DOI:
10.1039/B816161P
(Paper)
CrystEngComm, 2009,
11, 102-108
Open and closed copper chain coordination polymers with alternating ferromagnetic and antiferromagnetic interactions†
Received
15th September 2008
, Accepted 16th September 2008
First published on 8th October 2008
Abstract
Two copper(II) coordination polymers formulated {[Cu3(HL)2(MeOH)2(H2O)2]·2H2O}n (1) and {[Cu4(L)2(H2O)8]·5H2O}n (2) were obtained utilizing the polycarboxylate ligand 2-(bis(carboxymethyl)amino)terephthalic acid (H4L) and characterized by X-ray diffraction, FT-IR spectra, elemental analysis and EPR spectra. 1 crystallizes in P21/c space group and shows a closed 1D double zigzag chain composed of trinuclear building block [Cu3(HL)2(H2O)4]. 2 crystallizes in a chiral space groupP21 and exhibits an open chain structure with tetranuclear building block [Cu4(L)2(H2O)8]. Fitting the variable-temperature magnetic susceptibilities of two compounds gave g = 2.16, J = 4.25 cm−1, zJ′ = −0.88 cm−1 for 1, and g = 2.13, J1 = −2.61 cm−1, J2 = 2.97 cm−1, zJ′ = −0.12 cm−1 for 2, respectively, indicating the alternating ferromagnetic and antiferromagnetic interactions.
Introduction
More and more attention has been focused on the development of novel coordination polymers, due to the general and significant values such as for gas storage, catalysis, sensors, magnetic materials, host–guest chemistry, etc.1–5 To design such compounds, some known factors need to be mentioned. The topologies of diversified coordination polymers based on the molecular building block usually affected by the coordination geometry of the central metal ions, the organic ligands and even some subtle effects from solvent, template, concentration, temperature, counteranion, etc. So far, polycarboxylate compounds especially some with symmetric units serving as precursor ligands (or linkers) are extensively developing, which have been a class of recognized good candidates for assembling such coordination polymers.6–9 The preferred selection for them may be due to the diversity of coordination fashions of carboxylic group and their abundance, diversity, multifunctionality, and overall stability.10 Investigations also reveal that copper ions were relatively easy to be embedded in such system,11 owing to their diversity of coordination geometry and the special affinity to oxygen atoms. Up to now, the studies on magneto-structural relationship show that carboxylate bridged systems tend to be observed serving as a vital way for superexchange effects. Especially, various bridging modes such as syn–syn, syn–anti and anti–anti configuration and bridging tridentate to two metal centers, may lead to different magnetic interactions in paramagnetic metal ions.12,13 In contrast, polycarboxylate ligands with asymmetric structure are less utilized in constructing organic–inorganic coordination polymers.14
Attracted by the various frameworks and interesting magnetic behavior of polycarboxylate complexes, we introduced 2-(bis(carboxymethyl)amino)terephthalic acid (H4L) as a precursor ligand with the aim to explore some new magnetic systems. This ligand featuring a terephthalic acid adding an amino acetic acid at phenyl group, is first applied in the synthesis of coordination polymers, as shown in Scheme 1. Previous studies on this ligand lead to the thermodynamic aspect of some complexes without any definite structural information.15 Unlike most of the complexes containing carboxylate ligands, which were obtained under solvothermal conditions,16a intense environment is unfavorable for amino acetic compounds to assembly coordination polymers.16b,c Reacting with a copper salt under mild conditions, H4L gave rise to the coordination polymers {[Cu3(HL)2(MeOH)2(H2O)2]·2H2O}n (1) with a closed double zigzag chain and {[Cu4(L)2(H2O)8]·5H2O}n (2) with an open asymmetric chain. To our knowledge, these two chain models were rarely observed in previous Cu(II) complexes bridged by a carboxylate ligand. Both compounds have been characterized by X-ray diffraction analyses, variable temperature magnetic measurements, and X-band EPR analyses. In addition, the non-covalent force plays an important role in assembling the desired supramolecular structure.
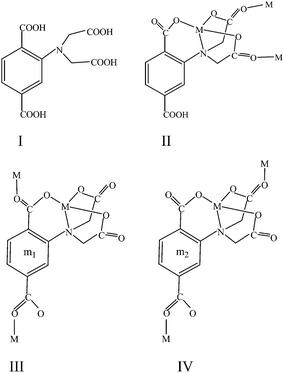 |
| | Scheme 1 Ligand H4L and its coordinate modes in complexes 1 (II) and 2 (III) and (IV) | |
Experiment
Materials and measurements
2-(bis(Carboxymethyl)amino)terephthalic acid (H4L) was prepared as in a similar method in literature.16a All other reagents were of A.R. grade obtained commercially and used without purification. Elemental analyses for carbon, hydrogen, and nitrogen were carried out on a Vario EL III elemental analyzer. The infrared spectrum was taken on an Bruker tensor 27 FT-IR spectrophotometer in the 4000–400 cm−1 region, using KBr pellets. The variable temperature magnetic susceptibility data for the crystalline samples of the two complexes were measured on a Quantum Design MPMS-7 SQUID magnetometer over a temperature range f 2–300 K with an applied field of 5 kG. Diamagnetic corrections were made with Pascal's constants for all of the constituent atoms. The EPR spectra were measured on a BRUKER EMX-6/1 EPR spectrometer.
