Inclusion of triphenylmethane derivatives by crown and linear O-containing molecules![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) : selective interactions and crystal structures†
: selective interactions and crystal structures†
Marina S.
Fonari
*a,
Yurii A.
Simonov
a,
Wen-Jwu
Wang
b,
Shang-Wei
Tang
b and
Eduard V.
Ganin
c
aInstitute of Applied Physics Academy of Sciences of Moldova, Academiei str., 5, MD-2028, Chisinau, R. Moldova. E-mail: fonari.xray@phys.asm.md; Fax: +373 22 72 58 87; Tel: +373 22 73 81 54
bDepartment of Chemistry Tamkang University, 151 Ying-Chuan Road, Tamsui, Taipei 25137, Taiwan (ROC). E-mail: wjw@mail.tku.edu.tw; Fax: +886-2-2620-9924; Tel: +886-2-26215656(2530)
cOdessa State Environmental University, Ministry of Education and Sciences of Ukraine, Lvovskaya str. 15, Odessa, 65016, Ukraine. E-mail: edganin@yahoo.com; Tel: +38-048-7852711
First published on 8th October 2008
Abstract
The sulfamide derivative of triphenylmethanol, 3-[hydroxy(diphenyl)methyl]benzenesulfonamide (H2NSO2Ph)Ph2COH was synthesized and, alongside with the parent triphenylmethanol and triphenylmethylamine, was investigated for selective interactions with crown ethers of different dimensionality (12-18-membered cycles). The molecule of 12-crown-4 (12C4) appeared to be the best candidate for Ph3COH, Ph3CNH2 and Ph3CNH3·NCS, while (H2NSO2Ph)Ph2COH forms the complex exclusively with 18-crown-6 (18C6). The triphenylammonia trifluoroacetate, Ph3CNH3·CF3COO, selectively forms the complex only with 2-methoxyethanol. The crystalline products of the compositions (Ph3COH)2·12C4, (Ph3CNH2)2·12C4, (Ph3CNH3·NCS)2·12C4, [(H2NSO2Ph)Ph2COH]2·18C6 and Ph3CNH3·CF3COO CH3OCH2CH2OH were obtained and studied by X-ray single crystal diffraction.
Introduction
Publication by Charles J. Pedersen of the synthesis of crown ethers and their property to form host–guest complexes with cations and comparatively small neutral guest molecules1,2 has initiated the search and investigation of new, functionally isomorphous to crown ethers host molecules,3–11 like cyclic cryptands, calixarenes and calixcrowns, cyclodextrines,12 and acyclic molecules like aromatic diols,8 arylureas,13,14 derivatives of triphenylmethane (tritane)7,9,14etc. The numerous studies revealed the low selectivity of triphenylmethanol (tritanol, Ph3COH) which binds in supramolecular complexes methanol, acetone, dimethyl formamide, dimethylsulfoxide, 1,4-dioxane, morpholine, piperidine, N-methylpiperazine, phenoxine,9,15tetrahydrofurane.16Triphenylsilanol Ph3SiOH, the precise analogue of triphenylmethanol, in its turn forms stable adducts with dimethylsulfoxide and 1,4-dioxane due to O–H⋯O and C–H⋯π(arene) interactions.17,18 Similarly, triphenylmethanaminium chloride forms an inclusion compound with acetone.19 Now we are witnesses to the growing numbers of successful application of bulky host molecules. Thus, using excellent inclusion properties of bulky bisphenols Csöregh and co-workers20 have stabilized carboxylic acid and ester molecules in a monomeric state for spectroscopic investigation. The recent research of Tohnai et al.21 has demonstrated the triphenymethylamine ability for an efficient and robust fabrication of [4 + 4] ion-pair clusters consisting of a wide range of sulfonic acids with an emphasis on the efficiency of the combination of sterically hindered triphenylmethylamine and sulfonate ions (complementarity in hydrogen bonding, steric effects of the substituents, acidity of the sulfonic acids). In 1998 Hayashi et al.22 showed on fluorosubstituted triphenylmethanol derivatives that the C–H⋯F(-C) interactions control the packing motif as well as the thermal stability of the crystal, in continuation in the hot CrystEngComm article Schollmeyer et al.23 demonstrated that the trityl alcohols bearing three bromine or three iodine atoms at the para-positions of the aromatic units, as well as the fluorosubstituted derivatives of triphenylmethanol may be a very useful platform for analysing OH⋯π and halogen–π interactions as driving forces in crystal packing.Surprisingly, apart from the two known examples of Ph3SiOH24 and Ph3CSH25 inclusion by 12-crown-4 (12C4), no systematic study of crown inclusion into the network of these bulky molecules has been carried out so far. This has motivated us to synthesize and study by X-ray diffraction the crystalline inclusion complexes of tritane derivatives with crown ethers of different dimensionality with an emphasis on the selectivity of interactions in these binary systems.
