar materials prepared by other methods are not included.47–51 The article begins with a discussion of the sol–gel process. The majority of the discussion is then devoted to reviewing studies of molecule–matrix interactions, molecular mobility and other silica materials properties by single molecule methods. The article concludes with a brief discussion of challenges and future directions.
2. Materials synthesis
2.1 The sol–gel process
Scheme 1 depicts a representative view of some of the processes that occur during the acid-catalyzed hydrolysis and condensation of TMOS.14,15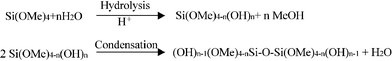 | ||
| Scheme 1 Salient processes involved in the acid-catalyzed hydrolysis and condensation of TMOS. | ||
Hydrolysis and condensation of the precursor begins immediately after preparation of the sol. As hydrolysis and condensation proceed, a rigid silica matrix, the gel, is formed. The final structure, morphology and porosity of the gel are highly dependent upon the ratio of precursor silane to water, the nature (acid or base) and concentration of the catalyst, and how the materials are processed and dried.14,15,52 In acid-catalyzed processes, the precursors tend to form highly crosslinked networks of relatively low porosity, while in base-catalyzed preparations, colloidal particles are produced that subsequently aggregate and react to give xerogels (dried gels) that have greater interstitial porosity.14 Two-step acid–base procedures have also been developed to take advantage of the benefits of both methods.53–56
2.2 Organically modified silicas (ORMOSIL)
ORMOSILs21–28 are typically prepared via the cohydrolysis and condensation of the alkoxysilanes (such as TMOS) with organoalkoxysilanes. In this article, the focus is entirely on these so-called Type II ORMOSILs.21 Typical precursors are represented as R′-Si(OR)3, where R is typically methyl or ethyl and R′ can be virtually any organic group, including methyl, ethyl, phenyl and acid- or amine-terminated substituents. Use of these precursors allows for control over the degree of crosslinking in the gel, and the reactivity, polarity, surface charge, acid/base properties, etc. of the pores found within.21–28 A distinct advantage of ORMOSIL methods is that the organic functional groups are covalently attached to and dispersed throughout the silicate framework. Covalent attachment of organic substituents to the external and internal (pore) surfaces can also be accomplished after synthesis by further reaction with species such as organically modified chlorosilanes.29 ORMOSILs have recently been shown to be useful in the preparation of superhydrophobic materials,57 and as platforms for biomolecule immobilization.1–3,172.3 Surfactant templated mesoporous materials
Mesoporous silica materials that contain pores of controlled size and organization can also be prepared by the sol–gel process by utilizing surfactants as “site-directing agents.”58,59 Lamellar, cubic or hexagonal ordering of ~2–50 nm sized pores/channels within the resulting gel can be achieved.60–66Pore size and organization are determined by the chemical structure and concentration of the specific surfactants employed.67Surfactant-containing mesoporous silica can be used and/or characterized without further modification, with the surfactant phase providing hydrophobic channels that facilitate transport of some molecules and strongly adsorb others.68–70 More commonly, they are calcined or Soxhlet-extracted to remove the surfactant, leading to formation of purely inorganic mesoporous silica.71 Removal of the surfactant may lead to pore collapse and an increase in materials disorder.60 The surfaces of mesoporous silicas can also be further derivatized with organic functional groupsin situ or ex situ to control pore surface properties.29
2.4 Silica monoliths and thin films
Sol–gel derived silica materials may be prepared as monoliths (bulk gels) or thin films. Monoliths are prepared by pouring the sol into a macroscopic vessel. Upon gelation a porous, solvent-filled gel is produced that is the same size and shape as the vessel. Upon drying, a xerogel is formed.52 Thin films are commonly prepared on planar substrates by aerosol methods,72spin coating,73dip coating,73,74 or electrochemical deposition,36 and are typically less than 1 μm thick.52Silica thin films are often significantly less porous than corresponding monoliths because gelation and evaporation in films occur simultaneously in a very short period of time (seconds).52 In monolith formation these processes take place over a substantially longer period (days to weeks).52 The thin film configuration is most often used in sensing applications because of the short path length for diffusion.52 Although exceptions exist,75 the vast majority of single molecule studies have been performed on silica films, and such films will be the primary focus of the discussion below.3. Single molecule studies of sol–gel-derived materials
Single molecule studies of sol–gel-derived materials provide distinct advantages over similar bulk investigations in that they allow for materials heterogeneity to be directly probed and individual chemical events and molecular-scale environments (including rare ones) to be observed and characterized. Taken together, the range of ongoing bulk and single molecule studies promises a more complete, in-depth understanding of molecule–matrix interactions, mass transport phenomena and matrix polarity, acidity and porosity properties of silica materials.3.1 Single molecule spectroscopic and microscopic methods
Single molecule fluorescence experiments are most commonly performed by sample-scanning or beam-scanning confocal microscopy or by wide-field imaging methods.41–46 To detect single molecules, the molecules must be spatially isolated from each other, although early work relied on spectral isolation.76,77 To achieve spatial isolation, the samples to be investigated are doped to very low dye concentrations (i.e., 0.01–1 nM). An ultra small detection volume (~1 fL) is then attained by using a high numerical aperture microscope objective for illumination of the sample and collection of the resulting fluorescence. The combination of low dye concentration and small detection volume makes it possible to detect the fluorescence from one molecule at a time. The use of thin silica films also facilitates single molecule detection by further restricting the volume probed.78 Out-of-focus background due to light scattering by the sample or emission from molecules at different sample depths is virtually eliminated with thin films. Background reduction in this case can easily exceed that afforded by confocal detection methods for thicker samples.44In a typical single molecule experiment, the individual molecules are first located by recording a fluorescence image of a portion (a few hundred square micrometers) of the sample. The single molecules frequently appear in such images as bright round fluorescent spots of diffraction-limited size and comprising Gaussian intensity profiles. In sample-scanning experiments, these spots (single molecules) are subsequently positioned one at a time in the detection volume of the microscope and their time-resolved and/or spectrally-resolved emission characteristics are recorded. In wide-field experiments, repeated images of the same area are recorded to follow the motions of the individual molecules in time and space. Both confocal and wide-field experiments can also be performed with spectral and polarization selectivity to monitor energy transfer between molecules (i.e. as in fluorescence resonance energy transfer experiments)79 and orientational motions of the individual molecules, respectively.70,80–82
Single molecule experiments require selection of appropriate dye molecules. The dye molecules to be employed must have large excitation cross-sections (~3 × 10−16 cm2 or larger) and high fluorescence quantum yields (~20% or larger). They must also be relatively photostable (i.e. capable of surviving millions of excitation/emission cycles). Finally, to serve as probes of materials properties, they must exhibit some sensitivity to the property of interest (i.e. pH, polarity, ionic interactions, pore size, etc.). Fig. 1 depicts the chemical structures of representative dyes that have been used in single molecule studies of sol–gel-derived silica materials to date.
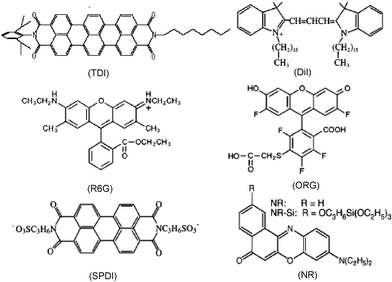 | ||
| Fig. 1 Chemical structures of dye molecules commonly used to probe silica materials. Shown are N-(2,6-diisopropylphenyl)-N′-(n-octyl)terrylene-3,4,11,12-tetracarboxylic diimide (TDI), 1,1′-dihexadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), rhodamine 6G (R6G), Oregon Green (ORG), N,N′-bis(3-sulfonatopropyl)perylene-3,4,9,10-tetracarboxylic diimide (SPDI), Nile Red (NR) and a silanized Nile Red (NR–Si) derivative. | ||
3.2 Noncovalent molecule–matrix interactions
Interactions between dopant molecules and the silica matrix are of relevance to a number of potential applications, including those in chemical sensing, separations and catalysis.1–8,83 Such interactions impede the diffusion of molecules through the matrix, slowing reaction rates and lengthening sensor response times. Generally, there are four possible types of noncovalent interactions that can occur between dopants and the silica matrix. These include ionic interactions, hydrogen bonding, hydrophobic interactions, and physical confinement (i.e. steric effects).17,38 Ionic interactions readily occur between cationic species and anionic sites (i.e., deprotonated silanols, Si–O−) on the silica surface, but they may also occur between dopants and other charged sites in ORMOSILs and surfactant-templated materials. Hydrogen bonding interactions can also occur between dopants and surface silanol (Si–OH) groups and similar functional groups in ORMOSILs.34,38 Hydrophobic interactions occur between nonpolar surface sites and nonpolar functional groups on dopant molecules and are of greatest importance in ORMOSILs and surfactant-containing mesoporous systems. Physical confinement becomes important when the pore size in a particular sample is similar to the size of the probe molecule. All of these forms of noncovalent interactions may lead to irreversible or periodic molecular adsorption or entrapment events or to partitioning of dopants into different sample regions.A number of bulk spectroscopic methods have been employed to better understand molecule/matrix interactions in sol–gel materials. Included are electrochemical methods,36FTIR,35fluorescence spectroscopy17,38,39 and anisotropy,84–87 and NMR.88–91 In many such studies, molecule/matrix interactions have been invoked to explain certain observations. For example, in electrochemical studies by the Collinson group,36,92–94 they noted that the apparent diffusion coefficients of redox molecules entrapped in silica or ORMOSIL monoliths was strongly dependent on the charge and the presence of hydrogen-bonding groups on the probe molecule. They postulated that specific molecule/matrix interactions influenced molecular diffusion. However, such interactions could not be directly observed from the electrochemistry alone. Single molecule methods provide the ability to directly observe such interactions and distinguish their contributions from those of slow diffusion due to increased local viscosity. Importantly, single molecule studies are also well suited to characterization of thin films, which are often technologically more useful, especially in sensing applications.