Preparation
Synthesis of {[Cu3(HL)2(MeOH)2(H2O)2]·2H2O}n1.
Method I.
Methanol solution (10 mL) of Cu(NO3)2·3H2O (1.5 mmol, 0.362 g) was mixed with 10 mL water solution containing (0.5 mmol, 0.149 g) H4L and 0.73 mL N(CH3)4OH, after stirring the mixture for about 2 h, then filtering it, and the filtration was allowed to stand in air for two days. A green precipitate was isolated and resolved with methanol and HCl at pH 3.5. Standing again for about two weeks, a green rectangular single crystal was obtained. Yield 15%. Elemental analysis, calc. C26H24N2O24Cu3 (%) C 33.22, H 2.56, N 2.98. Found (%), C 33.29, H 2.60, N 2.87. FT-IR spectra (KBr pellets), 3563 (bs), 1584(vs), 1538(m), 1409(vs), 1314(m), 1279(m), 1248(s), 1177(m), 963(w), 933(w), 840(m), 775(s), 705(w), 653(w), 621 (w), 549(w).
Method II.
When replacing Cu(NO3)2 and N(CH3)4OH for CuSO4 with excess amount of p-chloride amino phenyl, the yield went up to 58%.
Synthesis of {[Cu4(L)2(H2O)8]·5H2O}n2.
The first product of 2 was prepared by H4L (0.25 mmol, 0.074 g) and KOH (1.5 mmol, 0.084 g) in 10 mL MeOH was added to (1.25 mmol, 0.300 g) Cu(NO3)2·3H2O in 10 mL MeOH, following by filtering and allowing the filtrate to stand in air. A dark green block crystal was isolated after one month. The second method is similar to 1. A solution of H4L (0.3 mmol, 0.089 g) and LiOH (0.50 mmol, 0.222 g) in 10 mL mixture of MeOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to a solution of CuCl2·2H2O (1 mmol, 0.170 g) and NaClO4 (5 mmol, 0.700 g), and then stir it for 2 h. Green square crystal was obtained after leaving in air for one month. Yield (41%), elemental analysis, Calc C24H38N2O28Cu4 (%) C 27.25, H 3.60, N 2.65. Found (%), C 27.16, H 3.64, N 2.72. FT-IR spectra (KBr pellets), 3394, 1581(vs), 1406(s), 1361(s), 1312(m), 1266(m), 962(w), 935(w), 771(m), 606(w).
:1) was added to a solution of CuCl2·2H2O (1 mmol, 0.170 g) and NaClO4 (5 mmol, 0.700 g), and then stir it for 2 h. Green square crystal was obtained after leaving in air for one month. Yield (41%), elemental analysis, Calc C24H38N2O28Cu4 (%) C 27.25, H 3.60, N 2.65. Found (%), C 27.16, H 3.64, N 2.72. FT-IR spectra (KBr pellets), 3394, 1581(vs), 1406(s), 1361(s), 1312(m), 1266(m), 962(w), 935(w), 771(m), 606(w).
X-Ray crystallography
.
Single crystals of 1 and 2 with suitable dimensions were mounted onto thin glass fibers, all the intensity data were collected at 294 K on a Bruker SMART 1000-CCD diffractometer (Mo Kα radiation, λ = 0.71073 Å) in ω scan modes. The structures were solved by the direct method followed by difference Fourier syntheses, and then refined by full-matrix least squares refinement on F2 using SHELXTL.17 All nonhydrogen atoms were refined with anisotropic parameters while H atoms were placed in calculated positions and refined using a riding model. Crystallographic data and refinement parameters are presented in Table 1.
| |
1
|
2
|
| Empirical formula |
C25H26N2O24Cu3 |
C24H34N2O28Cu4 |
|
M
r
|
913.10 |
1058.69 |
|
T/K |
294(2) |
294(2) |
| Wavelength/Å |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21 |
|
a/Å |
8.903(5) |
7.6207(10) |
|
b/Å |
23.565(14) |
20.686(3) |
|
c/Å |
8.164(5) |
11.6092(16) |
|
β/° |
111.178(9) |
97.296 |
|
V/Å 3 |
1597.1(17) |
1815.3(4) |
|
Z
|
2 |
2 |
| D/Mg·m−3 |
1.899 |
1.926 |
| Absorption coefficient/mm−1 |
2.083 |
2.420 |
|
F(000)
|
922 |
1064 |
| Crystal size/mm |
0.20 × 0.18 × 0.14 |
0.16 × 0.24 × 0.36 |
|
θ/° |
1.73 to 25.02 |
1.77 to 25.02 |
| Limiting indices |
−10 ≤ h ≤ 10, −28 ≤ k ≤ 20, −9 ≤ l ≤ 9 |
−6 ≤ h ≤ 9 −23 ≤ k ≤ 24 −11 ≤ l ≤ 13 |
|
R(int)
|
0.0427 |
0.0276 |
| Refinement method |
Full-matrix least -squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints /parameters |
2826/5/268 |
5660/10/568 |
| GOF on F2 |
1.090 |
1.015 |
|
R1, wR2 [I > 2σ(I)] |
0.0372, 0.1159 |
0.0280, 0.0644 |
|
R1, wR2 (all data) |
0.0456, 0.1212 |
0.0310, 0.0657 |
| Absolute structure parameter |
|
0.001(9) |
| Largest diff. peak and hole/e Å−3 |
1.488, −0.483 |
0.413, −0.534 |
Result and discussion
Structural description
{[Cu3(HL)2(MeOH)2(H2O)2]·2H2O}n (1).
