Self-assembly of a novel alternant amphiphilic poly(OPE-alt-TEO) copolymer: from nanowires to twist fibrillar architectures with molecular dimensions†
Pei
Wang
a,
Zhun
Ma
a,
Yan-Lian
Yang
b,
Qu-Li
Fan
c,
Xin-Fei
Yu
a,
Chen
Wang
b,
Wei
Huang
*c and
Lian-Hui
Wang
*a
aLaboratory of Advanced Materials, Fudan University, Shanghai, 200433, P. R. China. E-mail: wlhui@fudan.edu.cn; Fax: 86-21-55664575
bNational Center of Nanoscience and Technology, Beijing, 100080, P. R. China
cInstitute of Advanced Materials, Nanjing University of Posts & Telecommunications, Nanjing, 210003, P. R. China. E-mail: wei-huang@njupt.edu.cn
First published on 31st October 2008
Abstract
A novel alternant amphiphilic polymer poly[1,4-bis(phenylethynyl)-2,5-bis(hexyloxy)benzene-alt-tetra(ethylene oxide)] was prepared. Atom force microscope (AFM) images showed that the molecular self-assembly morphologies changed from molecular nanowires to twist fibrillar architectures with the increase of the solution concentrations. Short and thin wires formed in dilute solution, while large bundles developed in relatively concentrated ones, shown by fluorescence microscope images. The photoluminescence (PL) spectra of the corresponding films indicate a self-assembly process of the polymers under slow solvents evaporation. Coplanar aggregation was confirmed through PL and photoluminescence excitation (PLE) spectra. Furthermore, the self-assembly process in polymer bulk was studied by wide-angle X-ray diffraction. To the best of our knowledge, it is the first time to reveal the change of the molecular morphologies with the altering concentration for the alternant amphiphilic conjugated polymers.
Introduction
The ability to dictate the three-dimensional shape and ordering of supramolecular structures by controlling the architecture of molecular building blocks has been demonstrated on many occasions.1–5Self-assembly behavior of block copolymers have been proved as a powerful route towards supramolecular objects with novel architectures, functions and properties.6–9 Especially, rod–coil block copolymers, which consist of conjugated segments connected with flexible segments, have been the subject of extensive interest due to their electroactive and optical properties.10 So far, the syntheses of amphiphilic rod–coil block copolymers containing the following conjugated oligomers have been reported: oligothiophene, oligo(p-phenylene), oligo(p-phenyleneethynylene), oligo(fluorene) and oligo(p-phenylenevinylene).11–17 Zonal, spherical, cylindrical, and lamellar aggregates have been found in these polymers, and a fine balance of π–π stacking interactions, multiple directional hydrogen bonds, entropic contributions, hydrophobic interactions, and steric constraints controlled by chemical architecture required to promote such self-assembling behavior are reasonably well established. However, compared with conventional coil–coil block copolymers, there has been limited success in control over the morphology change of conjugated rod–coil block copolymers through systematic variation of the chemical composition and/or utilization of external stimuli such as temperature, time and solvents.18 Our group is interested in this area because the self-assembling ability of the rod–coil copolymers represents an attractive approach to organize functional species into nanometer domains.19,20 This research area offers an opportunity to synthesize functional polymers with well-defined supramolecular architectures.In this paper, we designed and synthesized an alternant polymer which combined a strong polar tetra(ethylene oxide) (TEO) group with a fixed apolar oligo(p-phenyleneethynylene)s (OPEs) unit. Its alternate structure is distinct from conventional rod–coil di- or tri-block copolymers. OPEs and its derivatives are one of the main classes of molecules that have been proposed and demonstrated for their strong π–π stacking interactions and apolar units.13,14 We demonstrated that supramolecular nanowires were constructed viaself-assembly when a dilute solution of this amphiphilic rod–coil polymer was dropped onto the substrate. And most interestingly, these self-assembly structures could alter from nanowires to twist fibrillar architectures by increasing polymer concentration. Correspondingly, photoluminescence (PL) spectra also varied with the solution concentration, which attributed to a coplanar aggregation. These studies provide a new insight into the relationship between morphologies and concentrations, and make these alternant block copolymers more attractive for photoelectrical devices in the future.
