Time-dependent depolarization of aligned HD molecules
Received
13th August 2008
, Accepted 13th October 2008
First published on 25th November 2008
Abstract
An aligned sample of HD(v = 1, J = 2, MJ = 0) molecules is prepared under collision-free conditions using the S(0) stimulated Raman pumping transition. Subsequent coupling to the spins of the deuteronID and the proton IH causes the initial degree of alignment to oscillate and decrease as monitored over the time range from 0–13 μs via the O(2) line of the [2 + 1] REMPIE,F1Σ+g−X1Σ+g (0,1) band. The time dependence of the rotational alignment is also calculated using both a hierarchical coupling scheme in which the rotational angular momentum J is regarded first to couple to ID, and then the resultant Fi to couple to IH, to form the total angular momentum F and a non-hierarchical coupling scheme in which the HD energy level structure is not assumed to be diagonal in the |IH(JID)FiFMF> basis set. The experimental data is in good agreement with the non-hierarchical calculation but not with the hierarchical calculation, as expected for this system. Additionally, we calculate the time dependence of the H and D nuclear spin polarizations.
Introduction
Many experimental techniques have been developed to prepare oriented and aligned molecules, either for their use as reagents in scattering experiments, or for studying the behavior of such species in isolation. Two of the most common and successful methods are alignment by strong electric fields1–4 and optical pumping with polarized light.5,6–14 While the former is limited to molecules with a dipole moment, optical pumping can be applied to a much wider range of species and can be used to align or orient those with small or no dipole moments. Optical transitions induced with linearly polarized light can serve to align molecules while oriented samples can be produced with circularly polarized light.
The broad applicability of the pumping technique has allowed for the preparation of several aligned reagents in this manner by various groups. Production of aligned HF for the study of stereodynamical effects in the Sr + HF reaction has been reported by Karny et al.6 using one-photon absorption. The time dependence of HCl(v = 1, J) alignment, prepared with IR excitation, was studied by Orr-Ewing et al.7 The photolysis of aligned and oriented HCl (v = 2, J) was recently used by Rakitzis and coworkers8,9 to prepare spin-polarized H and Cl atoms by means of hyperfine coupling. Polarization spectroscopy was applied by McKendrick and co-workers10,11 to measure the time evolution of OH orientation and alignment following inelastic collisions with Ar and He. The two-photon stimulated Raman pumping (SRP) process has also become a popular method for polarizing samples of molecules having no dipole moments. Sitz and Farrow12 studied the time dependence of an aligned sample of room-temperature N2(v = 1, J = 0–14), as prepared by SRP. Kandel et al.13 prepared in this manner a sample of aligned HD (v = 1, J = 2) for subsequent use as a target in the Cl + HD hydrogen abstraction reaction. Recently, our group has reported the first preparation of an oriented sample of H2(v = 1, J = 2, MJ = 2) using this technique as well.14
Molecules aligned (or oriented) by optical pumping methods are subject to subsequent degradation of the initial alignment by the coupling of the rotational angular momentum J to other angular momenta, called “hidden” angular momenta, which are not affected by the electric dipole allowed transition. In the case of HD the hidden angular momenta are the nuclear spin of the H atom, IH = 1/2 and the nuclear spin of the D atom ID = 1. As the molecule rotates, there is a current loop from all the negatively charged electrons in the molecule and all the positively charged nuclei, and these two current loops are of different magnitudes, causing the rotation of the molecule to generate a magnetic field. The hyperfine coupling arises from the interaction of the nuclear spins with the magnetic field induced by the rotational motion of the molecule. This coupling serves to split the rotational levels into hyperfine levels. In addition nuclei with a nuclear spin greater than unity possess a quadrupole moment, which further splits and complicates the hyperfine energy level pattern. If the laser excitation of a molecule to a particular rotational sublevel |JMJ〉 occurs without hyperfine resolution (this is true if Δt≪ 1/Δν, where Δt is the duration of the laser pulse, and Δν is the magnitude of the hyperfine splitting), a coherent superposition of hyperfine sublevels associated with |JMJ〉 results. The result of the subsequent time evolution is oscillatory population transfer between the different MJ-sublevels, and this beating is referred to as hyperfine depolarization, as it serves to reduce the initial degree of polarization of the J vector distribution, although at sufficiently long times the degree of polarization will be restored (recurrence phenomenon). This effect has been previously studied in detail both theoretically and experimentally for many systems.2,15–18 We also note that an analogous depolarization effect can be observed in molecules with net non-zero electronic spin S, but we neglect this effect here because HD is in a singlet state (S = 0).