1 was synthesized by two methods. The structure of 1 has been characterized by X-ray diffraction analysis. The ORTEP structure and crystallographic data were shown in Fig. 1 and Table 1, respectively. As illustrated in Fig. 1, 1 was assembled by trinuclear [Cu3(HL)2(MeOH)2(H2O)2] building blocks with HL3− as skeleton ligands and two H2O molecules in a crystal lattice. The trinuclear block with a C2 symmetric unit is composed of three Cu2+ ions, two trivalent anionic HL3−, MeOH and two H2O molecules.
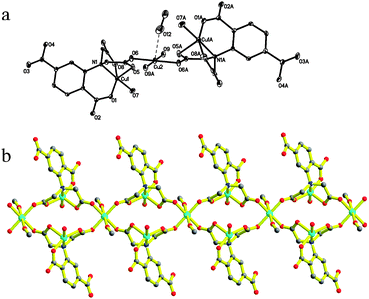 |
| | Fig. 1 The coordination of complex 1 with a numbering scheme (a), symmetry code, A: −x, 0.5 + y, 0.5 −z. (b) The diagram of complex 1. Carbon atoms are labeled with medium grey circles, red dots represent oxygen atoms, nitrogen atoms are blue and cyan represents the copper atoms. | |
The three copper centers are located in different coordinate environment. Cu1 and Cu3 ions were five-coordinated in square pyramids of NO4, and Cu2 was six-coordinated surrounded by an octahedron of O6. Two HL3− displayed the same tripodal coordination modes to seize the symmetry related Cu1 ions, in which the long arm acted by the phenyl carboxylate group offers one equatorial O atom. The residual chelating positions including two equatorial sites and one apical site were occupied by N1 and its two short arms contributing to two oxygen atoms, respectively. Then, a slightly distorted square pyramidal geometry around the centrosymmetric copper ion is completed along with the coordination of one H2O molecule. Cu1 had a deviation of about 0.1953 Å from its NO3 plane. The apical Cu1–O5 distance of 2.173(3) Å is 0.212 Å longer than the mean Cu–O distance of 1.951 Å in the equatorial plane, which is slightly shorter than 2.258 Å of the axial Cu–O distance in the square pyramidal geometry.13 Cu1–O and Cu1–N distances in the equatorial plane are in good agreement with those of the reported compounds.13,18 The residual two O atoms of the amino acetic groups bind to one intrablock and one interblock Cu2+ centers along their square plane, respectively. That is, each HL3− afforded one such carboxylic oxygen atom to link the adjacent trinuclear units through binding to the six coordinate copper(II) ions along their equatorial plane, meanwhile the two short arms at the N atom bridge both the intraunit and interunit Cu1 and Cu2viacarboxylate groups in a syn–anti fashion. One direct effect from the coordination mode is the formation of the perfect square plane CuO4, the other valuable result is that trinuclear units were linked into a 1D double zig-zag chain. The two axial positions of the Cu2 ion were occupied by two methanol molecules with a Cu–O distance of 2.568 Å, which is 0.610 Å longer than the mean Cu–O bond length of the equatorial plane. Obviously, the elongation should be owing to the Jahn–Teller effect of Cu2. The distances are 5.266, 5.338, 5.761 and 8.903 Å for Cu1⋯Cu2, Cu1⋯Cu2b, Cu1⋯Cu1b and Cu2⋯Cu2b, respectively. The terminal carboxylic group at the phenyl ring can not coordinate with copper(II) ions. The only role which it played in this compound is to link the neighboring chains with their O2 atoms viaH-bond and then extend to a 2D supermolecular framework as shown in Fig. 2. The O–H⋯O distance is 2.554 Å and the angle is 170.92°. The solvent water molecule was distributed in these tunnels staked by them.
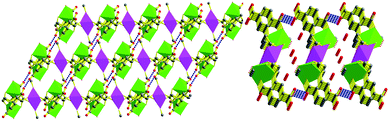 |
| | Fig. 2 The 2D supramolecular structure of 1 linked viaH-bonds viewed along the b (left, the water molecules were omitted for clarity) and the a axis (right). Carbon atom, medium grey; oxygen, red circle; copper atoms, green and purple polyhedra. | |
{[Cu4(L)2(H2O)8]·5H2O}n (2).