Experimental
Preparation of the crystalline inclusion compounds
It has been stated that spontaneous evaporation of solvents from the mixture of coronands I–III with triphenylmethanol (IVa), 1,1,1-triphenylmethanamine (IVb) and 1,1,1-triphenylmethanaminium thiocyanate (IVc) results in the crystalline molecular complexes of compositions (IVa)2.I (complex VI), (IVb)2.I (complex VII), (IVc)2.I (complex VIII), respectively (Scheme 1).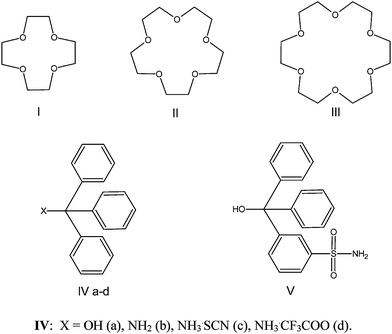 | ||
| Scheme 1 Formulae of the substances used. | ||
The precipitation of VI from methanol solution indicates its higher stability in comparison with the triphenylmethanol complex with methanol.9,15 The incorporation of the H-donor sulfamide group in the phenyl ring of IVa molecule yielding the 3-[hydroxy(diphenyl)methyl]benzenesulfonamideV drastically changes the mode of intermolecular recognition and results in the extraction of the largest 18C6 from the reaction mixture of I, II, III with the formation of the crystalline molecular complex of the composition (V)2.III (complex IX). In previously reported studies no selectivity in the interaction of neutral molecules with the titled crown ethers was registered.4 Moreover, the bulky di(benzenesulfonyl)amine HN(SO2C6H5)2 yields crystalline molecular complexes both with I26 and III.27–29 The replacement of the thiocyanate anion (IVc) by trifluoroacetate (IVd) dramatically changes the complexation process. From the methanolic mixture of I, II, III, compound IVd is reverted in an invariable form, while from the solution IVd–I–methanol–2-methoxyethanol the crystalline complex of triphenylmethanaminium trifluoroacetate with 2-methoxyethanol (compound X) is precipitated selectively. Thus, herein for the first time the selective interaction of bulky triphenylmethane derivatives with crown ethers is described with an emphasis on the possibility of crown ethers' separation30 similar to the procedure described earlier.31,32
The initial chemicals were used as received from Aldrich without further purification. The 1H NMR spectra were recorded with a Bruker AC 300 instrument at 300 MHz using tetramethylsilane as an internal reference. Thin-layer chromatography was conducted on Silufol plates with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 methanol–chloroform as eluent and ninhydrin as developer. The crown ethers appeared as gray spots against a pink background.
:8 methanol–chloroform as eluent and ninhydrin as developer. The crown ethers appeared as gray spots against a pink background.