Experimental
The polymer poly[1,4-Bis(phenylethynyl)-2,5-bis(hexyloxy)benzene-alt-tetra(ethylene oxide)] [poly(OPE-alt-TEO), P1] was synthesized by a Pd-catalyzed coupling as described in the ESI† (Scheme 1).21,22P1 was soluble in common organic solvents such as chloroform, toluene, and tetrahydrofuran (THF), but insoluble in water. Gel permeation chromatography (GPC) analysis indicated that the polymer had a number-average molecular weight (Mn) of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1, with a polydispersity index of 2.94. The optical properties of P1 both in dilute solution and thin film state, were investigated by absorption and photoluminescence (PL) spectra. Atom force microscope (AFM) images illustrated the detailed morphologies of this polymer under different conditions.
800 g mol−1, with a polydispersity index of 2.94. The optical properties of P1 both in dilute solution and thin film state, were investigated by absorption and photoluminescence (PL) spectra. Atom force microscope (AFM) images illustrated the detailed morphologies of this polymer under different conditions.
![Synthesis route of poly [1,4-bis(phenylethynyl)-2,5-bis(hexyloxy)benzene-alt-tetra(ethylene oxide)] (P1).](/image/article/2009/CP/b810630d/b810630d-s1.gif) | ||
| Scheme 1 Synthesis route of poly [1,4-bis(phenylethynyl)-2,5-bis(hexyloxy)benzene-alt-tetra(ethylene oxide)] (P1). | ||
Solutions with different concentrations (10−7 mg mL−1 as dilute one, 10−3 mg mL−1 as semi-dilute one and 10−1 mg mL−1 as concentrated one) of P1 in toluene were prepared and filtered. Molecular self-assembly was accomplished by applying one drop of dilute or semi-dilute solution onto a freshly cleaved highly orientated pyrolytic graphite (HOPG) surface and allowing the solvent to evaporate in air at room temperature. The HOPG plates were covered with 50 mL beakers to protect their surfaces from dust. This may also help create a relatively confined space and make the evaporation of solvents slower as compared to that in an open-air environment. For the measurement of fluorescence microscopy, semi-dilute and concentrated toluene solutions were used to cast the films. Spin coating was carried out at the rate of 500 rpm for first 10 s, followed by rotated at the rate of 1500 rpm for 30 s.
Results and discussion
A Self-assembly properties
In order to investigate the detailed microcosmic aggregation structures, AFM topographical images were recorded under different concentrations. The AFM image in Fig. 1(a) showed micrometer-long nanowires deposited on graphite surface from a dilute solution of OPE-alt-TEO with an average width of 10 nm. From the enlarged AFM image in Fig. 1(b), a nanowire can be clearly measured with a height of 1.2 ± 0.2 nm.23 Indeed, symmetric compounds with short TEO groups are found to organize easily into fibrillar assemblies.11 Considering the convolution of the AFM tips (tip radius = 10 nm), the true average width of the nanowires should be 1–2 nm, which is in agreement with the length of the OPE segments. Compared the present nanowire height with the length of the alkyl side chains in the 1,4-bis-heptyloxy-2,5-bis-phenylethynylbenzene units,24 it is feasible to propose the architecture of the wires with OPE segments packed parallel to each other with their long molecular axis perpendicular to the wire axis through π–π interactions (A tentative packing structure of the polymer is shown in Fig. 1(b)). Obviously, the length of the nanowire is much longer than a single polymer chain. It shall consist of many polymer chains which are packed together by π–π interactions among OPE segments.24 Meanwhile, a lot of spherical particles randomly dispersed on HOPG surface as shown in Fig. 1, which was a common feature observed in analogous polymers.24–26 It was explained that the liquid flow dragged solute toward the rim of a droplet and pinned the contact line, which defined the round shape after the solvent drying out, because the formation of such droplets usually with a circular shape could minimize surface energy. | ||