The hyperfine depolarization can also be understood classically using a vector model in which J couples to one or more hidden angular momenta that are unaffected by the electric dipole allowed transition to form the total angular momentum F, about which J precesses. To simplify this discussion, let us just consider the case where only one nucleus has the nuclear spin I. Then the coupling results in the time-dependent polarization transfer between J and I, causing the prepared alignment of the rotation to vary in time. As expected, the degradation of the initial polarization is significant when J and I are comparable in magnitude, whereas the depolarization effect becomes less important when the magnitude of J is much larger than that of I. When J and I are equal in magnitude, complete depolarization of J and complete polarization of I can be observed.
Thus, this mechanism can be used to transfer polarization from molecular rotation to produce large nuclear polarizations in molecules. This transfer was demonstrated directly by Sofikitis et al.8 by the production of highly polarized Cl atoms from the photolysis of state prepared HCl(v = 1, J = 1, MJ = 1) molecules, and followed over a timescale of about 200 ns.
Here, we report on the preparation of aligned HD(v = 1, J = 2, MJ = 0) under collision-free conditions by SRP and the subsequent time dependence of the alignment over the range from 0 to 13 μs, as probed by [2 + 1] REMPI. The theory necessary to understand and analyze the measurements is presented in the following section. The time dependences of both the rotational polarization and the polarization of the nuclei are calculated and the former is compared to the experimental results.
We demonstrate the ability to observe the polarization of the molecular rotation out to about 13 μs, which is more than a factor of 10 longer than previous measurements using this pump–probe laser technique. Our work may be regarded as a descendent of Ramsey’s original technique for precisely measuring magnetic moments in atomic or molecular beams by applying two separated oscillating electromagnetic fields that excite transitions between ground-state sublevels in a magnetic field.19 It might be asked how well can hyperfine structure be measured. High resolution is usually achieved by increasing the accuracy and precision to which the frequency of a molecular transition is determined. But another approach to high resolution exists, namely increasing the time of a measurement. Recently, a beam of Stark-decelerated OH molecules was slowed to a speed of 200 m s−1 allowing interaction with a microwave field for up to a millisecond, resulting in the measurement of the ground-state Λ-doublet splitting with an uncertainty of tens of hertz or less.20 It has been suggested that even longer interaction times approaching a second will be possible with “molecular fountains” in which molecules that are decelerated to a few meters per second prior to launching, fly upwards before falling back under gravity.21 In the present experiment, the observation time is limited by the construction of our experiment; simple modifications would allow significantly longer observation times, so that polarization transfer can be observed in other molecular systems for which the hyperfine coupling is still weaker. Polarization transfer in small molecules can be used to enhance gas-phase NMR spectra by orders of magnitude,22 allowing the observation of spectra at low enough pressures that intramolecular effects are negligible. The present study should be regarded primarily as a proof of principle of what could be achieved.