Fig. 2 displays the mobility distributions they obtained. The data depict a 7-fold increase in the population of freely tumbling ORG molecules (14%), relative to R6G (2%). The fewer tumbling R6G molecules were attributed to ionic interactions between the cationic dye and the anionic silica surface. The increased tumbling of anionic ORG was ascribed to repulsive interactions with the matrix. However, the relatively small populations of tumbling ORG and R6G molecules, and the relatively minor changes in mobility brought about by exposure of the matrix to water and pH 7 buffer, were taken as evidence that physical entrapment and hydrogen bonding also play major roles in governing mobility in this system.
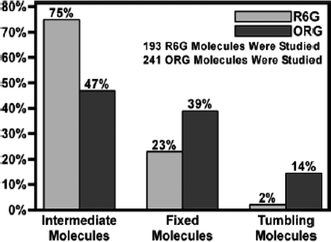 | ||
| Fig. 2 Mobility distributions of R6G and ORG encapsulated in dry sol–gel-derived silica films. (Reprinted with permission from ref. 80. Copyright 2004, American Chemical Society.) | ||
Ionic interactions with the silica framework and entrapped surfactants have also been explored by monitoring the translational diffusion of single molecules. The Higgins and Collinson groups have recently used single molecule methods to probe such interactions in surfactant (cetyltrimethylammonium bromide, CTAB)-containing mesoporous films.68,69 In one such study, cationic, anionic and neutral dyes were employed to probe molecular mobility under both dry and hydrated film conditions.69Dye diffusion was initially explored by fluorescence imaging in a sample-scanning confocal microscope. The images obtained provided evidence of the relative strengths of molecule–matrix interactions in each case. Those that interacted strongly with the matrix yielded fixed, round fluorescent spots having Gaussian intensity profiles, while mobile molecules produced fluorescent streaks in the images.
Individual sample regions were subsequently positioned within the focal volume of the microscope and single-point fluorescence time transients recorded. Observation of constant fluorescence signals, followed by clear bleaching events were attributed to entrapment of molecules at fixed sites, due to strong molecule–matrix interactions.69 In contrast, molecules that exhibited a high degree of mobility produced transients comprised of repeated fluorescence bursts, with little or no evidence of bleaching. However, even mobile molecules produced evidence of reversible molecule–matrix interactions via the occasional appearance of events in which the signal remained constant (and above background) for time periods too long to be explained by diffusion.48,50 Such events were ascribed to reversible molecular adsorption, and are typically masked in bulk studies of molecular mobility.50 More recently, Mason et al., have demonstrated temperature-dependent single molecule methods for measuring the enthalpy of these interactions.96
In the studies of CTAB-containing materials, an uncharged dye, Nile Red (NR, Fig. 1), was observed to be mobile under all conditions, while an anionic dye, a sulfonated perylene diimide (SPDI, Fig. 1), was found to be immobile.69 The latter observation was attributed to strong ionic interactions between the dye and cationic surfactant. Evidence for strong interactions between the cationic dye 1,1′-dihexadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, Fig. 1) and an anionic surfactant, sodium dodecyl sulfate (SDS), was also obtained. In contrast, SPDI and DiI were found to interact less strongly with SDS and CTAB, respectively. These results clearly demonstrate the importance of ionic molecule–matrix interactions in these films.
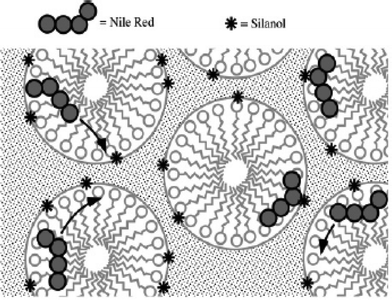 | ||
| Fig. 3 Model for diffusion and adsorption of Nile Red within CTAB-containing films. The dye moves relatively freely through the surfactant-filled and/or hydrated pores, periodically encountering surface silanols (stars) on the silica pore surfaces to which it can hydrogen bond, leading to reversible adsorption events.68 | ||
Some of the best demonstrations of the role played by physical confinement in governing molecular mobility come from work by the Bräuchle group.70,98 In a recent report,70 these researchers clearly showed that terrylenediimide (TDI, Fig. 1) molecules found within CTAB-templated mesoporous materials assumed a well-defined orientation, with their long-axes aligned parallel to the long axis of the mesopores. Importantly, alignment was maintained in both dry materials, where the molecules appeared completely immobile, and solvent-loaded films, in which the molecules diffused primarily along one dimension (through the pores), while remaining in the aligned state. The dimensions of TDI were noted to be 1.1 nm × 2.5 nm and the pores 2–3 nm in diameter. It was also noted that the surfactant found within the pores may further restrict molecular motions, leading to the dramatic orientational confinement effects observed.
3.3 Molecular mobility
Bulk measurements of the translational and rotational mobility of molecules doped into sol–gel materials have been made by a variety of techniques, including electrochemical methods,92–94,99fluorescence anisotropy84–87 and NMR.88,90 The Collinson group, for example, has used electrochemical methods to measure the apparent diffusion coefficients, D, of redox probes doped into sol–gel monoliths during the gelation, aging, and drying processes.92,93,100 The molecules employed were of similar sizes but carried different charges. The magnitude of D in each case and the rate at which it changed during drying were clearly dependent on the charge of the redox probe and its size, relative to the silica pore size.9,293,100 However, details such as the heterogeneity in D and the fraction of dopants interacting with the matrix could not be determined.Electrochemical methods have also been used to study the behavior of redox molecules trapped in thin silica films.101,102 However, it is much harder to measure D in thin films using electrochemistry because the redox current obtained depends on both D and the concentration of electroactive species. The research groups of DeArmond and Collinson showed that only a small fraction of the total concentration of a given redox probe is actually electroactive.101,102 Hence, only rough estimates of D could be obtained. No specifics regarding reversible surface adsorption or physical confinement could be extracted from the data. Since single molecule methods frequently provide the means to distinguish between diffusion phenomena and molecule/matrix interactions (see above), they allow for more accurate measurements of diffusion coefficients, determination of relative populations of mobile and immobile species, and assessment of adsorption/desorption dynamics.
As applied here, FCS involves the recording of single-point fluorescence time transients using a confocal microscope. Such transients usually incorporate signal fluctuations that may have a number of origins. In cases where these fluctuations are caused by diffusion of single molecules into and out of the microscope detection volume, the results can be used to determine the molecular diffusion coefficient. For this purpose, the time transients obtained are first autocorrelated to find the distribution of time scales over which the signal fluctuates.103
The properly normalized autocorrelation function, C(τ), is calculated from time transient data as follows:
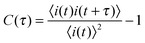 | (1) |
To determine the relevant diffusion coefficient(s) and to distinguish between signal fluctuations involving molecular diffusion and adsorption/confinement, the autocorrelated data are subsequently fit to an appropriate model decay function. The choice of the model is made based upon careful inspection of the time transient data. The appearance of fluorescence bursts exhibiting constant fluorescence (aside from shot noise) for time periods too long to be explained by rare diffusive events are usually attributed to the reversible (or irreversible) adsorption of the dye to the sample surface. In cases where such events are not apparent, it is usually concluded that observed variations in burst lengths arise from multicomponent diffusion, due primarily to sample heterogeneity. Single point time transients can be easily recorded with 50–100 μs time resolution, or they can be recorded on a single photon basis and subsequently analyzed with or without binning.110,111 Such high time resolution allows for diffusion coefficients to be measured even in bulk solution.
A variety of models have been used to explain results obtained from silica materials. For example, Dai and coworkers75 used a two component diffusional model to fit their autocorrelation data in single molecule studies of R6G diffusion in a commercial mesoporous glass. Similarly, Martin et al. utilized this model to fit data obtained from NR molecules diffusing through ORMOSIL films.112 In contrast, Fu and coworkers68 and Ye, et al.69 employed a model that includes both two-dimensional diffusion and reversible surface adsorption to fit data obtained from surfactant-containing and calcined mesoporous films.48–50 The mathematical expression of this model is as follows:
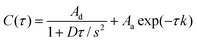 | (2) |
Single molecule tracking provides a more direct means for following single molecule diffusion.105 In SMT, the sample is subjected to wide-field illumination by a laser source. The illuminated field of view is subsequently collected and imaged onto a charge-coupled device (CCD) detector, where a “movie” depicting sample emission in time is recorded. The single molecules in each image appear as diffraction-limited fluorescent spots that may move from frame to frame.105 The fluorescent spots observed are fit to two-dimensional Gaussian profiles to precisely determine the molecular position in each frame. Under high signal-to-noise conditions, the molecular position can readily be determined to better than 10 nm.108 The motion of the individual molecules is then reconstructed by determining the relative positions of each molecule in many successive frames.
SMT methods have the distinct advantage that the actual path (or trajectory) taken by a molecule can be recorded and long-term adsorption or entrapment events directly visualized. While SMT is a very powerful method in these respects, the results obtained are sometimes limited by the finite trajectory lengths that can be recorded, due to photobleaching. With currently available detectors, SMT studies are also limited to relatively slow diffusion, since data cannot be recorded with the same time resolution as in single point time transients.