2 can also be obtained through two methods at room temperature. The coordination mode and the crystallographic data were exhibited in Fig. 3 and Table 1, respectively. The X-ray diffraction analysis reveals that 2 is also a 1D coordinate polymer. Different from 1, this complex presents an open single chain with a second building unit of [Cu4(L)2(H2O)8]. As illustrated in Fig. 3, two tetravalent anionic L4− acting as hexadentate ligands, four Cu2+ ions and eight coordinated H2O molecules are involved in the unit. The environments around the four copper(II) ions are all square pyramidal. Two are in O5 and half in NO4 environment. For convenient description, exhibited in Scheme 1, we entitle the two coordination modes m1 and m2 shown by the L4−, respectively. In m1, the carboxylic group bridges the Cu1 and Cu2 centers along the basal planes in a syn–anti fashion. The basal positions of Cu1 were occupied by two opposite H2O molecules and two oxygen atoms from the carboxylic group. A water molecule occupies the apical site of Cu1. Besides being linked by such three-atom bridge with Cu1, the Cu2 center was connected to one amino N atom along the basal plane and the two carboxylic oxygen atoms O5 and O3 along the basal and axial directions. The coordination of O20 (H2O) ultimately completed the Cu2 geometry. The environment composition of the Cu3 and Cu4 centers is similar to Cu1 and Cu2, respectively, except for the bridging group and bridging sites. L4− linking the Cu3 and Cu4 ions was named m2. In this mode, Cu3 and Cu4 were bridged together by the syn–anti bridged O11C22O22 (from amino acetic group) along the basal and axial directions, respectively. As a result, the square pyramidal Cu3 were rotated by 143.93° with respect to Cu1 geometry, while there is only an angle of 7.30° between Cu2 and Cu4 geometries. Furthermore, as a bridging unit, the delocalization within O11C22O22 made less distortion of Cu4 geometry than Cu2 geometry, which can be concluded from the crystallographic data. The change of bridging groups led to different inter-metallic distances as well, 4.816, 5.293, 9.086 and 8.980 Å for Cu1⋯Cu2, Cu3⋯Cu4, Cu2⋯Cu3 and Cu4⋯Cu1, respectively. Connected by the ending carboxyl groups, the second building blocks were extended to a 1D open chain structure.
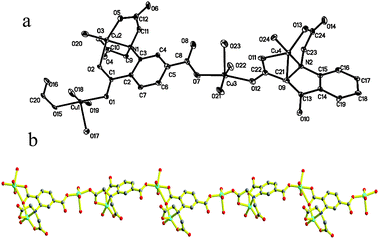 |
| | Fig. 3 The ORTEP diagram of complex 2 with a numbering scheme. Hydrogen atoms have been omitted for clarity (a), and ball and stick diagram of 2 (b). Carbon atom, medium grey; oxygen, red circle; nitrogen atoms, blue circles; copper atoms, cyan circle. | |
An interesting feature of 2 is the supramolecular framework. Neighboring chains arranged at parallel directions are linked together via O20⋯O4 H-bonds and form a 2D net framework with rectangular grids of about 23.72 × 7.62 Å. As illustrated in Fig. 4, the grids are so large that they can allow other parallel chains to cross through, and result in an interpenetrating structure. In fact, the 2D interpenetrating nets were also linked by two nonconvalent forces, one is O–H⋯O H-bonds formed between the coordinate water of the basal plane in Cu1 and the uncoordinated carboxyl oxygen atom at the first site of m2. All hydrogen bond distances and angles were listed in the ESI.† Another interaction is π⋯π stacking interactions. The interpenetration of the net and the existence of H-bonds shorten the distances and increase the overlap between the phenyl planes in m1 and m2. The calculation results show that upper overlapped m1phenyl rings with m2 have data as centroid–centroid 3.726 Å, inter-planar distance 3.681 Å and 3.531 Å, and dihedral angle 10.4°, respectively. The lower one with m2 has data as centriod–centriod 3.963 Å, inter-planar distance 3.557 Å and 3.793 Å and the same dihedral angle as the former. All the above data fall in the normal range for π⋯π stacking,19 suggesting the existence of the π⋯π stacking interactions shown in Fig. 5.
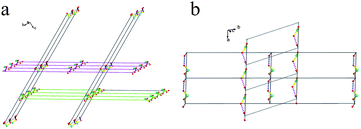 |
| | Fig. 4 The interpenetrating framework of 2, (a) and (b): The topology framework. The long lines represent one repeated Cu4 unit which interacts with the neighbouring parallel chain via an H-bond labelled with a short line. | |
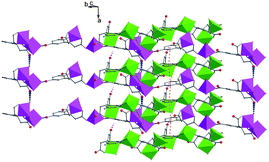 |
| | Fig. 5 The polyhedral framework of 2 with π⋯π stacking interactions labeled with orange dashes. | |
The thermal weight measurements for the two compounds were carried out in the range 25–700 °C. 1 lost weight gradually. From 25 to 157 °C, the weight loss of 15.85% agrees well with the loss of two lattice waters, two coordinate waters and two methanol molecules (15.97%). The following weight loss in the range of 157–251 °C with a value of 12.96% was attributed to the rest coordinate water molecules and the part of broken carboxylic acid groups. Upon heating, the mass experienced abrupt and slow loss from 251 to 477 °C which corresponds to the decomposition of the organic ligand with the sum value of 40.40% (calc. 40.76%). The thermal analysis for 2 showed that the lattice water and coordinate water molecules were lost successive until 180 °C with a value of 15.56% (less than the calc. 17.05% because of the slow breaking of the short Cu–O bond in the Cu3 basal plane). Higher than 180 °C the tetravalent organic ligand was decomposed rapidly with a max rate at 280 °C, the total weight loss is 46.84% and finished at about 419 °C.