Theory
The spatial distribution of angular momenta J of an atomic or molecular ensemble may be described by the (2J + 1)2 density matrix elements ρM′JMJ, or by the (2J + 1)2 multipole moments Aq(k)(J), where k≤ 2J. The two descriptions are equivalent and related by the following expressions:23| | 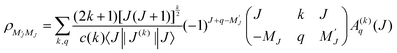 | (1) |
| | 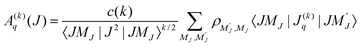 | (2) |
where the c(k) are normalization constants: c(0) = 1, c(1) = 1, c(2) = 61/2, c(3) = (5/2)1/2, c(4) = (35/8)1/2, etc. When the MJ-sublevel populations, defined with respect to a laboratory axis, are not all equal, the ensemble is said to be polarized. Consider such a sample which, for example, has been prepared by pulsed-laser excitation with linearly or circularly polarized light. We assume that the initial spatial distribution of nuclear spins is random, and that it is unaltered by the optical excitation process. The subsequent coupling of two nuclear spins to the rotational angular momentum J, leads to a time dependence of a molecule’s rotational polarization, and thus each moment Aq(k)(J,t = 0), becomes multiplied by a depolarization factor G(k) (J,t)| | | A(k)q(J,t) = A(k)q(J,t = 0) G(k)(J,t) | (3) |
The depolarization factors have been derived previously by Altkorn, Greene, and Zare15 for both the cases where a hierarchical coupling scheme does and does not apply. These are expressed, respectively, as| | 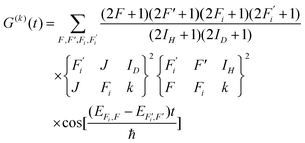 | (4) |
| | 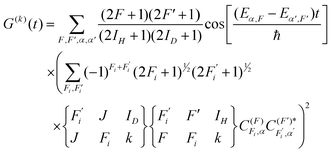 | (5) |
Here F is the quantum number associated with the total angular momentum of the system, and Fi is an intermediate quantum number resulting from the coupling Fi = ID + J (see Fig. 1 for an illustration of the coupling scheme). Fi is a good quantum number in the limit of hierarchical coupling so that the |IH(IDJ)FiFMF〉 basis is used in this case to calculate the hyperfine energy splittings. Typically this coupling scheme is valid when the coupling of one nuclear spin to the rotation is much stronger than that of the other, so that polarization transfer from the rotation to that nucleus is much faster than to the other. When hierarchical coupling cannot be used, the hyperfine Hamiltonian written in the |IH(IDJ)FiFMF〉 basis must be diagonalized, which yields hyperfine energies that depend not on Fi but on α, a quantum number that results from general, non-hierarchical coupling. Finally, note that the eigenvectors of the Hamiltonian 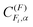 only appear in the non-hierarchical expression. This is because
only appear in the non-hierarchical expression. This is because 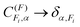 , when hierarchical coupling applies and this substitution yields eqn (4). The beating frequencies are thus determined by the energy differences between the hyperfine levels while their amplitudes depend on the magnitudes of ID, IH, and J.
, when hierarchical coupling applies and this substitution yields eqn (4). The beating frequencies are thus determined by the energy differences between the hyperfine levels while their amplitudes depend on the magnitudes of ID, IH, and J.
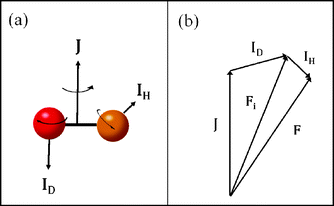 |
| | Fig. 1 Illustration of (a) the three angular momenta of the hydrogen deuteride molecule and (b) the vector model of the angular momentum coupling. | |
In addition, analogous expressions for the nucleus have been derived by Rubio-Lago et al.24 The time-dependent polarization factors (non-hierarchical) are given by
| | 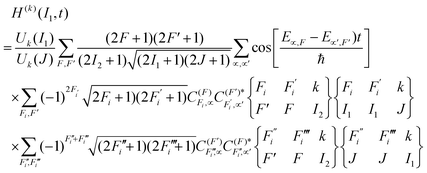 | (6) |
| | 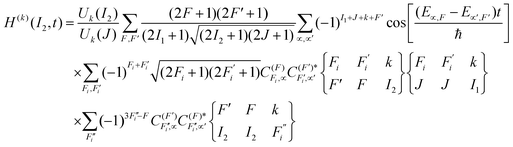 | (7) |
and we relate the spatial distribution of each nuclear spin
I1 and
I2 at time
t to the initial spatial distribution of the rotation
J through the expression
| | | A(k)q(I,t) = H(k)(I,t) ×A(k)q(J,t = 0) | (8) |
In the above,
Uk(
I) and
Uk(
J) are normalization constants that allow moments with differing values of
I and
J to be related, and are reciprocals of the
Vk(
J) defined elsewhere.