A common method to determine diffusion coefficients from SMT data is to plot the mean square displacement (MSD) of each molecule vs. time. According to the Einstein equation for single-component two-dimensional (2D) diffusion, this plot should exhibit a linear dependence on time:
| MSD = 〈r2(t)〉 = 4Dt | (3) |
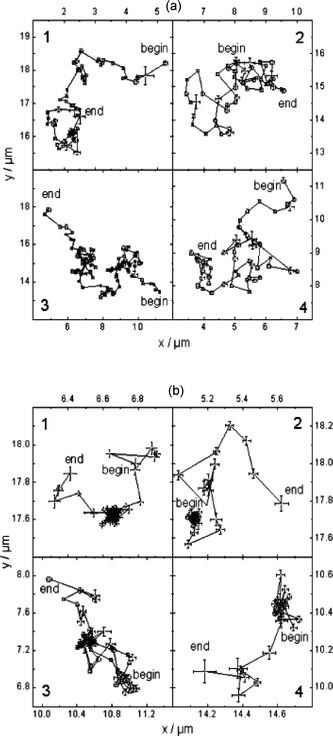 | ||
| Fig. 4 Four representative single molecule transients obtained from M22 (a) and M3 (b) samples. The data are reflective of Brownian motion in M22 and free diffusion coupled with trapping (dense clouds of data points) in M3. (Reprinted with permission from ref. 98. Copyright 2004, American Chemical Society.) | ||
Single molecule diffusion in surfactant-templated mesoporous materials has been studied by the Higgins and Collinson groups68,69 and by the Bräuchle group.70,82 The former employed single point fluorescence time transients and FCS analysis to measure molecular diffusion coefficients. Both CTAB-containing and calcined thin films (with 770 nm and 400 nm thicknesses, respectively) were studied. Molecular mobility in these films was investigated under both dry and hydrated (20–50% relative humidity, RH) conditions. Three different dye molecules (NR, DiI and SPDI, Fig. 1) were employed to probe the effects of molecule–matrix interactions, as discussed above.
In the CTAB-containing films, anionic SPDI molecules were found to adsorb at fixed locations under all conditions, due to strong interactions with the cationic surfactant (see above). In stark contrast, neutral NR molecules exhibited facile diffusion under all conditions (20–50% RH), yielding an average D of 2.4 × 10−10 cm2 s−1 in dry (20% RH) films and a slightly larger D of 2.7 × 10−10 cm2 s−1 in hydrated (50% RH) films. NR mobility was attributed to its partitioning into the hydrophobic inner regions of the surfactant micelles. Among the three dyes employed, DiI was found to be most sensitive to changes in film hydration, with average D values of 0.9 × 10−10, 1.3 ×10−10, and 3.0 ×10−10 cm2 s−1 obtained in films exposed to 30%, 40%, and 50% RH environments, respectively, as shown in the histograms in Fig. 5.
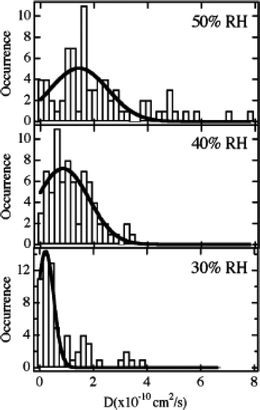 | ||
| Fig. 5 Histograms showing the distribution of diffusion coefficients measured for DiI-doped CTAB-containing mesoporous silica films at the relative humidities (RH) shown. The solid lines depict the Gaussian fits to each histogram. (Reprinted with permission from ref. 69. Copyright 2007, American Chemical Society.) | ||
Single molecule diffusion studies also provide information on the relative importance of diffusion and reversible surface adsorption or physical entrapment in a given system. In the studies by the Higgins and Collinson groups68,69 information on the relative contributions of adsorption and diffusive events to observed signal fluctuations was obtained from the relative amplitudes of these two components in fits of the autocorrelation decays to eqn (2). Fitting of these data also provides information on the amount of time the molecules spend adsorbed to the surface. In these studies, signal fluctuations from diffusing NR molecules were found to comprise only 43% of the autocorrelation decay amplitude under dry (20% RH) conditions and 58% in hydrated films (50% RH). Similarly, NR molecules were found to spend an average of 25 s in an adsorbed state in dry materials and about half as long under hydrated conditions. These observations indicate that water facilitates NR diffusion and desorption from the silica surface.
In calcined films, all three dyes were found to be immobile under dry conditions. Immobility in these samples was attributed to strong dye–matrix interactions that occur in the absence of solvent (water). In contrast, all three dyes were found to be mobile in films probed at 50% RH. The average diffusion coefficients were measured to be 4.0 × 10−10, 3.1 × 10−10, and 2.9 × 10−10 cm2 s−1, for SPDI, DiI, and NR, respectively. These results show SPDI is the most mobile of the three, likely because of repulsive interactions between the anionic dye and anionic silica surface, which may cause its exclusion from the smallest silica pores.
Dai and coworkers75 used FCS to study three-dimensional diffusion of R6G in mesoporous glass with a pore size of 13 nm and negligible micropores. Three models describing single component diffusion, two component diffusion and diffusion coupled with adsorption were tested in fitting the autocorrelation data. Fig. 6 shows a representative example, depicting the correlation curve obtained at a distance of 10 μm into the mesoporous glass. The data are shown fit to the three different models. The single component model (Fig. 6a) failed to fit the data properly, while the two-component models (Fig. 6b,c) proved much better. Assuming two-component diffusion, the results obtained indicated that 38% of the molecules moved with D1 = 6.81 × 10−7 cm2 s−1 and 62% of the time they exhibited D2 = 4.84 × 10−8 cm2 s−1. It was concluded that the slower component may arise from multiple contacts between the molecules and the pore walls. Fitting with the diffusion/adsorption two-component model indicated that 80% of the molecules moved freely through the materials with D = 4.89 × 10−7 cm2 s−1, while the remainder reversibly adsorbed to the silica surface, exhibiting a mean desorption time of 67 ms. While both two-component models fit the data, it was concluded the latter was more plausible, due to the possibility for ionic interactions between positively charged R6G and the negatively charged silica surface.75
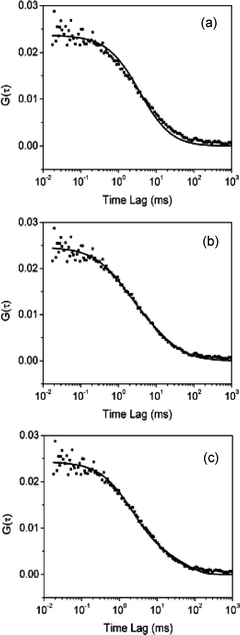 | ||
| Fig. 6 Autocorrelation data obtained for R6G diffusion in mesoporous glass fitted with models for (a) single component diffusion, (b) two component diffusion and (c) combined diffusion and adsorption. (Reprinted with permission from ref. 75. Copyright 2003, American Chemical Society.) | ||
Some of the most dramatic results on single molecule diffusion in mesoporous silica have come from recent SMT studies by the Bräuchle group.70,82,98,108,113,114 In one of their studies,70 this group investigated the mobility of TDI molecules in CTAB-containing silica held under dry air and under a saturated chloroform atmosphere. The TDI molecules were found to be immobile under dry conditions and mobile when exposed to chloroform vapor. Most interestingly, the trajectories obtained conclusively demonstrated that the molecules migrated along one dimension (1D) within the mesopores (i.e., along the cylindrical channels). The 1D trajectories spanned distances of several micrometers, and the molecular long axis of each dye molecule was found to orient parallel to the channel axis even while the molecules were moving. As noted above, the physical shape of TDI (1.1 nm × 2.5 nm) and steric interactions with the matrix likely prevented the molecules from freely rotating in the channels.
Fig. 7 depicts some of the results from these studies. Best fits to these data were obtained using a function that accounted for both 1D diffusion and adsorption. Furthermore, as shown in Fig. 7b, their MSD results were not perfectly linear in time, as would be expected for a random walk, but rather tended towards smaller values at longer times. The latter observation is good evidence for confined diffusion and was attributed to the presence of “dead ends” in the channels of these materials. The data shown in Fig. 7c were used to arrive at an average D value of 3.9 × 10−12 cm2 s−1, while that in Fig. 7d demonstrated that the average molecule spent 18% of its time adsorbed to the silica surface. These plots also clearly depict the distribution in D and hence, the level of materials heterogeneity.
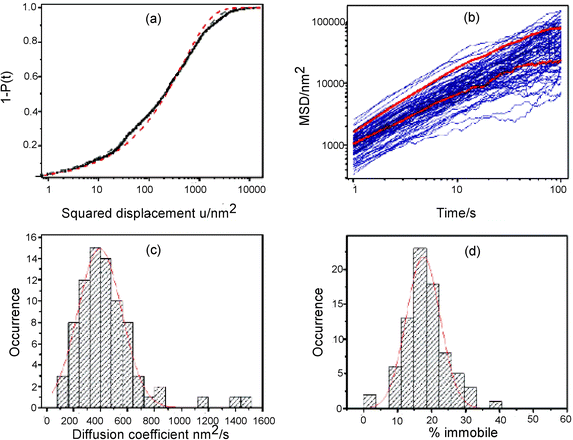 | ||
| Fig. 7 (a) Cumulative probability distribution for a 1 s image frame time from a trajectory shown in the original article.70 (b) Mean squared displacement (MSD) for linear diffusion as a function of time. (c) Histogram of the diffusion coefficients obtained from the MSD plots. The solid line shows a Gaussian fit to the distribution yielding a mean diffusion coefficient of 390 nm2 s−1. (d) Histogram of the percentage of time each molecule spends in an adsorbed state. The solid line shows a Gaussian fit to the distribution, yielding a mean of 18%. (Reprinted with permission from ref. 70. Copyright 2008, American Chemical Society.) | ||
In another study by the Bräuchle group,82TDI diffusion in hexagonal, lamellar and mixed hexagonal/lamellar templated mesoporous films was investigated. These materials were prepared by controlling the amount of a longer-chain surfactant (Brij 56) used in sol preparation. In pure lamellar films, doughnut-shaped images of TDI emission in SMT images indicated that the molecules again assumed a constant orientation while moving laterally in two dimensions through the lamellar phase. In contrast, the results obtained from pure hexagonal films showed that the TDI molecules were rotating rapidly as they moved in down the mesoporous channels. In mixed films, both doughnut-shape and Gaussian fluorescence patterns appeared. The results obtained also showed that the lamellar and hexagonal channels were interconnected, as evidenced by time dependent changes in the fluorescence patterns obtained from each molecule and from alternation between 2D and 1D motions during single tracking periods. Other studies by this same group114 employed both transmission electron microscopy (TEM)115 and SMT to prove the molecules were moving along the mesopore channels. Fig. 8 depicts single molecule diffusion trajectories overlayed on a TEM image of the same sample region. It was concluded that there were five structural elements in two-dimensional hexagonal mesoporous films: straight segments, curved segments, domain boundaries, “disordered” regions and connective channels.