EPR spectra and magnetic properties
To ensure the purity of the crystals, PXRD measurements were performed, and the result agrees well with their structures ESI).† The X-band EPR spectra of 1 and 2 are displayed in Fig. 6.
1 shows a three-line well resolved feature. The broad resonance lines represent a compromise between two lineshapes, which is typical of exchange-coupled extension.20 Temperature dependence behavior of 1 was shown at 120 and 298 K. The signal at 2974 G became weak and broad as the temperature decreased, meanwhile the signals at 3099 and 3236 G got sharper than room temperature and accompanied the vanishing of a weak signal at 3145 G, indicating the existence of a magnetic coupling interaction. Compared to 1, 2 shows little change between 298 and 120 K with a three resonance line spectra as well. Cooling the samples only made the three peaks sharpen. The spectra were simulated by assuming S = 1/2 for compounds 1 and 2, respectively. The fitting procedure yields gav = 2.16 and gav = 2.15 for 1 and 2, respectively.
The temperature dependence of χmT (the χm was used to express the molar magnetic susceptibilities of the chain system) for 1 is presented in Fig. 7. The χmT value is 1.35 cm3K mol−1 at 300 K, which is slightly larger than the expected value of 1.13 cm3K mol−1 for three uncoupled Cu2+ ions (S = 1/2, g = 2.0). Upon cooling, the χmT values remain about the same value in the range of 300–100 K, then the χmT begin to rise up to the maximum at 16 K, indicating ferromagnetic behavior. As the temperature lowers, the plots decrease abruptly until reaching the minimum of 0.79 cm3K mol−1 at 2 K, which may be a feature of antiferromagnetic behavior in this region.
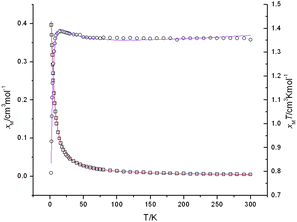 |
| | Fig. 7 Temperature dependence of the magnetic susceptibilities χmvs. T (empty circle) and χmTvs. T (empty quadrangle) plots of 1 under an applied field of 5 kOe with the best-fitting data as red and purple lines, respectively. | |
From the structure of 1, the 1D double zigzag chain was assembled by centrasymmetric trimeric blocks, Cu2 ion was linked with the surrounding Cu1 centers via carboxylic groups in syn–anti fashions and both along the equatorial direction. However, the Cu3 unit in each close ring was distinct. Besides the bridging fashion mentioned above, there is a syn–anti bridging mode along the equatorial and axial direction, respectively. As per the description above, such bridges link linear Cu3 fragment together and complete the double zigzag chain. In view of this case, two different exchange coupling constant J values were considered, the intra-trinuclear can be expressed by J = J12 = J23, the other interactions was considered as an inter-unit coupling constant zJ′, applying the molecular-field approximation.21 The coupling mode was illustrated in Scheme 2. The exchange Hamiltonian used the modified form H = −2J(S1·S2 + S2·S3. The corresponding molar magnetic susceptibilities are expressed by eqn (1) and (2)20b
| |  | (1) |
| | | χm = χtri/[1−χtri(2zJ′/Ng2β2)] | (2) |
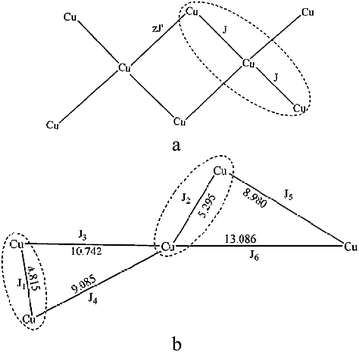 |
| | Scheme 2 The magnetic coupling modes of 1 (a) and 2 (b). | |
As a result, the fitting gave good agreement with the experimental plots with g = 2.16, J = 4.25 cm−1, zJ′ = −0.88 cm−1, Nα = 180 × 10−6 and R = 9.0 × 10−5(R = ∑│(χmT)exp − (χmT)calc│2/∑(χmT)exp2), in which the value of g fully complies with that obtained from EPR analysis. The different coupling constants can be interpreted according to the Hatfield theory,22 and the plot details under the condition of J/zJ′ > 1 correspond to the rule drawn by Coronado.23 The results revealed intrachain alternating ferromagnetic and antiferromagnetic interactions.24a According to the structural information, the magneto-structural relations could be concluded as follows. The carboxylate group bridging pair of copper ions at the equatorial planes forced the two Cu ions out of the related carboxylate plane (0.1992 Å for Cu1 and 0.1195 Å for Cu2), which therefore led to ferromagnetic interactions.24b However, the bridge from a basal position to an axial position resulted in antiferromagnetic interactions between the two copper ions owing to a nearly planar ⋯Cu⋯O⋯C⋯O⋯Cu⋯ unit (0.0439 Å for Cu1 and 0.0506 Å for Cu2). As a result, an alternating F/AF magnetic behavior was exhibited in this system. Syn–anticarboxylate bridged copper compounds tend to predict weak magnetic coupling. Although less than J = 12 for dominating ferromagnetic interactions in a copper compound containing a monoatomic bridge,25 the main ferromagnetic coupling in 1 is also important.