25 Again, the corresponding expressions when hierarchical coupling is valid can be obtained by letting the
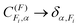
.
Finally, we combine conservation of the projection of the total angular momentum
| | | 〈MJ(t)〉 + 〈MI1(t)〉 + 〈MI2(t)〉 = 〈MJ(t = 0)〉 | (9) |
with expressions for each angular momentum projection term on the left-hand side
| | 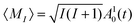 | (10) |
to obtain
| | 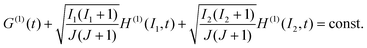 | (11) |
This condition allows us to check our calculated expressions for the rotational depolarization and nuclear polarization factors, which is illustrated in the Discussion section.
Experimental
Here we present only a concise summary of the experimental techniques used in this study and refer the reader to a preceding publication14 for a detailed description. A mixture of 10% HD (Cambridge Isotopes, 97% purity) in Ar was supersonically expanded into the extraction region of a Wiley–McLaren time-of-flight (TOF) spectrometer, described in detail elsewhere,26,27 through a pulsed nozzle (General Valve) with a backing pressure of 500–1000 Torr. This produces almost exclusively HD(v = 0) in low rotational levels. The HD(v = 0, J = 0) molecules in the expansion are subsequently excited to the HD(v = 1, J = 2, M = 0) state via the S(0) SRP transition using linearly polarized light, producing an aligned sample. The pump and Stokes wavelengths necessary for SRP, 532 and 670.640 nm respectively, are produced using the second harmonic of an injection-seeded Nd3+:YAG laser (Continuum PL9020) and the output of a dye laser (Continuum ND6000, LDS698 dye), respectively. Upon entering the chamber both SRP beams are perpendicular to the TOF axis, and because these beams create the alignment of the molecules, we choose their polarization direction to define the z axis of our laboratory frame. The excited-state HD molecules are state-specifically ionized after a variable pump–probe time delay by [2+1] REMPIvia the O(2) line of the E,F1Σ+g−X1Σ+g (0,1) band using linearly polarized light at 209.572 nm. The UV light for the REMPI is generated by frequency tripling the output of a second Nd3+:YAG-pumped dye laser (Lambda Physik LPD3000, DCM dye) using two BBO crystals in series. The linear polarization of the probe light was alternated on a shot-to-shot basis so that its electric field vector is either parallel or perpendicular to the laboratory z axis. The HD+ ions were detected with the TOF spectrometer for pump–probe delays of 0–13 μs producing probe polarization-dependent signals denoted by I|| and I⊥. The experimental setup is shown in Fig. 2 and the SRP and REMPI scheme is shown in Fig. 3.
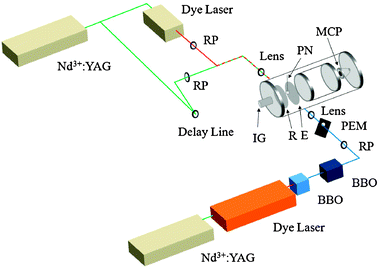 |
| | Fig. 2 Schematic diagram of experimental apparatus. Abbreviations are as follows: BBO = β-barium borate crystal; E = extractor grid; IG = (nude) ion gauge; MCP = microchannel plates; PEM = photoelastic modulator; PN = pulsed nozzle; R = repeller plate; RP = Rochon prism (linear polarizer). | |
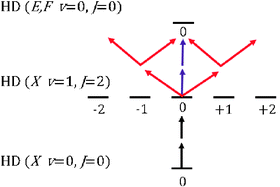 |
| | Fig. 3
SRP and REMPI schemes used to prepare and detect HD(v = 1, J = 2, MJ = 0) from HD (v = 0, J = 0) using linearly polarized light. Purple and red arrows represent the electric field vector of the probe radiation parallel and perpendicular to the pump radiation (black arrows). | |
Discussion
In this section we evaluate the rotational depolarization factors G(k) and nuclear polarization factors H(k) that describe the time-dependent behavior of the HD molecule following excitation to the HD(v = 1, J = 2, MJ = 0) state. To do so, it is necessary to calculate the energies of the hyperfine levels, which is done by diagonalizing the hyperfine Hamiltonian for the HD molecule in the absence of external fields28| | 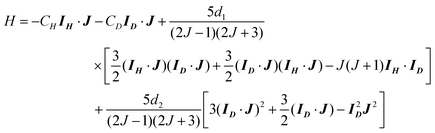 | (12) |