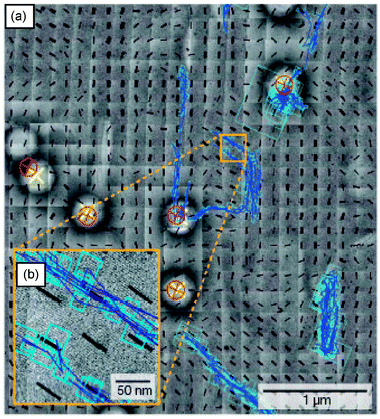 | ||
| Fig. 8 (a) Overlapped single molecule trajectories on TEM images. (b) The inset more clearly shows that the trajectories run along the channels. The black lines show the orientation of the pores from TEM data. The red and yellow crosses depict the positions of the same polystyrene bead in the wide field fluorescence and TEM images, respectively. The blue lines show the single molecule trajectories. (Reprinted with permission from ref. 114. Copyright 2007 Nature Publishing Group.) | ||
For example, Harris and coworkers117 have used total internal reflection fluorescence methods (TIR-FCS) to probe the diffusion of dye-labeled poly(amidoamine) (PAMAM) dendrimers in silica films prepared by the Stöber process.118 PAMAM dendrimers were selected as probes due to their monodispersity and spherical structure, ensuring that dispersion observed in the data arose primarily from silica heterogeneity. The dependence of molecular diffusion on the size of the dendrimers (three generations were employed) in relation to the size of the pores was studied. The studies revealed that the large dendrimers were excluded from pores formed between small (27 nm) particles, suggestive of a low number of pore defects. Smaller dendrimers were found to explore a wider range of pore structures, experiencing a greater level of pore tortuosity and reflecting the level of heterogeneity in pore size/shape in these materials.
Martin-Brown, et al.112 have also used single molecule methods (primarily FCS) to study dye diffusion within ORMOSIL films. Commercial NR and a silanized NR derivative (NR-Si, Fig. 1)112 were used as probes in this study. ORMOSIL films were prepared from acid-catalyzed sols containing varying mole ratios of TEOS and isobutyltrimethoxysilane (BTMOS). Images and single point fluorescence time transients with FCS analysis provided evidence of molecule mobility. Fig. 9 shows representative data from 33% and 90% BTMOS content films. In 33% BTMOS films, discrete, permanent photobleaching events were observed, consistent with immobile molecules. In contrast, transients recorded in 90% BTMOS films provided clear proof of translational diffusion for bothdyes. This result was most surprising for the case of NR-Si, which was expected to covalently attach to the silica matrix.
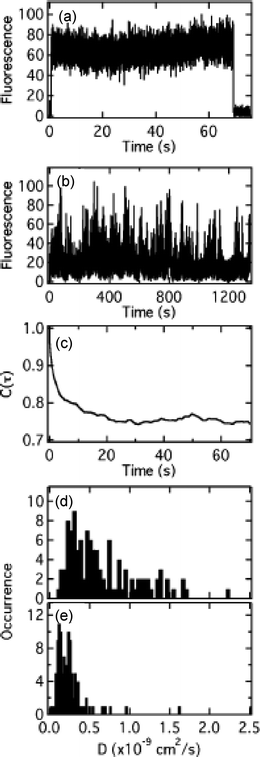 | ||
| Fig. 9 (A) Single point fluorescence time transient obtained from a 33% BTMOS film doped with NR. (B) Fluorescence transient recorded in a NR–Si-doped 90% BTMOS film. (C) Autocorrelation of the transient shown in B. (D and E) Histograms of the diffusion coefficients measured from ~100 sites in 90% BTMOS films doped with NR and NR–Si, respectively.112 | ||
FCS analysis procedures were applied to the time transients and the resulting autocorrelations fit to a 2D, two-component diffusion model.112 Both molecules yielded fast and slow diffusion components. The fast components for NR and NR-Si yielded, on average, D = 1.2 × 10−9 cm2 s−1 and 2.9 × 10−10 cm2 s−1, respectively, with both slow components 100-fold smaller. The ~4-fold difference in the diffusion coefficients for NR and NR-Si indicated that the NR–Si molecules were indeed bound to a component of the silica matrix, but this component was comprised of liquid-like silica oligomers. It was concluded these were organically-modified silica oligomers that had phase separated from the inorganic components of the film. Imaging results119 showed these components could be spatially resolved under certain circumstances.
3.4 Polarity properties
An in-depth understanding of the polarity properties of silica materials is required for many of their applications.38 The polarity of the matrix plays a role in governing partitioning of molecules into and out of silica materials. It can also impact chemical reactions and charge transfer processes occurring within the matrix. Bulk fluorescence and absorption spectroscopies have long been used to probe the average polarity of silica materials,38–40 with perhaps the most significant scientific questions arising in ORMOSILs.38,40,116 For example, Brennan, et al.120 have used 7-azaindole (7AI) and 6-propionyl-2-dimethylaminonaphthalene (PRODAN), both solvatochromic dyes, to examine ORMOSILs prepared from TEOS and a minority fraction of methyltriethoxysilane (MTES), propyltrimethoxysilane (PTMOS) or dimethyldimethoxysilane (DMDMS). The results showed that internal materials polarity was dominated by the solvent composition from immediately after gelation until ~5–7 days later for MTES and DMDMS containing films. Afterwards, a point was reached in some of the materials where the polarity properties of the organic component began to take over. Both blue shifts in the emission maxima and broadening of the spectra were observed. The latter was attributed to formation of at least two different classes of environments having distinct polarity properties, while the former could reflect partitioning of the dyes into regions of high organic content.While the spectral linewidths obtained from bulk studies are reflective of sample heterogeneity, they only provide an average view of materials properties and are difficult to quantitatively interpret in terms of the actual distribution of environments present. Single molecule methods provide a means to investigate the polarity properties of individual nanometer-scale environments, thus allowing for the entire inhomogeneous distribution probed by a given dye to be mapped. Such methods also provide the means to characterize sample heterogeneity through a number of different statistical parameters: the mean, median, and most common environments can be determined, as can the width and skewness (if present) of the distribution. Finally, the populations and properties of rare environments can also be assessed.
The Higgins group has recently described single molecule methods that are useful for characterizing the polarity properties of organic polymers.121 These same methods were also applied to ORMOSIL thin films.97,119 In these studies, NR was loaded into the ORMOSILs at nanomolar concentrations and used to sense materials polarity. NR was selected because it is one of the most solvent-sensitive fluorescent dyes known.122 Its solvent sensitivity arises from the large change in dipole moment associated with its lowest-energy electronic transition, which has been classified as an intramolecular charge-transfer transition.123,124
In these single molecule studies, fluorescence spectra were acquired and analyzed using a modified form of Marcus theory for charge-transfer transitions.125–127 Of the range of available polarity models,128,129 the charge-transfer model was selected because it allows for the most information to be derived from single-molecule data. Specifically, information on both the static and dynamic polarity properties of the materials can be obtained. These properties are reflected in the environment-dependent shift in the transition energy, ΔΔG°, and the reorganization energy, λ, respectively, of the matrix (and solvent) dipoles surrounding each molecule.121 Polar environments yield larger ΔΔG° values, while nonpolar regions give smaller values. Likewise, more dynamic environments yield relatively larger λ values and rigid environments smaller values. The approximations and assumptions involved in this analysis are described in the original manuscripts.97,119,121
The ORMOSIL films studied97,119 were derived from binary sol mixtures containing either 3-(triethoxysilyl)propionitrile (CNS) and TEOS or BTMOS and TEOS. Sols having different compositions (defined by the mole percent of organic precursor in the sol) were prepared, doped with NR, and spin cast as thin films on glass substrates. Fig. 10 shows histograms of the static (ΔΔG°) and dynamic (λ) polarity properties obtained as a function of organic precursor content.119
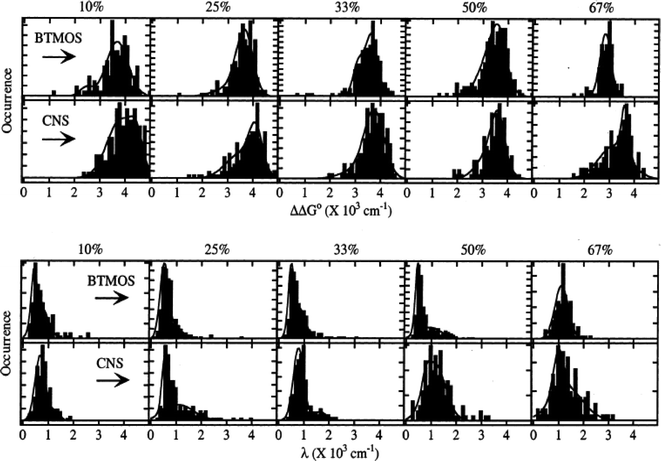 | ||
| Fig. 10 Histograms of ΔΔG° and λ polarity parameters measured for ORMOSIL films derived from BTMOS/TEOS and CNS/ TEOS composite sols. The mole percent organic precursor (BTMOS or CNS) used in each sol is given at the top of each column. (Reprinted with permission from ref. 119. Copyright 2002, American Chemical Society.) | ||
Most of the distributions depicted could be classified as bimodal, multimodal or simply non-Gaussian, providing clear evidence for nonrandom variations in the film properties. The presence of the associated classes of environments was attributed in part to hydrogen bonding of some NR molecules to the matrix. However, the results were more generally attributable to (nonequilibrium) phase separation of organic- and inorganic-rich film domains, as had been previously proposed from bulk studies.120,130,131 Phase separation could have resulted from differences in the hydrolysis and condensation rates of the precursors132 or from the mutual immiscibility of the precursors. They could also reflect variations in the properties of any solvent confined within the matrix.133 As with all dye-based polarity studies, preferential partitioning of the dye into certain environments could have biased the observed distributions, as addressed by Bardo, et al.97 Nevertheless, these distributions prove that in some cases the “average” environments reflected in bulk spectroscopic results may not adequately reflect the actual materials properties.