| Fig. 1 AFM phase images of P1 droplet from toluene on HOPG surface: (a) dilute solution, 10−7 mg mL−1, the image size is 5 × 5 μm2. (b) Magnified AFM image of the region indicated by the black square in Fig. 1(a), the image size is 1.26 × 1.26 μm2. (c) Concentrated solution, 10−3 mg mL−1, the image size is 5 × 5 μm. (d) High resolution image for self-assembly of P1 from semi-dilute solutions, 10−3 mg mL−1, the image size is 1.0 × 0.5 μm2. (e) Height profile of the section chosen in Fig. 1(c). | ||
In Fig. 1(c), fibril-like architectures self-assembled on graphite surface by depositing semi-dilute solutions of P1, with the width distribution from 25 nm to around 65 nm and the corresponding height from 10 nm to 15 nm. The fibrillar morphology of similar molecular system have been reported,15 however, such a long-distance linear arrangement of nano-scale fibrillar structure has never been found for these alternant amphiphilic molecules containing hydrophilic and hydrophobic segments.13,14,27 Loop structures were clearly discriminated in these fibrillar architectures in Fig. 3(d). We discretionarily selected one section to investigate the detailed configuration of these twist fibrillar structures. Fig. 1(e) is height profile of the section chosen from Fig. 1(c), it showed a wave-shaped curve, which represented a regular undulate period about 40 nm width, but the undulate height varying from 2.5 nm to 5 nm. Based on these results, it can be concluded that this helix structure should be attributed to a result of different twists. These helix structures might be fabricated by the intermolecular π–π stacking of the OPE segments with slight incline between the neighbors and/or the intertwist of those nanowires originally existed in the dilute solution.28,29 And such a twist can occur if compression is slightly nonaxial, and the stress is directed at an angle, and a deformation releases the stress associated with the transversal component of the biaxial compression.30 Thus, the different dimensions of fibers should be due to different numbers of nanowires that twist together.
Moreover, comparing the fluorescence microscope images in Fig. 2, we found that morphologies and dimensions of these nano-scale thread structures could be altered with the solution concentrations. In order to gain more information of the correlation between the spectra and the self-assembly structures, the corresponding PL spectra of the aggregate in films were also investigated. Two samples were dropped onto clean quartz glass as substrates from the polymer toluene solutions, and the other two were prepared by spin-coating the polymer solution on substrate. All the molecules emitted strong green fluorescence, which originated from the aggregated OPE block. The spin-coating samples exhibited random globelet structures. The corresponding PL spectra showed two peaks at 496 nm and 532 nm in Fig. 2(b). However, images of dropping samples showed a more regulated molecular arrangement, as illustrated in Fig. 2(c) and (d), respectively. Short and thin wires were shaped in semi-dilute polymer solution, while large bundles were constructed from the concentrated one. It was also demonstrated that the dimension of these fibrillar self-assembly structures changed with the solution concentrations, and formed only through a slow solvent evaporation. At the same time, only one emission peak at 532 nm in Fig. 2(d) represented a more regular homogeneous state. And according to the literature report,31 a segmented donor–acceptor polyimide with hexa(ethylene oxide) as coil block on the backbone, are long enough to fold back on themselves, aided by intrachain aromatic charge–transfer interaction and metal–ion-complexation. Similarly, the alternant TEO spacers increase the possibility of the rod segments (OPEs) folding back with each other. Therefore, such corresponding spectral features are expected for π-stacked chromophores that occur as chain folding-back formation. It is worth noting that this concentration dependence of formed chain morphologies is common in polymeric system but this is the first report on an alternant rod–coil amphiphilic polymer. The fundamental of such a dependence is attributed to the interaction and aggregation of thin poly(OPE-alt-TEO) nanowires which exist in dilute solution lead them to twist and form helical fibers from semi-dilute solution as well as extending to many bundles of larger fibers from concentrated solution.