in the |IH(IDJ)FiFMF〉 basis. When a hierarchical coupling scheme is employed, it is assumed that the Hamiltonian is already diagonal in this basis. Here, CH and CD are the spin-rotational interaction constants whose values signify the strength of the magnetic field at the hydrogen and deuterium nuclei, respectively, arising from rotation of the molecule. The constants d1 and d2 are the nuclear spin–nuclear spin magnetic interaction constant and the quadrupole interaction constant. Note that a nuclear quadrupole term is only present for molecules which possess nuclei of spin I≥ 1, and that IH = 1/2 and ID = 1. The hyperfine constants have been measured by Ramsey and co-workers28 for HD(v = 0, J = 1) and they found CH = 85.600 kHz, CD = 13.122 kHz, d1 = 17.761 kHz, and d2 = −22.454 kHz. We use the ground-state hyperfine constants in our calculation of the time dependence of the rotational and nuclear polarizations, i.e. we assume that the ground-state hyperfine constants are slowly varying functions of the vibrational level.
Fig. 4 displays the signal ratio I(0)/I(90) for the detection of HD(v = 1, J = 2, MJ = 0) as well as the theoretical predictions for both non-hierarchical and hierarchical coupling schemes. The intensities for the signals may be expressed in terms of a multipole expansion29:
| | | I = I0[1 + s2A(2)0G(2)(t)P2(cos θ) + s4A(4)0G(4)(t)P4(cos θ)] | (13) |
where the
A(k)0 describe the initial alignment of the system and the
sk are the [2 + 1]
REMPI sensitivity factors for the corresponding
A(k)0. The
Pk (cos
θ) are
kth order Legendre polynomials, where
θ is the angle between the laboratory frame
z axis and the polarization direction of the linearly polarized probe laser light. Note that detection using linearly polarized light is sensitive only to the even moments of the angular momentum distribution.
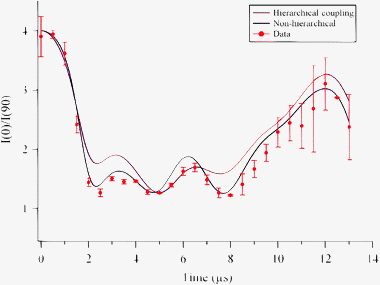 |
| | Fig. 4 Experimentally measured and calculated time evolution of the HD rotational polarization. The data are plotted as the ratio of the parallel to the perpendicular integrated ion signal I||/I⊥ as a function of SRP-REMPI time delay from 0–13 μs. The error bars are calculated as 2σ from three measurements at each time delay. | |
From eqn (13) we obtain
| | | I|| = I0[1+s2A(2)0G(2)(t)P2(1) + s4A(4)0G(4)(t)P4(1) = I0[1 + s2A(2)0G(2)(t)+s4A(4)0G(4)(t)] | (14a) |
and
| | 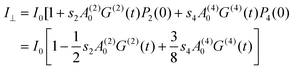 | (14b) |
The
A(k)0 depend only on the values of
J and
MJ of the prepared state, while the
sk depend only on the initial and final values of
J in the 2 + 1
REMPI step, and the polarization of the light. For the
O(2) line of the
E,F1Σ
+g−
X1Σ
+g (0,1) [2 + 1]
REMPI band using linearly polarized light and assuming 100% population in the
MJ = 0 sublevel,
s2 = −10/7,
s4 = 72/7,
A(2)0 = −1 and
A(4)0 = 1/4. We find that the ground-state hyperfine constants produce rotational depolarization factors which, when non-hierarchical coupling is used, show good agreement with the experimental data for HD(
v = 1,
J = 2). This result indicates that the HD(
v = 1) hyperfine constants are close enough in magnitude to those of the ground state that our experimental sensitivity does not allow us to observe the vibrational dependence. The poor agreement between the experimental data and the hierarchical calculation is expected because the hydrogen and deuterium coupling constants are similar in magnitude; a hierarchical coupling scheme describes the system well when one
nucleus couples much more strongly to the molecular rotation than does the other.