The inhomogeneous distributions observed also allow for more detailed classification of the sample environments. For example, from the results shown in Fig. 10, it can be concluded that the most common environments probed were significantly more polar and less dynamic than the average environments. This observation indicates that the aforementioned phase separation may occur only in rare circumstances, leading to the observed tails to lower and higher values of ΔΔG° and λ, respectively. It is also of interest that the static and dynamic polarity properties for BTMOS-containing films change abruptly at about 50% BTMOS, while the properties of CNS-containing samples change gradually. The incorporation of liquid-like oligomers in films of high BTMOS content is the likely cause,112 suggesting the properties of the BTMOS films may be closer to their equilibrium values than in films of lower organic content, where the properties may be kinetically controlled.
3.5 Matrix acidity
The hydrogen bonding and ionic molecule–matrix interactions discussed above arise from the presence of protonated and deprotonated silanol species in the silica materials.134 These silanols are weakly acidic and the extent to which they are protonated or deprotonated depends upon their individual pKas and on the pH of the environment in which each is found. The pKa of silanol groups on silica surfaces is known to vary between ~4.5 and 9.134,135Silanol pKas in silica films will vary with the density of silanol groups present and the organic content of the environment (e.g. in ORMOSILs or due to solvent).The acidity properties of the silanols found in/on sol–gel-derived materials have been studied by several bulk methods, including temperature-programmed desorption (TPD),58,136FTIR,137,138 Raman34 and NMR spectroscopies.58,136 Though these studies have provided valuable information on the average acidity of the relevant surface sites, they provide little information on the level of heterogeneity present. Sample heterogeneity is known to be important in several applications of sol–gel-derived materials, including in the response of silica-based pH sensors. Representative bulk work in this area is taken from the Saavedra group, in which sol–gel methods were used to deposit thin silica films as waveguiding and indicator layers in pH sensing devices.139 In this study, it was found that the pH indicator dye responded over an unexpectedly broad range of pH values. It was concluded that the sensing dye was entrapped in “chemically inequivalent microenvironments” that caused the dye to respond differently in each.139 In the same study, hysteresis in the sensor data was attributed to the presence of indicator molecules that responded slowly to pH changes.
Single molecule methods have the potential to provide valuable new insight into the origins of such observations. As with all single molecule studies, the initial selection of an appropriate pH-sensitive fluorescent dye is required. The dye selected should be highly fluorescent and exhibit clear pH-dependent changes in its emission and/or excitation spectra, but little or no change in emission yield. It should also have a pKa similar to that of the species to be probed. C.SNARF-1, which has a pKa of 7.5, is a nearly ideal probe for studying the effects of silanol site properties and was employed by Fu, et al. in recent studies of silica film acidity.140 A slightly different form of this dye was used in earlier single molecule studies of agarose gels.141 The protonated form of C.SNARF-1 emits in a band centered at 580nm, while the deprotonated form emits near 640 nm. The ratio of single molecule emission in these two bands (R = I580/I640) can be used as a probe of the pH of the local environment surrounding each molecule.
In the studies by Fu, et al., the local acidity properties of films prepared via the acid-catalyzed hydrolysis and condensation of TEOS were investigated.140 It was concluded the local environments in “as-prepared” films had an average pH of ~4.8. The range of R values obtained was consistent with a 1–2 pH unit variation in the local acidity. Variations in the concentration of residual acid catalyst and in the local silanol pKas were the likely cause of these pH variations.
The extent to which local film acidity could be altered/controlled by exposure to external solutions of known pH was also explored. In these studies, the silica films were treated by immersion in various pH solutions for different lengths of time (1 h and 8 h). Histograms of the R values obtained are shown in Fig. 11. Each distribution appears to be Gaussian in shape and monomodal, as evidenced by the fits appended to each data set. This observation suggests that variations in the acidity properties are random. Hence, the mean acidity properties could have been accurately determined by bulk spectroscopic methods. However, such a conclusion could not have been drawn without first recording these data.
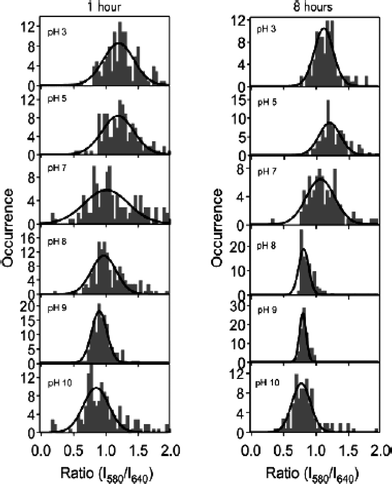 | ||
| Fig. 11 Histograms of the emission intensity ratios (R = I580/I640) from C.SNARF-1 for silica films treated at the specified pH for 1 h (left) and 8 h (right), respectively. (Reprinted with permission from ref. 140. Copyright 2004, American Chemical Society.) | ||
The mean acidity values obtained by fitting the R distributions provide an initial view of film acidity properties. These data are plotted vs. treatment pH and fit to a Henderson–Hasselbalch expression as a means to characterize the apparent pH dependent response in each series of experiments.142 As depicted in Fig. 11, the R values showed the same qualitative trend to smaller values with increasing treatment pH, as observed in bulk solution phase experiments. However, the trend was much more gradual in the films treated for only 1 h, and was most similar to bulk solution results for films treated for 8 h, exhibiting a sharp transition around the pKa of the dye. Importantly, the R values obtained at high pH in the 1 h treated films were significantly larger than in the 8 h treated samples, indicating that some molecules remained protonated even after treatment in high-pH solutions for 1 h. This observation indicated that some environments were inaccessible on this time scale, a conclusion that is consistent with those from bulk studies,143,144 and which is also reflective of sample heterogeneity.
The widths of the R distributions provide the best view of heterogeneity in terms of film acidity properties. The results obtained by Fu and coworkers140 (see Fig. 11) demonstrated that film heterogeneity was strongly dependent on treatment time, as reflected in the uniformly narrower distributions obtained from films treated for 8 h, relative to those obtained after only 1 h. These differences were again attributed to kinetic limits to changes in the local pH, due to entrapment of some molecules in tightly constrained pores within the film.
The distribution widths also provide a means for probing the influence of the surface silanols on the local pH. As shown in Fig. 11, the distributions obtained after treatment at pH 8 and 9 were the most narrow (near the noise-limited width), whereas those obtained after treatment at pH 7 were broadest. At pH 7, the average pH is close to the pKa of the dye, where it is most sensitive to changes in local acidity.141 The distribution widths at pH < 7 and pH > 9 were therefore concluded to reflect significant heterogeneity in the film acidity properties, likely resulting from the presence of residual acid and silanol pKa variations. In contrast, the much narrower distributions observed at pH 8 and 9 indicated the local environments were very homogeneous in this pH range. Enhanced materials homogeneity was attributed to buffering of the local pH by the majority surface silanols, which likely have pKa values of 8.5–9.134,135
4. Single nanoparticle studies
A significant limitation of the single molecule experiments described above is the short lifetime of the dyes employed. Under ambient conditions, such dye molecules usually photobleach after only a few seconds of continuous illumination,145 limiting the number of spectroscopic measurements that can be made, and the precision of the results obtained. One possible route to improved measurements would be to instead employ strongly luminescent or scattering metal33,146–149 or inorganic nanoparticles,150 or luminescent organic polymer nanoparticles.151Nanoparticles that are sensitive to and report on their chemical environment are now being developed.152Gold nanoparticles have been employed previously as probes of sol–gel-derived silica materials. For example, Akbarian, et al. have demonstrated the photochemical production and optimization of gold nanoparticles in a silica matrix for surface enhanced Raman scattering (SERS) experiments.33 They used their silica-based SERS substrates to detect pyrazine molecules diffusing into the matrix. Such measurements could also readily be performed at the single particle level146,147,153 to obtain information on materials heterogeneity.