 | ||
| Fig. 2 Fluorescence microscopy images of P1 prepared by spin-coating from toluene solution of 10−3 mg mL−1 (a), 10−1 mg mL−1 (b); and dropped from the toluene solution of 10−3 mg mL−1 (c), 10−1 mg mL−1 (d). | ||
B Optical properties
Excitation and emission fluorescence spectra in toluene for these rigid–flexible alternant block copolymers are presented in Fig. 3. According to the report of Bunz,32 the excitation spectra are employed instead of the UV spectra. The dilute solution with 10−7 mg mL−1 concentration has almost identical excitation spectra, containing three absorption bands with peaks at 274, 287, and 366 nm, respectively.33 The peak at 366 nm is red shifted about 44 nm compared with that of (1,4-diphenylethynylene)benzene, due to the electron-donating effect of the alkoxy groups. The corresponding PL band at 410 nm referred to chromophores as a single OPE unit. This specific peak was also observed by existence of the donating alkyloxy substituents which increased the band gap of the polymer by lowering the HOMO.34 And on the same spectrum, emission peak at 434 nm should be due to the coplanar structure.32 Two broad absorption bands with peaks at 313 and 448 nm and the corresponding emission peak which red-shifted to 476 nm were observed when the concentration increased to 10−3 mg mL−1. In the previous literature report, Bunz has shown that absorption and emission spectra of 1, 4-bis(phenylethynyl)benzene red-shifted more than 60 nm in solid state due to a co-planar aggregation.32 Here, this large red-shifted emission peak at 476 nm with a shoulder at 520 nm in a semi-dilute solution were also supposed to be due to this aggregate formation. Simultaneously, the spectra of much more concentrated solution (10−1 mg mL−1) showed a new emission band at 522 nm with a more red-shifted absorption band at 479 nm, close to the film state due to more prominent aggregation. The previous literatures also reported that the aggregates displayed a significant loss of fine vibrational resolution. A similar feature in the solution state was revealed, which was attributed to the OPE units aligning in register to each other. Finally, it should be noted that a strong overlap between excitation and emission bands may be responsible for some distortion on the blue edge of the emission as a result of self-absorption. From these PL spectra, we concluded that 522 nm emission peak at the concentration of 10−1 mg mL−1 represented a more prominent aggregation over those initially-formed π–π stacked OPE segments. And fluorescence microscope images (Fig. 2) showed fibrillar assembles with an emission band at 532 nm. It is well known that the PL spectra in the solid-state are generally red-shifted slightly as in the solution states by more considerable coplanar effect, so the molecule morphology in solution referring to this peak at 522 nm was close as it in solid state.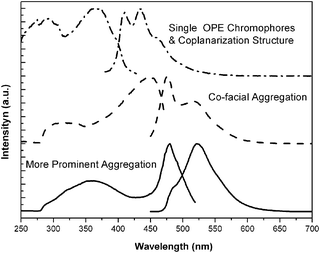 | ||
| Fig. 3 Excitation and emission spectra of P1 are obtained in toluene with three different concentrations 10−7 mg mL−1 (dash dot line), 10−3 mg mL−1 (dash line) and 10−1 mg mL−1 (solid line). The excitation spectra are acquired, from top to bottom, by detecting the emission at λem = 410, 476 and 522 nm. The corresponding emission spectra are obtained with excitation wavelength at λex = 368, 448 and 479 nm. | ||
C WAXD study
All of the P1assemblies in AFM images, fluorescence microscopy images and PL spectra demonstrate a self-assembly process from dilute solution to concentrated one. It is necessary for us to investigate the bulky properties of this polymer. Wide-angle X-ray diffraction is employed to examine these OPE-alt-TEO samples. In order to get a more ordered structure sample, we prepared a P1 sample by slow evaporation of toluene solution at 0 °C. The diffraction profiles of the polymer are shown in Fig. 4. In the spectra, both P1 powder and model compound show reflections at 2θ = 12.8°,35 corresponding to the d spacing at 6.88 Å, which is in excellent agreement with the periodicity expected from