In Fig. 5 we display the A(2)0 alignment moments for both the rotation and the deuterium nuclear spin over the time range from 0 to 20 μs following the initial alignment of the HD molecule. As the molecular rotation becomes depolarized, the deuterium nuclear spin becomes aligned. We find that the strongest deuterium alignment occurs near 3 μs with a value of A(2)0 = −0.271.
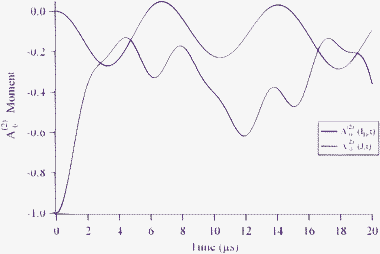 |
| | Fig. 5 The A(2)0 alignment parameter for the HD rotation and deuterium nuclear spin. Maximal alignment for ID occurs with a value of A(2)0 = −0.271 near approximately 3 μs following optical preparation of HD(v = 1,J = 2, MJ = 0). | |
The experiments we have described concern linearly polarized light, for which the rotational angular momenta can only be aligned. Briefly, we consider the excitation of HD with circularly polarized light for which the rotational angular momenta can also be oriented. In particular, the HD molecules can be prepared in the (v = 1, J = 2, MJ = 2) state using circularly polarized light in SRP excitation step, in the same manner14 as was previously reported for H2(v = 1, J = 2, MJ = 2). We calculate the time dependence of the MJ sublevel expectation values for the rotation and nuclei using eqn (10) and display the results in Fig. 6. The sum of the projections of the three angular momenta is constant and is equal to the initially prepared projection, 〈MJ〉 + 〈MH〉 + 〈MD〉 = 2. We see that the polarization of the H and D nuclei reach 60% of their maximal values at times of about 2 and 4.5 μs, respectively, producing highly polarized H and D nuclei.
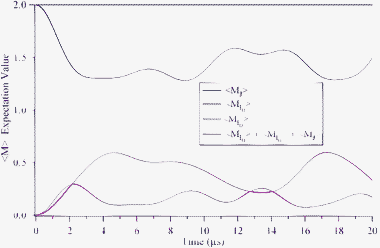 |
| | Fig. 6 Time dependence of 〈MI〉 for the rotational and nuclear spin angular momenta along with their sum, following optical preparation of oriented HD in the (v = 1, J = 2, MJ = 2) level. | |
We demonstrate the ability to monitor the degree of molecular rotational and nuclear polarization on a timescale of at least 13 μs (and longer times should be feasible). The treatment we have presented above is quite general. It applies to other J,MJ levels as well as other molecules that have different hyperfine coupling constants.
Acknowledgements
T. P. R. and D. S. gratefully acknowledge support from the GSRT (Greek General Secretariat for Research and Technology) Bilateral Collaboration grant Greece-USA 05NON-EU-68. N. C. M. B., D. J. M., and R. N. Z. thank the US National Science Foundation for their support of this work through Grant No. NSF CHE-0650414.
References
- R. J. Beuhler, Jr, R. B. Bernstein and K. H. Kramer, J. Am. Chem. Soc., 1966, 88, 5331 CrossRef CAS.
- P. R. Brooks and E. M. Jones, J. Chem. Phys., 1966, 45, 3449 CrossRef CAS.
- D. Van Den Ende and S. Stole, Chem. Phys., 1984, 89, 121 CrossRef CAS.
- H. Jalink, D. H. Parker, K. H. Meiwes-Broer and S. Stolte, J. Phys. Chem., 1986, 90, 552 CrossRef CAS.
- R. N. Zare, Ber. Bunsenges. Phys. Chem., 1982, 86, 422 CAS.
- Z. Karny, R. C. Estler and R. N. Zare, J. Chem. Phys., 1978, 69, 5199 CrossRef CAS.
- A. J. Orr-Ewing, W. R. Simpson, T. P. Rakitzis and R. N. Zare, Isr. J. Chem., 1994, 34, 95 CAS.