Plots of χmvs. T and χmTvs. T of 2 were represented in Fig. 8. Measurement at room temperature gave a χmT value of 1.71 cm3K mol−1 which slightly was larger than the four Cu2+ ions with a spin only value S = 1/2 (the theoretical value is 1.50 cm3K mol−1). The χmTvs. T plot shows that χmT is a nearly constant at 1.70 cm3K mol−1 with a tiny increase from 300 to 50 K, and then decreases slowly until 10 K, indicating the simultaneous presence of two independent magnetic interactions and reaching a similar balance in this temperature range. Reducing the temperature successively, the χmT value dropped abruptly to reach 1.08 cm3K mol−1 at 2 K, suggesting that antiferromagnetic coupling strived for the absolute advantage at low temperature. The observation is in concordance with the fact that complex 2 contains two different (μ-carboxyl)dicopper(II) fragments, Cu1–COO–Cu2 and Cu3–COO–Cu4. Although both units show five-coordinate tetragonal pyramidal geometry and are bridged by the three atomic carboxylates, respectively, the different positions of the bridging groups, result in distinct angles of the axial z orbitals in the two metal centers of the two fragments. This phenomenon is similar to the cases reported by Gutiérrez-Zorrilla and Lezama26 which also contain two kinds of independent magnetic interactions resulting from different structural conformations. An exact theoretical treatment of the experimental data for 2 is extremely complicated due to the highly asymmetric structure of this complex. Therefore, some approximations and evaluations are needed to obtain an operative expression for the magnetic susceptibilities. For reducing the number of adjustable parameters, all the interfragment magnetic interactions (J3, J4, J5 and J6) are not included owing to their long approaches. The Hamiltonian for this type of exchange is H = −2J1S1S2 − J2S3S4. The magnetic behavior of 2 can be regarded as the sum of the independent contributions of two dimmer fragments.26 Thus, the χm can be expressed as eq.(3)
| |  | (3) |
| |  | (4) |
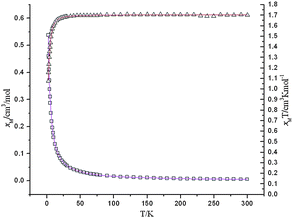 |
| | Fig. 8 Temperature dependence of the magnetic susceptibilities χmvs. T (empty triangle) and χmTvs. T plots (empty quadrangle) of 2 under an applied field of 5 kOe with the best-fitting data as purple lines and red lines, respectively. | |
Interchain exchange was accounted for the addition of a mean field correction term to eqn (3), the equation for the susceptibilities then has the form as eqn (4). J′ is the interchain exchange and z is the number of the nearest neighbors. The best fit of eqn (4) to the data in the range 2–300 K yielded g = 2.13, J1 = −2.61 cm−1, J2 = 2.97 cm−1, zJ′ = −0.12 cm−1 and R = 1.0 × 10−5 (R = ∑│(χmT)exp − (χmT)calc│2/∑(χmT)exp2). All magnetic parameters were allowed to vary freely in the fitting calculations. Consequently, the results confirm the assumption of F/AF behavior in 2.
Copper(II) complexes with F/AF alternating interactions are not often documented. To the best of our knowledge, most cases took place in some azide bridged copper(II) coordinate polymers.21,27,28 There are some common features for these compounds with one particular example excluded:28 (i) coexistence of two kinds of bridge modes EE and EO for the azide anion; (ii) closed chain with a dimeric copper unit; (iii) copper(II) ions alternatively bridged by double EE and EO arranged in cis-mode, being almost perpendicular to each other and therefore present in a μ-1,1,3- or μ2-1,3-azide. Besides these species, several O bridged or mixed O and bpm bridged copper chains23,25,29 have also been reported, characterized by an alternating chain with a dinuclear unit. All metal centers were located either in square pyramidal or octahedral geometry, whereas no hints of carboxylic acid (syn–anti) bridged copper(II) complexes with such character were observed previously in literature. The two compounds in this work are the first example of a double zig-zag chain and a rare case of open chains with alternating F/AF magnetic behavior
Conclusions
In conclusion, two copper coordination polymers have been obtained using several methods with a asymmetric polycarboxylate ligand and copper salt, realizing the closed double zig-zag chain which is unusually observed in copper chains reported previously, and an open asymmetric chain. The transformation from a closed to open structure not only changed the bridge group but shortened the intermetallic distances. Fitting of the temperature dependence of the magnetic susceptibility plots reveals that either the closed chain or the open one exhibited two magnetic coupling interactions within the chains, which offers two novel copper chain models with F/AF (ferromagnetic/antiferromagnetic) systems.
Acknowledgements
The authors gratefully acknowledge helpful discussion with Prof. Li-Cun Li. This work was supported by the National Natural Science Foundation of China (Grants 20631030 and 20425103), the NSF of Tianjin (Grant 06YFJZJC009000), the State Key Project of Fundamental Research of MOST (Grant 2007CB815305) and MOE (Grant 20060055039) of China
References
-
(a) S. Takamizawa, E. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 42, 4331 CrossRef CAS;
(b) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS;
(c) Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278 CrossRef CAS.