Compound V, 3-[hydroxy(diphenyl)methyl]benzenesulfonamide: a mixture of 1,1′,1″-(chloromethanetriyl)tribenzene (2.788 g, 10 mmol) and chlorosulfonic acid (2.35 g, 20 mmol) was heated for 8 h at 150 °C and then cooled and poured onto 50 g of ice. The crystals were separated, placed into 40 ml of 25% aqueous ammonia, and heated with stirring for 12 h. The crystals that formed at 20 °C were washed with water, dried in air, and recrystallized from acetone–hexane (1:1). Yield: 1.80 g (53%), mp 188–190 °C, found, %: C 67.21; H 5.09; N 4.19; S 9.55, required for C19H17NO3S: C 67.24; H 5.05; N 4.13; S 9.45. 1H NMR (CDCl3), δ, ppm: 7.27m, 7.44m (14H, CH).
Compound VI, triphenylmethanol–1,4,7,10-tetraoxacyclododecane, 2:1:triphenylmethanol (260 mg, 1 mmol) and a mixture of 1,4,7,10-tetraoxacyclododecane (176 mg, 1 mmol), 1,4,7,10,13-pentaoxacyclopentadecane (220 mg, 1 mmol) and 1,4,7,10,13,16-hexaoxacyclooctadecane (264 mg, 1 mmol) were dissolved in methanol (10 ml) The colorless, transparent crystals were obtained by slow evaporation of solvents at room temperature for several days. Yield: 593 mg (85%), mp 147–148 °C, found, %: C 79.28; H 6.94, required for C46H48O6: C 79.33; H 6.90. 1H NMR (CDCl3), δ, ppm: 1.62 (s 2H, OH), 3.70 (s, 16H, CH2), 7.24–7.35 (m, 30H, CH).
Compound VII, 1,1,1-triphenylmethanamine–1,4,7,10-tetraoxacyclododecane, 2:1:1,1,1-triphenylmethanamine (259 mg, 1 mmol) and a mixture of 1,4,7,10-tetraoxacyclododecane (176 mg, 1 mmol), 1,4,7,10,13-pentaoxacyclopentadecane (220 mg, 1 mmol) and 1,4,7,10,13,16-hexaoxacyclooctadecane (264 mg, 1 mmol) were dissolved in a mixture of benzene/diethyl ether (1:2, 5 ml). The colorless, transparent crystals were obtained by slow evaporation of solvents at room temperature for several days. Yield: 495 mg (71%), mp 94–95 °C, found, %: C 79.51; H 7.25; N 4.03, required for C46H50N2O4: C 79.53; H 7.24; N 4.05. 1H NMR (CDCl3), δ, ppm: 1.86 (s, 4H, NH), 3.70 (s, 16H, CH2), 7.20–7.32 (m, 30H, CH).
Compound VIII, triphenylmethanaminium thiocyanate–1,4,7,10-tetraoxacyclododecane, 2:1:triphenylmethanaminium thiocyanate (318 mg, 1 mmol) and a mixture of 1,4,7,10-tetraoxacyclododecane (176 mg, 1 mmol), 1,4,7,10,13-pentaoxacyclopentadecane (220 mg, 1 mmol) and 1,4,7,10,13,16-hexaoxacyclooctadecane (264 mg, 1 mmol) were dissolved in methanol (15 ml). The colorless, transparent crystals were obtained by slow evaporation of solvents at room temperature for several days. Yield: 650 mg (80%), mp 170–172 °C, found, %: C 70.85; H 6.48; N 6.93; S 7.89, required for C48H52N4O4S2: C 70.90; H 6.45; N 6.89; S 7.93. 1H NMR (CDCl3), δ, ppm: 3.70 (s, 16H, CH2), 7.09–7.45 (m, 30H, CH).