- D. Sofikitis, L. Rubio-Lago, M. R. Martin, D. J. Ankeny Brown, N. C. -M. Bartlett, A. J. Alexander, R. N. Zare and T. P. Rakitzis, J. Chem. Phys., 2007, 127, 144307 CrossRef.
- D. Sofikitis, L. Rubio-Lago, M. R. Martin, D. J. A. Brown, N. C.-M. Bartlett, R. N. Zare and T. P. Rakitzis, Phys. Rev. A, 2007, 76, 012503 CrossRef.
- S. Marinakis, G. Paterson, J. Kłos, M. L. Costen and K. G. McKendrick, Phys. Chem. Chem. Phys., 2007, 9, 4414 RSC.
- G. Paterson, S. Marinakis, M. Costen, K. G. McKendrick, J. Klos and R. Tobola, J. Chem. Phys., 2008, 129, 074304 CrossRef.
- G. O. Sitz and R. L. Farrow, J. Chem. Phys., 1994, 101, 4682 CrossRef CAS.
- S. A. Kandel, A. J. Alexander, Z. H. Kim, R. N. Zare, F. J. Aoiz, L. Bañares, J. F. Castillo and V. Sáez Rábanos, J. Chem. Phys., 2000, 112, 670 CrossRef CAS.
- N. C.-M. Bartlett, D. J. Miller, R. N. Zare, D. Sofikitis, T. P. Rakitzis and A. J. Alexander, J. Chem. Phys., 2008, 129, 084312 CrossRef.
- R. Altkorn, R. N. Zare and C. H. Greene, Mol. Phys., 1985, 55, 1 CAS.
- M. Rutowski and H. Zacharias, Chem. Phys., 2004, 301, 189 CrossRef; M. Rutowski and H. Zacharias, Chem. Phys., 2005, 310, 321 CrossRef (erratum).
- A. D. Rudert, J. Martin, W. B. Gao, J. B. Halpern and H. Zacharias, J. Chem. Phys., 1999, 111, 9549 CrossRef CAS.
-
D. Sofikitis, PhD Thesis, University of Crete, 2006.
-
N. F. Ramsey, Molecular Beams, Clarendon, Oxford, 1956 Search PubMed.
- E. R. Hudson, H. J. Lewandowski, B. C. Sawyer and J. Ye, Phys. Rev. Lett., 2006, 96, 143004 CrossRef.
- S. Y. T. Van de Meerakker, H. L. Bethlem and G. Meijer, Nature Physics, 2008, 4, 595 Search PubMed.
- M. S. Albert, G. D. Cates, W. Happer, B. Saam, C. S. Springer and A. Wishnia, Nature, 1994, 370, 199 CrossRef CAS.
- A. J. Orr-Ewing and R. N. Zare, Annu. Rev. Phys. Chem., 1994, 45, 315 CrossRef CAS.
- L. Rubio-Lago, D. Sofikitis, A. Koubenakis and T. P. Rakitzis, Phys. Rev. A, 2006, 74, 042503 CrossRef.
- G. G. Balint-Kurti, A. J. Orr-Ewing, J. A. Beswick, A. Brown and O. S. Vasyutinskii, J. Chem. Phys., 2002, 116, 10760 CrossRef CAS.
- W. R. Simpson, A. J. Orr-Ewing, T. P. Rakitzis, S. A. Kandel and R. N. Zare, J. Chem. Phys., 1995, 103, 7299 CrossRef CAS.
- W. R. Simpson, T. P. Rakitzis, S. A. Kandel, A. J. Orr-Ewing and R. N. Zare, J. Chem. Phys., 1995, 103, 7313 CrossRef CAS.
- W. E. Quinn, J. M. Baker, J. T. LaTourrette and N. F. Ramsey, Phys. Rev., 1958, 112, 1929 CrossRef CAS . The sign of d2 is incorrectly stated as positive in the abstract but negative in the text of this paper.
- R. N. Dixon, J. Chem. Phys., 1986, 85, 1866 CrossRef CAS.
|
| This journal is © the Owner Societies 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.