one phenylene–ethynylene repeat unit along the main polymeric chain,36 The pre-prepared sample of P1 did not show this diffraction peak in Fig. 4(A), however, in Fig. 4(B) a shoulder peak at the beginning of the diffraction image refers to this distance. The closest distance d1 between two main chains is 3.54 Å, which is a little smaller than the model compound (3.8 Å).35 The favorable packing of the π–π interaction accords with this distance, which demands for the OPE units to align in register with each other because of the alternant TEO groups. The distance (d2) 4.66 Å and 4.04 Å of the pre-prepared P1 samples are assigned to the TEO units.37 The side chain arrangement reinforces the natural preference of the conjugated backbones to assume a regular conformation between two OPE segments of different layers, d3 = 6.06 Å and 5.08 Å elucidate this distance with different arrangements of interdigitated configurations.36 Diffraction images also shows some weak signals, which should refer to some sub-diffraction peaks. Compared with the pre-prepared P1 sample diffraction image, P1 powder did not show any regular pattern above 20°, which indicates that the powder sample cannot form a self-assembly structure, and those regular structures should be constructed in solutions and evaporating process. | ||
| Fig. 4 (A) WAXS diffractions of (a) P1 powder, (b) P1 disposed sample, (c) model compound, and (d) OEO monomer. (B) Diffraction image of P1 disposed sample and (C) corresponding schematic representation of a proposed packing structure of the polymers in the solid state. | ||
Conclusions
In summary, the self-assembly structure of a tailor-made polymer—poly(OPE-alt-TEO)—may alter from nanowires to twist fibrillar architectures as a function of solution concentrations. The tendency to obtain the nanowires by the slow evaporation of the solvent and, consequently, the self-assembly process indicates that the growth of these nanostructures is a kinetically-dominant phenomenon. Photophysical behaviors were also investigated, and the corresponding morphology changes in the width and the orientation of these anisotropic molecular assemblies with different solution concentrations will render them candidates as molecular nanowires in molecular electronic devices. More detailed and systematic research on the morphologies of these amphiphilic polymers with different molecular weight and relative molar ratio of polar segment to apolar one is in progress, and will be reported elsewhere.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China under Grants 60537030, 90406021, 20504007 and 30425020, as well as Shanghai Leading Academic Discipline Project, Project No. B113. The authors thank Dr Ting-Cheng Li for providing WAXS data of P1.References
- S. I. Stupp, V. LeBonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstutz, Science, 1997, 276, 384 CrossRef.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H. W. Spiess, S. D. Hudsonk and H. Duank, Nature, 2002, 419, 384 CrossRef.
- X. Zeng, G. Ungar, Y. Liu, V. Percec, A. E. Dulcey and J. K. Hobbs, Nature, 2004, 428, 157 CAS.
- V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, H. Duan, S. N. Maganov and S. A. Vinogradov, Nature, 2004, 430, 764 CrossRef CAS.
- J. T. Chen, E. T. Thomas, C. K. Ober and G. P. Mao, Science, 1996, 273, 343 CrossRef CAS.
- S. Jain and F. S. Bates, Science, 2003, 300, 460 CrossRef CAS.
- Z. Li, E. Kesselman, Y. Talmon, A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Mater. (Weinheim, Ger.), 2001, 13, 1217 CrossRef CAS.
- S. Lu, T. X. Liu, L. Ke, D. G. Ma, S. J. Chua and W. Huang, Macromolecules, 2005, 38, 849.
- P. Leclère, M. Surin, P. Viville, R. Lazzaroni, A. F. M. Kilbinger, O. Henze, W. J. Feast, M. Cavallini, F. Biscarini, A. P. H. J. Schenning and E. W. Meijer, Chem. Mater., 2004, 16, 4452 CrossRef.
- B. S. Kim, D. J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333 CrossRef CAS.
- W. Y. Huang, S. Matsuoka, T. K. Kwei, Y. Okamoto, X. Hu, M. H. Rafailovich and J. Sokolov, Macromolecules, 2001, 34, 7809 CrossRef CAS.