The intrinsic light scattering properties of silica nanoparticles can also be used to probe matrix properties. For example, in bulk studies, Nakanishi et al. have employed scattering methods to follow the sol–gel process in pore-forming materials154 prepared by polymerization-induced phase separation methods.56,155,156 The macroporous sol–gel-derived monoliths obtained are an intriguing class of materials that have attracted considerable attention owing to their unique structural features and potential applications in catalysis, separations, superhydrophobic materials, drug delivery, chemical sensors, and optics.155–158
Dong, et al.159 have recently used in situ microscopic methods and confocal correlation spectroscopy (CCS)160 to follow the formation of thin macroporous monoliths of MTMOS-derived materials from before phase separation to well after gelation at the single-pore/single-nanoparticle level. The acid catalyzed sol–gel process employed led to formation of silica nanoparticles very early after sol preparation. These nanoparticles were found to efficiently scatter light in a confocal microscope, providing an intrinsic probe of the sol–gel process and the growth and aggregation of the individual silica nanoparticles. Confocal images of the evolving materials allowed for direct observation of phase separation in the sol, yielding two distinct phases, one high in silica polymer/nanoparticle concentration (i.e., regions exhibiting relatively strong light scattering, the matrix) and one of low silica content (regions exhibiting relatively weak scattering, the pores). The time evolution in the size, shape, and position of these domains was followed by optical microscopy, through gelation.
Single point time transients recorded in various regions of interest (i.e. within individual pores and in the matrix) were recorded and used to follow the silica nanoparticle growth and aggregation processes. Autocorrelation data obtained were fit to a two-component diffusion model and used to determine the nanoparticle diffusion coefficients. Prior to phase separation, all of the data could be fit to a single diffusional component, consistent with a monomodal, random distribution of MSQ particle sizes. After phase separation, many of the autocorrelation data showed clear evidence for a bimodal distribution of particles, reflecting the presence of some small particles having relatively large diffusion coefficients, while the majority of particles were larger, yielding smaller D values.
Fig. 12a shows the D values obtained for the majority particles as a function of time. The dashed and solid vertical lines indicate the phase separation and gelation times, respectively. Prior to 10 h, the average D values remained constant at ~2 × 10−7 cm2 s−1, suggesting the MSQ nanoparticles grew rapidly to ~30 nm in size, at which point the growth process was temporarily interrupted. The measured D values subsequently began to decrease dramatically after 10 h. The observed decrease was concluded to result either from a change in the viscosity of the liquid within the pores or from a resumption of particle growth. Fig. 12b,c depict the amplitude of the autocorrelation data and the observed signal burst rate in the time transients. The decrease observed in these data, both of which are expected to track particle concentration161 provided strong evidence that the decrease in D after 10 h reflected a resumption of particle growth. It was concluded that initial particle nucleation and growth resulted from reactions between MTMOS monomers and small oligomers, which were largely consumed early in the process, while this later growth phase involved aggregation of the previously-formed nanoparticles. The delay in aggregation was attributed to the kinetics of intraparticle and surface reactions required to facilitate particle aggregation (i.e. via a reduction in nanoparticle polarity).
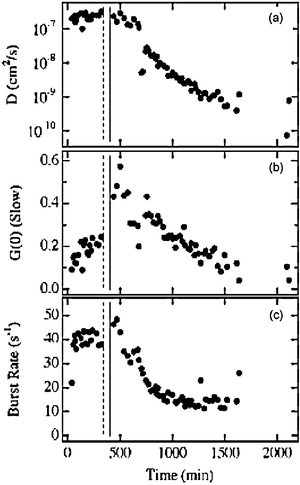 | ||
| Fig. 12 (A) Diffusion coefficient (slow component) as a function of time for silica nanoparticles within single pores. (B) Amplitude of the slow diffusion component. (C) Burst rate as a function of time. The vertical dashed and solid lines depict the phase separation and gelation times. (Reprinted with permission from ref. 159. Copyright 2007, American Chemical Society.) | ||
5. Conclusions and future directions
This article has reviewed some of the recent single molecule studies of sol–gel derived silica materials. Single molecule methods for probing molecule/matrix interactions in dye-doped silica materials have been described. Studies of dopant mobility within silica materials using FCS and SMT methods have also been covered and the relevance of the results, especially with respect to the quantitative measurement of diffusion coefficients in the presence of host–guest interactions, have been highlighted. Likewise, recent single molecule studies of sol–gel matrix polarity and acidity properties have been reviewed. The ability of all such studies to provide detailed and valuable new information on materials heterogeneity was demonstrated.The studies described above, along with other studies not described in this review, prove that single molecule methods represent an invaluable set of tools for the study of silica materials. They have already provided a better understanding of the nanoscale properties of these technologically useful and fundamentally interesting materials and will continue to do so for the foreseeable future. However, improvements to the methods presently employed are still required to fully understand silica materials. For example, dyes that better probe a single class of materials properties, without yielding data perturbed by other effects are required. Likewise, dye molecules that are more photostable are also required so that more spectroscopic information can be derived from them at higher signal-to-noise ratios. Luminescent or strongly-scattering nanoparticles may serve as substitutes for dyes in the future,33,148,152 assuming such particles can be tailored to exhibit sensitivities to specific materials parameters, as can already be achieved using fluorescent dyes. Improvements to the spatial resolution achieved in single molecule studies are also required to fully understand silica properties variations on relevant nanometer length scales. To this end, sub-diffraction-limited single molecule methods such as stochastic optical reconstruction microscopy (STORM, and related methods)162,163 and stimulated emission depletion microscopy (STED)164 are sure to be applied in studies of silica materials in the very near future. The latter methods will be especially important in single molecule fluorescence studies of the pores and chemically active surface sites165 of silica-based zeolites.166–168
Acknowledgements
The authors gratefully acknowledge the support of the National Science Foundation (CHE-0316466, CHE-0647849 and CHE-0648716) in these studies. The reviewers of this manuscript are thanked for their helpful contributions.References
- V. B. Kandimalla, V. S. Tripathi and H. X. Ju, Crit. Rev. Anal. Chem., 2006, 36, 73–106 CrossRef CAS.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- P. C. A. Jeronimo, A. N. Araujo and B. S. M. Montenegro, Talanta, 2007, 72, 13–27 CrossRef CAS.
- U. Schubert, New J. Chem., 1994, 18, 1049–1058 Search PubMed.
- G. Frenzer and W. F. Maier, Annu. Rev. Mater. Res., 2006, 36, 281–331 CrossRef CAS.
- A. M. Siouffi, J. Chromatogr., A, 2003, 1000, 801–818 CrossRef CAS.
- L. A. Colon, T. D. Maloney, J. Anspach and H. Colon, in Advances in Chromatography, 2003, vol 42, pp. 43–106 Search PubMed.
- W. Li, D. P. Fries and A. Malik, J. Chromatogr., A, 2004, 1044, 23–52 CrossRef CAS.
- M. D. Petit-Dominguez, H. Shen, W. R. Heineman and C. J. Seliskar, Anal. Chem., 1997, 69, 703–710 CrossRef CAS.
- H. Wei and M. M. Collinson, Anal. Chim. Acta, 1999, 397, 113–121 CrossRef CAS.
- P. Innocenzi and B. Lebeau, J. Mater. Chem., 2005, 15, 3821–3831 RSC.
- H. Liu, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, J. Solid State Electrochem., 2004, 8, 450–466 CrossRef CAS.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, Chem. Rev., 2004, 104, 4463–4492 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing, Academic Press, Boston, 1990 Search PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- G. Carturan, R. Dal Toso, S. Boninsegna and R. Dal Monte, J. Mater. Chem., 2004, 14, 2087–2098 RSC.
- B. Dunn and J. I. Zink, Acc. Chem. Res., 2007, 40, 747–755 CrossRef CAS.
- J. D. Brennan, Acc. Chem. Res., 2007, 40, 827–835 CrossRef CAS.
- M. Pagliaro, R. Ciriminna and G. Palmisano, Chem. Soc. Rev., 2007, 36, 932–940 RSC.
- H. Schmidt, J. Sol–Gel Sci. Technol., 1994, 1, 217–231 CrossRef CAS.
- S. Sanchez and F. Ribot, New J. Chem., 1994, 18, 1007–1047 Search PubMed.
- U. Schubert, N. Huesing and A. Lorenz, Chem. Mater., 1995, 7, 2010–2027 CrossRef CAS.
- J. Wen and G. L. Wilkes, Chem. Mater., 1996, 8, 1667–1681 CrossRef CAS.
- D. Avnir, L. C. Klein, D. Levy, U. Schubert and A. B. Wojcik, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport, and Y. Apeloig, John Wiley & Sons, 1998, pp. 2317–2362 Search PubMed.
- M. M. Collinson, TrAC Trends Anal. Chem., 2002, 21, 30–38 CrossRef CAS.
- C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559–3592 RSC.
- A. Walcarius, D. Mandler, J. A. Cox, M. M. Collinson and O. Lev, J. Mater. Chem., 2005, 15, 3663–3689 RSC.
- M. Pagliaro, R. Ciriminna, M. W. C. Man and S. Campestrini, J. Phys. Chem. B, 2006, 110, 1976–1988 CrossRef CAS.
- L. Nicole, C. Boissiere, D. Grosso, A. Quach and C. Sanchez, J. Mater. Chem., 2005, 15, 3598–3627 RSC.
- C. Bonhomme, C. Coelho, N. Baccile, C. Gervais, T. Azais and F. Babonneau, Acc. Chem. Res., 2007, 40, 738–746 CrossRef CAS.
- N. Hüsing, U. Schubert, K. Misof and P. Fratzl, Chem. Mater., 1998, 10, 3024–3032 CrossRef.
- Y. Lu, G. Cao, R. P. Kale, S. Prabakar, G. P. Lopez and C. J. Brinker, Chem. Mater., 1999, 11, 1223–1229 CrossRef CAS.