Compound IX, 3-[hydroxy(diphenyl)methyl]benzenesulfonamide–1,4,7,10,13,16-hexaoxacyclooctadecane, 2:1:3-[hydroxy(diphenyl)methyl]benzenesulfonamide (340 mg, 1 mmol) and a mixture of 1,4,7,10-tetraoxacyclododecane (176 mg, 1 mmol), 1,4,7,10,13-pentaoxacyclopentadecane (220 mg, 1 mmol) and 1,4,7,10,13,16-hexaoxacyclooctadecane (264 mg, 1 mmol) were dissolved in a mixture of acetone/hexane (1:1, 15 ml). The colorless, transparent crystals were obtained by slow evaporation of solvents at room temperature for several days. Yield: 835 mg (98%), mp 174–176 °C, found, %: C 63.71; H 6.24; N 2.99; S 6.85, required for C50H58N2O12S2: %: C 63.67; H 6.20; N 2.97; S 6.85. 1H NMR (CD3OD), δ, ppm: 3.62 (s, 24H, CH2), 7.27–7.44 (m, 28H, CH).
Compound X, triphenylmethanaminium trifluoroacetate–2-methoxyethanol, 1:1:triphenylmethanaminium trifluoroacetate (373 mg, 1 mmol) (373 mg, 1 mmol)) and 1,4,7,10-tetraoxacyclododecane (176 mg, 1 mmol) were dissolved in a mixture of methanol/2-methoxyethanol (1:1, 10 ml). The colorless, transparent crystals were obtained by slow evaporation of solvents at room temperature for several days. Yield: 380 mg (85%), mp 155–156 °C, found, %: C 64.13; H 5.80; N 3.19; F 12.64, required for C24H26F3NO4: %: C 64.18; H 5.83; N 3.12; F 12.68. 1H NMR (DMSO), δ, ppm: 2.49 (s, 3H, CH3), 3.49 (s, 4H, CH2), 7.05–7.44 (m, 30H, CH).
Crystallographic studies
The X-ray intensity data for VI–X were recorded at room temperature on a Bruker SMART 1000 CCD area detector diffractometer employing graphite monochromatized Mo Kα radiation (λ = 0.71073 Å) in ϕ and ω scan mode. Final unit cell dimensions and positional data were obtained and refined on an entire data set. Integration and scaling resulted in a data set corrected for Lorentz and polarization effects using DENZO.33 The scaling as well as global refinement of the crystal parameters were performed by SCALEPACK.33 The absorption correction for VI, VIII, IX was applied using SADABS.34 The structure solution and refinement proceeded similarly using SHELX-97 program package35 for all structures. Direct methods yielded all non-hydrogen atoms of the asymmetric unit. Some disorder was found in IX and X: in IX the O6 and C24 atoms of the macrocyclic ring are disordered over two positions with partial occupancies of 0.90(1) and 0.10(1), and only the major component was refined in an anisotropic approximation. In X the triflate anion, CF3COO− and 2-methoxyethanol molecule were modeled in such a way that anionic F and O atoms and CH3–O–CH2–CH2− fragment of the 2-methoxyethanol molecule were disordered over two positions with partial occupancies of 0.58(1) and 0.42(1), respectively; both positions were refined in an anisotropic approximation. In all structures C–bound H atoms were placed in calculated positions and were treated using a riding-model approximation, with Uiso(H) = 1.2Ueq(C). The O- and N-bound H-atoms were determined from a difference Fourier map and were then allowed to refine isotropically with Uiso(H) = 1.5Ueq(O,N). DFIX restraints were applied to the N–H distances (d = 0.86 Å) in VII and VIII. Crystal data together with further details of data collections and refinement calculations are given in Table 1. CCDC reference numbers 690773–690777.| Compound | VI | VII | VIII | IX | X |