- C. D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS.
-
(a) H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844 CrossRef CAS;
(b) B. D. Chandler, A. P. Côté, D. T. Cramb, J. M. Hill and G. K. H. Shimizu, Chem. Commun., 2002, 1900 RSC.
-
(a)
R. L. Carlin, Magnetochemistry, Springer-Verlag, Berlin, 1986 Search PubMed;
(b)
O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed;
(c)
Molecular Magnets: Recent Highlights, ed. W. Linert and M. Verdaguer, Springer-Verlag, Wien, 2003 Search PubMed.
-
(a) B. X. Dong, J. Peng, C. J. Gómez-García, S. Benmansour, H. Q. Jia and N. H. Hu, Inorg. Chem., 2007, 15, 5933 CrossRef;
(b) S. Hermes, D. Zacher, A. Baunemann, C. Wöll and R. A. Fischer, Chem. Mater., 2007, 19, 2168 CrossRef CAS;
(c) C. Janiak, Dalton Trans., 2003, 2781 RSC.
- D. Cheng, M. A. Khan and R. P. Houser, J. Chem. Soc., Dalton Trans., 2002, 4555 RSC.
-
(a) S. K. Ghosh and P. K. Bharadwaj, Inorg. Chem., 2004, 43, 5180 CrossRef CAS;
(b) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef.
- K. Kanamori, K. Ino, H. Maeda, K. Miyazaki, M. Fukagawa, J. Kumada, T. Eguchi and K. Okamotos, Inorg. Chem., 1994, 33, 5547 CrossRef CAS.
- S. Dominguez, A. Mederos, P. Gili, A. Rancel, A. E. Rivero, F. Birito, F. L. Ioret, X. Solans, C. Ruiz-Perez, M. L. Rodriguez and I. Birito, Inorg. Chim. Acta, 1997, 255, 367 CrossRef CAS.
-
(a) K. E. Holmes, P. F. Kelly, S. H. Dale and M. R. J. Elsegood, CrystEngComm, 2005, 7, 202 RSC;
(b) B. Paul, B. Zimmermann, K. M. Fromm and C. Janiak, Z. Anorg. Allg. Chem., 2004, 630, 1650 CrossRef CAS;
(c) Y.-F. Zhou, F. L. Jiang, D. Q. Yuan, B. L. Wu, R. H. Wang, Z. Z. Lin and M. C. Hong, Angew. Chem., Int. Ed., 2004, 43, 5665 CrossRef CAS;
(d) P. Cheng, S. P. Yan, C. Z. Xie, B. Zhao, X.
Y. Chen, X. W. Liu, C. H. Li, D. Z. Liao, Z. H. Jiang and G. L. Wang, Eur. J. Inorg. Chem., 2004, 2369 CrossRef CAS;
(e) J. Yang, J. F. Ma, Y. Y. Liu, J. C. Ma, H. Q. Jia and N. H. Hu, Eur. J. Inorg. Chem., 2006, 1208 CrossRef CAS;
(f) R. Gheorghe, P. Cucos, M. Andruh, J. P. Costes, B. Donnadieu and S. Shova, Chem.–Eur. J., 2006, 12, 187 CrossRef CAS;
(g) P. Wang, C. N. Moorefield, M. Panzer and G. R. Newkome, Chem. Commun., 2005, 465 RSC.
-
(a) M. Murugesu, R. Clérac, C. E. Anson and A. K. Powell, J. Phys. Chem. Solids, 2004, 65, 667 CrossRef CAS;
(b) P. R. Levstein and R. Calvo, Inorg. Chem., 1990, 29, 1581 CrossRef CAS.
- M. L. Kirk, W. E. Hatfield, M. S. Lah, D. Kessissoglou, V. L. Pecoraro, L. W. Morgan and J. D. Petersen, J. Appl. Phys., 1991, 69, 6013 CrossRef CAS.
-
(a) Y. Y. Zhang, B.-Q. Ma, S. Gao, J. R. Li, Q. D. Liu, G. H. Wen and X. X. Zhang, J. Chem. Soc., Dalton Trans., 2000, 2249 RSC;
(b) Y. Yan, C. D. Wu, X. He, Y. Q. Sun and C. Z. Lu, Cryst. Growth Des., 2005, 2, 821 CrossRef.
-
(a) R. Krannich and E. Uhlig, Z. Anorg. Allg. Chem., 1973, 402, 285 CrossRef CAS;
(b) A. Martin and E. Uhlig, Z. Anorg. Allg. Chem., 1970, 376, 282 CrossRef CAS.
-
(a) Y. Q. Xu, D. Q. Yuan, B. L. Wu, L. Han, M. Y. Wu, F. L. Jiang and M. C. Hong, Cryst. Growth Des., 2006, 5, 1168 CrossRef;
(b) E. F. K. McCandlish, T. K. michael, J. A. Neal, E. C. Lingafelter and N. J. Rose, Inorg. Chem., 1978, 17, 1383 CrossRef;
(c) J. Sanchiz, S. Dominguez, A. Mederos, F. Brito and J. M. Arrieta, Inorg. Chem., 1997, 26, 4108 CrossRef.