- F. Akbarian, B. S. Dunn and J. I. Zink, J. Phys. Chem., 1995, 99, 3892–3894 CrossRef CAS.
- A. Anedda, C. M. Carbonaro, F. Clemente, R. Corpino and P. C. Ricci, J. Phys. Chem. B, 2003, 107, 13661–13664 CrossRef CAS.
- D. Rivera and J. M. Harris, Anal. Chem., 2001, 73, 411–423 CrossRef CAS.
- M. M. Collinson, Acc. Chem. Res., 2007, 40, 777–783 CrossRef CAS.
- D. Avnir, Acc. Chem. Res., 1995, 28, 328–334 CrossRef CAS.
- B. Dunn and J. I. Zink, Chem. Mater., 1997, 9, 2280–2291 CrossRef CAS.
- B. Dunn and J. I. Zink, J. Mater. Chem., 1991, 1, 903–913 RSC.
- T. Keeling-Tucker and J. D. Brennan, Chem. Mater., 2001, 13, 3331–3350 CrossRef CAS.
- P. Tamarat, A. Maali, B. Lounis and M. Orrit, J. Phys. Chem. A, 2000, 104, 1–16 CrossRef CAS.
- W. E. Moerner, J. Phys. Chem. B, 2002, 106, 910–927 CrossRef CAS.
- A. M. Kelley, in Encyclopedia of Chemical Physics and Physical Chemistry, ed. J. H. Moore and N. D. Spencer, Institute of Physics Publishing, Bristol, UK, 2001, vol. 3, pp. 2199–2228 Search PubMed.
- W. E. Moerner and D. P. Fromm, Rev. Sci. Instrum., 2003, 74, 3597–3619 CrossRef CAS.
- D. A. Higgins and Y. Hou, in Encyclopedia of Nanoscience and Nanotechnology, ed. J. A. Schwartz, C. Contescu and K. Putyera, Marcel-Dekker, New York, 2004, p. 3575 Search PubMed.
- D. A. Higgins and M. M. Collinson, Langmuir, 2005, 21, 9023–9031 CrossRef CAS.
- M. J. Wirth and D. J. Swinton, Anal. Chem., 1998, 70, 5264–5271 CrossRef CAS.
- M. J. Wirth, M. D. Ludes and D. J. Swinton, Appl. Spectrosc., 2001, 55, 663–669 CrossRef CAS.
- M. J. Wirth and D. J. Swinton, J. Phys. Chem. B, 2001, 105, 1472–1477 CrossRef CAS.
- M. J. Wirth, D. J. Swinton and M. D. Ludes, J. Phys. Chem. B, 2003, 107, 6258–6268 CrossRef CAS.
- Z. Zhong, M. Lowry, G. Wang and L. Geng, Anal. Chem., 2005, 77, 2303–2310 CrossRef CAS.
- M. M. Collinson, in Chalcogenide Glasses and Sol–Gel Materials, ed. H. S. Nalwa,Academic Press, 2001, vol. 5,pp. 163–194 Search PubMed.
- A. H. Boonstra and T. N. M. Bernards, J. Non-Cryst. Solids, 1988, 105, 207–213 CrossRef CAS.
- T. N. M. Bernards, M. J. Van Bommel, E. W. J. L. Oomen and A. H. Boonstra, J. Non-Cryst. Solids, 1992, 147, 13–17 CrossRef.
- T. N. M. Bernards, E. W. J. L. Oomen, M. J. Van Bommel and A. H. Boonstra, J. Non-Cryst. Solids, 1992, 142, 215–224 CAS.
- H. Dong and J. D. Brennan, Chem. Mater., 2006, 18, 4176–4182 CrossRef CAS.
- X. Zhang, F. Shi, J. Niu, Y. G. Jiang and Z. Q. Wang, J. Mater. Chem., 2008, 18, 621–633 RSC.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS.
- U. Ciesla and F. Schuth, Microporous Mesoporous Mater., 1999, 27, 131–149 CrossRef.
- W. H. Zhang, X. B. Lu, J. H. Xiu, Z. L. Hua, L. X. Zhang, M. Robertson, J. L. Shi, D. S. Yan and J. D. Holmes, Adv. Funct. Mater., 2004, 14, 544–552 CrossRef CAS.
- A. Taguchi and F. Schuth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS.
- S. Fujita and S. Inagaki, Chem. Mater., 2008, 20, 891–908 CrossRef CAS.
- M. Tiemann, Chem. Mater., 2008, 20, 961–971 CrossRef CAS.
- P. C. A. Alberius, K. L. Frindell, R. C. Hayward, E. J. Kramer, G. D. Stucky and B. F. Chmelka, Chem. Mater., 2002, 14, 3284–3294 CrossRef.
- Y. Fu, F. Ye, W. G. Sanders, M. M. Collinson and D. A. Higgins, J. Phys. Chem. B, 2006, 110, 9164–9170 CrossRef CAS.
- F. Ye, D. A. Higgins and M. M. Collinson, J. Phys. Chem. C, 2007, 111, 6772–6780 CrossRef CAS.
- C. Jung, J. Kirstein, B. Platschek, T. Bein, M. Budde, I. Frank, K. Muellen, J. Michaelis and C. Bräuchle, J. Am. Chem. Soc., 2008, 130, 1638–1648 CrossRef CAS.
- P. Selvam, S. K. Bhatia and C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237–3261 CrossRef CAS.
- J. D. Jordan, R. A. Dunbar, D. J. Hook, H. Zhuang, J. A. Gardella, L. A. Colon and F. V. Bright, Chem. Mater., 1998, 10, 1041–1051 CrossRef CAS.
- C. J. Brinker, A. J. Hurd, P. R. Schunk, G. C. Frye and C. S. Ashley, J. Non-Cryst. Solids, 1992, 147/148, 424–436.
- C. J. Brinker, G. C. Frye, A. J. Hurd and C. S. Ashley, Thin Solid Films, 1991, 201, 97–108 CrossRef CAS.
- S. M. Mahurin, S. Dai and M. D. Barnes, J. Phys. Chem. B, 2003, 107, 13336–13340 CrossRef CAS.
- M. Orrit and J. Bernard, Phys. Rev. Lett., 1990, 65, 2716–2719 CrossRef CAS.
- W. P. Ambrose and W. E. Moerner, Nature, 1991, 349, 225–227 CrossRef CAS.
- C. Julien, A. Debarre, D. Nutarelli, A. Richard and P. Tchenio, J. Phys. Chem. B, 2006, 110, 3902–3909 CrossRef CAS.
- T. Ha, Methods, 2001, 25, 78–86 CrossRef CAS.
- J. W. Gilliland, K. Yokoyama and W. T. Yip, Chem. Mater., 2004, 16, 3949–3954 CrossRef CAS.
- J. W. Gilliland, K. Yokoyama and W. T. Yip, Chem. Mater., 2005, 17, 6702–6712 CrossRef CAS.
- J. Kirstein, B. Platschek, C. Jung, R. Brown, T. Bein and C. Bräuchle, Nat. Mater., 2007, 6, 303–310 CrossRef CAS.
- R. Gupta and N. K. Chaudhury, Biosens. Bioelectron., 2007, 22, 2387–2399 CrossRef CAS.
- U. Narang, R. Wang, P. N. Prasad and F. V. Bright, J. Phys. Chem., 1994, 98, 17–22 CrossRef CAS.
- D. S. Gottfried, A. Kagan, B. M. Hoffman and J. M. Friedman, J. Phys. Chem. B, 1999, 103, 2803–2807 CrossRef CAS.
- M. H. Huang, H. M. Soyez, B. S. Dunn and J. I. Zink, Chem. Mater., 2000, 12, 231–235 CrossRef CAS.
- G. Hungerford, A. Rei, M. I. C. Ferreira, K. Suhling and C. Tregidgo, J. Phys. Chem. B, 2007, 111, 3558–3562 CrossRef CAS.
- F. Stallmach, J. Karger, C. Krause, M. Jeschke and U. Oberhagemann, J. Am. Chem. Soc., 2000, 122, 9237–9242 CrossRef CAS.
- F. Cros, J. P. Korb and L. Malier, Langmuir, 2000, 16, 10193–10197 CrossRef CAS.
- M. Okazaki and K. Toriyama, J. Phys. Chem. B, 2003, 107, 7654–7658 CrossRef CAS.
- I. G. Shenderovich, G. Buntkowsky, A. Schreiber, E. Gedat, S. Sharif, J. Albrecht, N. S. Golubev, G. H. Findenegg and H. H. Limbach, J. Phys. Chem. B, 2003, 107, 11924–11939 CrossRef CAS.
- A. R. Howells, P. J. Zambrano and M. M. Collinson, Anal. Chem., 2000, 72, 5265–5271 CrossRef CAS.
- M. Kanungo and M. M. Collinson, Anal. Chem., 2003, 75, 6555–6559 CrossRef CAS.
- M. Kanungo and M. M. Collinson, Langmuir, 2005, 21, 827–829 CrossRef CAS.
- J. W. Gilliland, K. Yokoyama and W. T. Yip, J. Phys. Chem. B, 2005, 109, 4816–4823 CrossRef CAS.
- R. Kennard, W. J. DeSisto, T. P. Giririjan and M. D. Mason, J. Chem. Phys., 2008, 128, 134710 CrossRef.
- A. M. Bardo, M. M. Collinson and D. A. Higgins, Chem. Mater., 2001, 13, 2713–2721 CrossRef CAS.
- C. Hellriegel, J. Kirstein, C. Bräuchle, V. Latour, T. Pigot, R. Olivier, S. Lacombe, R. Brown, V. Guieu, C. Payrastre, A. Izquierdo and P. Mocho, J. Phys. Chem. B, 2004, 108, 14699–14709 CrossRef CAS.