- W. Y. Huang, W. Gao, T. K. Kwei and Y. Okamoto, Macromolecules, 2001, 34, 1570 CrossRef CAS.
- M. Surin, D. Marsitzky, A. C. Grimsdale, K. Müllen, R. Lazzaroni and P. Leclère, Adv. Funct. Mater., 2004, 14, 708 CrossRef CAS.
- H. Wang, H. H. Wang, V. S. Urban, K. C. Littrell, P. Thiyagarajan and L. Yu, J. Am. Chem. Soc., 2000, 122, 6855 CrossRef CAS.
- H. Wang, W. You, P. Jiang, L. Yu and H. H. Wang, Chem.–Eur. J., 2004, 10, 986 CrossRef CAS.
- K. Li and Q. Wang, Chem. Commun., 2005, 38, 4786 Search PubMed.
- S. Lu, Q. L. Fan, S. J. Chua and W. Huang, Macromolecules, 2003, 36, 304 CrossRef CAS.
- S. Lu, Q. L. Fan, S. Y. Liu, S. J. Chua and W. Huang, Macromolecules, 2002, 35, 9875 CrossRef CAS.
- U. Ziener and A. Godt, J. Org. Chem., 1997, 62, 6137 CrossRef.
- C. Z. Zhou, T. Liu, J. M. Xu and Z. K. Chen, Macromolecules, 2003, 36, 1457 CrossRef CAS.
- P. Samorí, N. Severin, K. Müllen and J. P. Rabe, Adv. Mater. (Weinheim, Ger.), 2000, 12, 579 CrossRef CAS.
- P. Samorí, V. Francke, K. Müllen and J. P. Rabe, Chem.–Eur. J., 1999, 5, 2312 CrossRef CAS.
- J. Xu, C. Z. Zhou, L. H. Yang, N. T. S. Chung and Z. K. Chen, Langmuir, 2004, 20, 950 CrossRef CAS.
- P. Samorí, I. Sikharulidze, Francke, K. Müllen and J. P. Rabe, Nanotechnology, 1999, 10, 77 CrossRef CAS.
- V. Francke, H. J. Räder, Y. Geerts and K. Müllen, Macromol. Rapid Commun., 1998, 19, 275 CrossRef CAS.
- W. Y. Yang, E. Lee and M. Lee, J. Am. Chem. Soc., 2006, 128, 3484 CrossRef CAS.
- P. H. Leclère, M. Surin, O. Henze, P. Jonkheijm, F. Biscarini, M. Cavallini, W. J. Feast, A. F. M. Kilbinger, R. Lazzaroni, E. W. Meijer and A. P. H. J. Schenning, J. Mater. Chem., 2004, 14, 1959 RSC.
- K. L. Genson, J. Holzmueller, M. Ornatska, Y. S. Yoo, M. H. Par, M. Lee and V. V. Tsukruk, Nano Lett., 2006, 6, 435 CrossRef CAS.
- S. Ghosh and S. Ramakrishnan, Angew. Chem., Int. Ed., 2004, 43, 3264 CrossRef CAS.
- M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. G. Garibay, J. Am. Chem. Soc., 2001, 123, 4259 CrossRef CAS.
- H. Li, D. R. Powell, R. K. Hayashi and R. West, Macromolecules, 1998, 31, 52 CrossRef CAS.
- C. A. Breen, S. Rifai, V. Bulovic and T. M. Swager, Nano Lett., 2005, 5, 1597 CrossRef CAS.
- T. Yasuda, T. Imase, Y. Nakamura and T. Yamamoto, Macromolecules, 2005, 38, 4687 CrossRef CAS.
- U. H. F. Bunz, V. Enkelmann, L. Kloppenburg, D. Jones, K. D. Shimizu, J. B. Claridge, H. C. Loye and G. Lieser, Chem. Mater., 1999, 11, 1416 CrossRef CAS.
- J. F. Hulvat and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 366 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b810630d |
| This journal is © the Owner Societies 2009 |