|---|---|---|---|---|---|
| Composition | 2C19H16O·C8H16O4 | 2C19H17N C8H16O4 | 2(C19H18N·NCS)·C8H16O4 | 2C19H17 NO3S·C12H24O6 | C19H18N·C2F3O2 C3H8O2 |
| CCDC number | 690773 | 690777 | 690776 | 690775 | 690774 |
| Formula weight | 696.84 | 694.88 | 813.06 | 943.10 | 449.46 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P1 | P1 | P21/c | P1 | C2/c |
| a /Å | 8.3614(7) | 8.4675(8) | 14.7301(15) | 8.4707(6) | 17.069(4) |
| b /Å | 10.4963(9) | 10.2973(13) | 16.1984(16) | 12.4699(9) | 10.694(3) |
| c /Å | 11.710(1) | 11.943(2) | 9.729(1) | 12.8600(9) | 26.520(7) |
| α/° | 82.114(2) | 84.058(9) | 90.0 | 115.372(1) | 90.0 |
| β/° | 86.716(2) | 88.008(12) | 103.97(2) | 97.929(1) | 107.367(5) |
| γ/° | 66.740(2) | 67.966(8) | 90.0 | 99.540(1) | 90.0 |
| V /Å3 | 935.25(14) | 960.1(2) | 2252.7(4) | 1176.63(14) | 4620(2) |
| Z | 1 | 1 | 2 | 1 | 8 |
| D c /Mg m−3 | 1.237 | 1.202 | 1.199 | 1.331 | 1.292 |
| μ(MoKα)/mm−1 | 0.081 | 0.076 | 0.165 | 0.179 | 0.103 |
| F(000) | 372 | 372 | 864 | 500 | 1888 |
| Reflections collected / unique | 5310/ 3605 [R(int) = 0.020] | 3625 / 3373 [R(int) = 0.026] | 12820 / 4413 [R(int) = 0.043] | 6829 / 4541 [R(int) = 0.015] | 12877 / 4554 [R(int) = 0.024] |
| Reflections with I>2σ(I) | 2978 | 2622 | 1462 | 3154 | 3066 |
| Goodness-of-fit | 1.060 | 1.042 | 0.831 | 0.925 | 1.011 |
| R, wR [I>2σ(I)] | 0.048, 0.138 | 0.041, 0.111 | 0.054, 0.117 | 0.039, 0.096 | 0.043, 0.121 |
Results and discussion
Crystallographic description of the complexes
Compounds VI and VII crystallize in the same triclinic space group P1 and have close unit cell dimensions, as is evident from Table 1. For both compounds the asymmetric unit comprises one half of the 12C4 molecule that resides on an inversion center and one Ph3COH or Ph3CNH2 molecule in general position (Fig. 1). The geometry of the Ph3COH (Ph3CNH2) and 12C4 molecules is in agreement with the literature data.24,25,36–38 The components are associated in the 1:2 molecular complexes via two single OH⋯O (NH⋯O) hydrogen bonds (Table 2).
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | Symmetry transformation for acceptor |
|---|---|---|---|---|---|
| VI | |||||
| O(3)–H(3A)⋯O(1) | 0.89(2) | 1.94(2) | 2.814(2) | 169(2) | x, y, z |
| C(19)-H(19A)⋯O(2) | 0.93 | 2.61 | 3.380(2) | 141 | x, y, z |
| C(8)-H(8A)⋯O(2) | 0.93 | 2.59 | 3.482(2) | 162 | x, y − 1, z |
| VII | |||||
| N(1)–H(1N)⋯O(1) | 0.89(2) | 2.22(2) | 3.087(2) | 168(2) | x, y, z |
| C(19)-H(19A)⋯O(2) | 0.93 | 2.66 | 3.426(2) | 140 | x, y, z |
| C(8)-H(8A)⋯O(2) | 0.93 | 2.65 | 3.493(2) | 152 | x, y − 1, z |
| VIII | |||||
| N(1)–H(2N)⋯O(1) | 0.89(2) | 1.92(2) | 2.790(3) | 166(3) | x, y, z |