- A. Walcarius, C. Delacote and S. Sayen, Electrochim. Acta, 2004, 49, 3775–3783 CrossRef CAS.
- M. M. Collinson, P. Zambrano, H. Wang and J. Taussig, Langmuir, 1999, 15, 662–668 CrossRef CAS.
- O. Dvorak and M. K. De Armond, J. Phys. Chem., 1993, 97, 2646–2648 CrossRef CAS.
- M. M. Collinson, C. G. Rausch and A. Voigt, Langmuir, 1997, 13, 7245–7251 CrossRef CAS.
- E. L. Elson and D. Magde, Biopolymers, 1974, 13, 1–27 CrossRef CAS.
- H. Qian, M. P. Sheetz and E. L. Elson, Biophys. J., 1991, 60, 910–921 Search PubMed.
- T. Schmidt, G. J. Schuetz, W. Baumgartner, H. J. Gruber and H. Schindler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2926–2929 CrossRef.
- D. Wöll, H. Uji-i, T. Schnitzler, J. Hotta, P. Dedecker, A. Herrmann, F. C. De Schryver, K. Müllen and J. Hofkens, Angew. Chem., Int. Ed., 2008, 47, 783–787 CrossRef.
- K. S. McCain, D. C. Hanley and J. M. Harris, Anal. Chem., 2003, 75, 4351–4359 CrossRef CAS.
- C. Hellriegel, J. Kirstein and C. Bräuchle, New J. Phys., 2005, 7, 1–14 CrossRef.
- C. Jung, B. K. Mueller, D. C. Lamb, F. Nolde, K. Muellen and C. Bräuchle, J. Am. Chem. Soc., 2006, 128, 5283–5291 CrossRef CAS.
- M. Wahl, I. Gregor, M. Patting and J. Enderlein, Opt. Express, 2003, 11, 3583–3591 Search PubMed.
- K. Zhang and H. Yang, J. Phys. Chem. B, 2005, 109, 21930–21937 CrossRef CAS.
- S. A. Martin-Brown, Y. Fu, G. Saroja, M. M. Collinson and D. A. Higgins, Anal. Chem., 2005, 77, 486–494 CrossRef CAS.
- C. Jung, C. Hellriegel, J. Michaelis and C. Bräuchle, Adv. Mater., 2007, 19, 956–960 CrossRef CAS.
- A. Zuerner, J. Kirstein, M. Doeblinger, C. Bräuchle and T. Bein, Nature, 2007, 450, 705–708 CrossRef CAS.
- Y. Sakamoto, M. Kaneda, O. Terasaki, D. Y. Zhao, J. M. Kim, G. Stucky, H. J. Shin and R. Ryoo, Nature, 2000, 408, 449–453 CrossRef CAS.
- D. Avnir, S. Braun, O. Lev, D. Levy and M. Ottolenghi, in Sol–Gel Optics Processing and Applications, ed. L. C. Klein, Kluwer Academic Publications, Massachusetts, 1994, pp. 539–582 Search PubMed.
- K. S. McCain, P. Schluesche and J. M. Harris, Anal. Chem., 2004, 76, 939–946 CrossRef CAS.
- K. S. McCain and J. M. Harris, Anal. Chem., 2003, 75, 3616–3624 CrossRef CAS.
- D. A. Higgins, M. M. Collinson, G. Saroja and A. M. Bardo, Chem. Mater., 2002, 14, 3734–3744 CrossRef CAS.
- J. D. Brennan, J. S. Hartman, E. I. Ilnicki and M. Rakic, Chem. Mater., 1999, 11, 1853–1864 CrossRef CAS.
- Y. Hou, A. M. Bardo, C. Martinez and D. A. Higgins, J. Phys. Chem. B, 2000, 104, 212–219 CrossRef CAS.
- J. F. Deye, T. A. Berger and A. G. Anderson, Anal. Chem., 1990, 62, 615–622 CrossRef CAS.
- A. K. Dutta, K. Kamada and K. Ohta, J. Photchem. Photobiol. A, 1996, 93, 57–64 Search PubMed.
- N. Sarkar, K. Das, D. N. Nath and K. Bhattacharyya, Langmuir, 1994, 10, 326–329 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 1261–1274 CrossRef CAS.
- B. S. Brunschwig, S. Ehrenson and N. Sutin, J. Phys. Chem., 1987, 91, 4714–4723 CrossRef CAS.
- R. A. Marcus, J. Phys. Chem., 1990, 94, 4963–4966 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 2003 Search PubMed.
- H. Frenkel-Mullerad and D. Avnir, Chem. Mater., 2000, 12, 3754–3759 CrossRef CAS.
- Y. Tang, E. C. Tehan, Z. Tao and F. V. Bright, Anal. Chem., 2003, 75, 2407–2413 CrossRef CAS.
- C. A. Fyfe and P. P. Aroca, J. Phys. Chem. B, 1997, 101, 9504–9509 CrossRef CAS.
- X. Feng and W. H. Thompson, J. Phys. Chem. C, 2007, 111, 18060–18072 CrossRef CAS.
- R. K. Iler, The Chemistry of Silica, John Wiley and Sons, New York, 1979 Search PubMed.
- S. Ong, X. Zhao and K. B. Eisenthal, Chem. Phys. Lett., 1992, 191, 327–335 CrossRef CAS.
- M. J. Meziani, J. Zajac, D. J. Jones, S. Partyka, J. Roziere and A. Auroux, Langmuir, 2000, 16, 2262–2268 CrossRef CAS.
- D. Rivera and J. M. Harris, Langmuir, 2001, 17, 5527–5536 CrossRef CAS.
- Y. Inaki, H. Yoshida, T. Yoshida and T. Hattori, J. Phys. Chem. B, 2002, 106, 9098–9106 CrossRef CAS.
- L. Yang and S. S. Saavedra, Anal. Chem., 1995, 67, 1307–1314 CrossRef CAS.
- Y. Fu, M. M. Collinson and D. A. Higgins, J. Am. Chem. Soc., 2004, 126, 13838–13844 CrossRef CAS.
- S. Brasselet and W. E. Moerner, Single Mol., 2000, 1, 17–23 CrossRef CAS.
- J. E. Whitaker, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1991, 194, 330–344 CrossRef CAS.
- J. E. Lee and S. S. Saavedra, Anal. Chim. Acta, 1994, 285, 265–269 CrossRef CAS.
- L. Yang and S. S. Saavedra, Anal. Chem., 1995, 67, 1307–1314 CrossRef CAS.
- E. Mei, A. M. Bardo, M. M. Collinson and D. A. Higgins, J. Phys. Chem. B, 2000, 104, 9973–9980 CrossRef CAS.
- S. M. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- Y. Kobayashi, M. A. Correa-Duarte and L. M. Liz-Marzan, Langmuir, 2001, 17, 6375–6379 CrossRef CAS.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef.
- R. C. Somers, M. G. Bawendi and D. G. Nocera, Chem. Soc. Rev., 2007, 36, 579–591 RSC.
- C. Szymanski, C. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543–8546 CrossRef CAS.
- M. Tomasulo, I. Yildiz and F. M. Raymo, J. Phys. Chem. B, 2006, 110, 3853–3855 CrossRef CAS.
- A. M. Michaels, M. Nirmal and L. E. Brus, J. Am. Chem. Soc., 1999, 121, 9932–9939 CrossRef CAS.
- H. Kaji, K. Nakanishi, N. Soga, T. Inoue and N. Nemoto, J. Sol–Gel Sci. Technol., 1994, 3, 169–188 CrossRef CAS.
- K. Nakanishi and K. Kanamori, J. Mater. Chem., 2005, 15, 3776–3786 RSC.
- K. Nakanishi and N. Tanaka, Acc. Chem. Res., 2007, 40, 863–873 CrossRef CAS.
- H. Dong, M. A. Brook and J. D. Brennan, Chem. Mater., 2005, 17, 2807–2816 CrossRef CAS.
- S. Hartmann, D. Brandhuber and N. Hüsing, Acc. Chem. Res., 2007, 40, 885–894 CrossRef CAS.
- H. Dong, F. Ye, D. A. Higgins and M. M. Collinson, Chem. Mater., 2007, 19, 6528–6535 CrossRef CAS.
- C. L. Kuyper, K. L. Budzinski, R. M. Lorenz and D. T. Chiu, J. Am. Chem. Soc., 2006, 128, 730–731 CrossRef.
- F. Ye, M. M. Collinson and D. A. Higgins, Anal. Chem., 2007, 79, 6465–6472 CrossRef CAS.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CAS.
- M. J. Rust, M. Bates and X. W. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS.
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS.
- M. B. J. Roeffaers, B. F. Sels, H. Uji-i, F. C. De Schryver, P. A. Jacobs, D. E. De Vos and J. Hofkens, Nature, 2006, 439, 572–575 CrossRef CAS.
- S. Hashimoto, K. Uehara, K. Sogawa, M. Takada and H. Fukumura, Phys. Chem. Chem. Phys., 2006, 8, 1451–1458 RSC.
- M. B. J. Roeffaers, R. Ameloot, M. Baruah, H. Uji-i, M. Bulut, G. De Cremer, U. Müller, P. A. Jacobs, J. Hofkens, B. F. Sels and D. E. De Vos, J. Am. Chem. Soc., 2008, 130, 5763–5772 CrossRef CAS.
- M. Busby, C. Blum, M. Tibben, S. Fibikar, G. Calzaferri, V. Subramaniam and L. De Cola, J. Am. Chem. Soc., 2008, 130, 10970–10976 CrossRef CAS.
| This journal is © the Owner Societies 2009 |