-
(a)
G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structuture solution, Göttinen University, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick, SHELXL-97, Progrm for X-ray Crystal Structure Refinement, Göttingen University, Germany, 1997 Search PubMed.
-
(a) L. Antolini, L. P Battaglia, G. B. Gavioli, A. B Corradi, G. Grandi, G. Marcotrigiano, L. Menabue and G. C. Pellacani, J. Am. Chem. Soc., 1983, 105, 4327 CrossRef CAS;
(b) M. J. Roman-Alpiste, J. D. Martin-Ramos, A. Castineiras-Campos, E. Bugella-Allamirano, A. G. Sicilia-Zafra, J. M. Gonzalez-Perez and J. Niclos-Gutierrez, Polyhedron, 1999, 18, 3341 CrossRef.
-
(a) D. L. Reger, J. R. Gardinier, R. F. Semeniuc and M. D. Smith, Dalton Trans., 2003, 1712 RSC;
(b) J. C. Liu, G. C. Guo, J. S. Huang and X. Z. You, Inorg. Chem., 2003, 42, 235 CrossRef CAS.
-
(a)
A. Bencini, D. Gatteschi. EPR of exchange coupled systems. Springer-Verlag. Berlin, Heildelberg, Germany, 1990, p. 39 Search PubMed;
(b) A. Tamayo, J. Casabó, L. Escriche, P. González, C. Lodeiro, A. C. Rizzi, C. D. Brondino, M. C. G. Passeggi, R. Kivekäs and R. Sillanpä, Inorg. Chem., 2007, 46, 5665 CrossRef CAS.
- E. Q. Gao, S. Q. Bai, C. Fe. Wang, Y. F. Yue and C. H. Yan, Inorg. Chem., 2003, 42, 8456 CrossRef CAS.
- D. K. Towle, S. K. Hoffmann, W. E. Hatfield, P. Singh and P. Chaudhuri, Inorg. Chem., 1988, 27, 394 CrossRef CAS.
- J. J. Borras-Almenar, E. Coronado, J. Curely, R. Georges and J. C. Gianduzzo, Inorg. Chem., 1994, 33, 5171 CrossRef CAS.
-
(a) A. Rodríguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Chem.–Eur. J., 2001, 7, 627 CrossRef CAS;
(b) E. Colacio, J. M. Dominguez-Vera, J. P. Costes, R. Kivekäs, J. P. Laurent, J. Ruiz and M. Sundberg, Inorg. Chem., 1992, 31, 774 CrossRef CAS.
- P, S. Mukherjee, T. K. Maji, G. Mostafa, J. Ribas, M. S. E. Fallah and N. R. Chaudhuri, Inorg. Chem., 2001, 40, 928 CrossRef CAS.
- S. Reinso, P. Vitoria, J. M. Gutiérrez-Zorrilla, L. Lezama, J. M. Madariaga and L. S. Felices, Amaia Iturrospe, Inorg. Chem., 2007, 46, 4010 CrossRef.
-
(a) G. D. Munno, M. G. Lombardi, M. Julve, F. Lloret and J. Faus, Inorg. Chim. Acta, 1998, 282, 82 CrossRef;
(b) T. K. Maji, P. S. Mukherjee, S. Koner, G. Mostafa, J. P. Tuchagues and N. R. Chaudhuri, Inorg. Chim. Acta, 2001, 314, 111 CrossRef CAS;
(c) Z. Shen, J. L. Zuo, Z. Yu, Y. Zhang, J. F. Bai, C. M. Che, H. K. Fun, J. J. Vittal and X. Z. You, J. Chem. Soc., Dalton Trans., 1999, 3393 RSC;
(d) Y. S. You, C. S. Hong and K. M. Kim, Polyhedron, 2005, 2, 249 CrossRef;
(e) Y. S. Xie, Q. L. Liu, H. Jiang, C. X. Du, X. L. Xu, M. G. Yu and Y. Zhu, New J. Chem., 2002, 26, 176 RSC.
- H. Y. Shen, W. M. Bu, E. Q. Gao, D. Z. Liao, Z. H. Jiang, S. P. Yan and G. L. Wang, Inorg. Chem., 2000, 39, 396 CrossRef CAS.
-
(a) G. D. Munno, M. Julve, F. Lloret, J. Faus, M. Verdaguer and A. Caneschi, Inorg. Chem., 1995, 34, 157 CrossRef CAS;
(b) I. Vasilevesky, N. R. Rose, R. Stenkamp and R. D. Willett, Inorg. Chem., 1991, 30, 4082 CrossRef CAS;
(c) S. Dalai, P. S. Mukherjee, G. Rogez, T. Mallah, M. G. B. Drew and N. R. Chaudhuri, Eur. J. Inorg. Chem., 2002, 12, 3292 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond length and angles of 1 and 2 (Tables S1 and S2); H-bond data in 2 (Table S3); TGA and DTA analysis of 1 and 2 (Fig. S1); Experimental and simulated PXRD patterns for 1 and 2 (Fig. S2 and S3). CCDC reference numbers 656885 and 656886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b816161p |
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 