| N(1)–H(3N)⋯N(2) | 0.90(2) | 2.01(2) | 2.892(4) | 166(3) | x, y, z |
| N(1)–H(1N)⋯S(1) | 0.90(2) | 2.35(2) | 3.246(3) | 170(3) | −x + 1, −y + 2, −z + 1 |
| IX | |||||
| O(3)–H(1O)⋯O(2) | 0.77(2) | 2.16(2) | 2.917(2) | 169(2) | –x, −y, –z |
| N(1)–H(1N)⋯O(5) | 0.82(2) | 2.37(2) | 3.029(2) | 138(2) | x, y − 1, z |
| N(1)–H(1N)⋯O(6) | 0.82(2) | 2.51(2) | 3.086(2) | 129(2) | x, y − 1, z |
| N(1)–H(2N)⋯O(4) | 0.88(2) | 2.25(2) | 3.013(2) | 145(2) | –x, −y, –z |
| N(1)–H(2N)⋯O(5) | 0.88(2) | 2.41(2) | 3.135(2) | 140(2) | –x, −y, –z |
| X | |||||
| N1–H(2N)⋯O(1) | 0.94(2) | 1.95(2) | 2.877(5) | 174(2) | x, y, z |
| N1–H(2N)⋯O(1′) | 0.94(2) | 1.98(2) | 2.886(6) | 162(2) | x, y, z |
| N1–H(1N)⋯O(2) | 0.93(2) | 1.93(2) | 2.862(2) | 177(2) | −x, y, −z + 1/2 |
| N1–H(3N)⋯O(3) | 0.95(2) | 1.80(2) | 2.752(11) | 179(2) | x, y, z |
| N1–H(3N)⋯O(3′) | 0.95(2) | 1.82(2) | 2.766(14) | 174(2) | x, y, z |
| O(2)–H(2B)⋯O(4) | 0.95(2) | 1.71(3) | 2.658(10) | 173(2) | x, y, z |
| O(2)–H(2B)⋯O(4′) | 0.95(2) | 1.69(3) | 2.640(10) | 171(2) | x, y, z |
 | ||
| Fig. 1 ORTEP drawing (a) for VI, (b) for VII with a partial numbering scheme. Thermal ellipsoids are drawn at 50% probability level. | ||
The crystal packing in VI and VII is dictated by the combination of conventional OH⋯O (NH⋯O) hydrogen bonds that combine the molecules in the 1:2 complex, weak CH(arene)⋯O(crown) interactions that combine the complexes in the tape with the hydrophobic exterior formed by the phenyl rings (Fig. 2), and the concerted sextuple phenyl embrace (SPE)39 between the Ph3COH (Ph3CNH2) molecules in the neighboring tapes. Four host molecules surround each 12C4 molecule in the tape. The packing in VII is characterized by slightly increased distances analogous to those shown in Fig. 2a for VI with the second hydrogen of the amino group being free of any involvement in hydrogen bonding (very similar to the pure phase of Ph3CNH2).37
 | ||
| Fig. 2 Crystal packing for VI and VII, (a) fragment of tape in VI with the shortest C–H⋯O distances shown by dashed lines, (b) fragments of adjacent tapes with SPE between two Ph3COH molecules, (c) fragment of tape in VII with the shortest N–H⋯O and C–H⋯O distances shown by dashed lines. | ||
This ordering function of 12C4 molecule in VI and VII is looking very amazing in confrontation with the relative although rather restrictive examples available in CSD:40 thus, inclusion of a dioxane molecule in the network of isostructural Ph3COH36 or Ph3SiOH17 molecules results in the 1:1 adduct in the first case and 1:2 adduct in the second one with pronounced changes in the crystal packing, although both compounds crystallize in the same triclinic crystal system. On the other hand, the same hosts yield the isostructural 2:1 aggregates with dimethylsulfoxide, both being crystallized in the same C2/c space group with close unit cell dimensions.15,17 Ferguson and co-workers36 analysing the series of the group 14 Ph3MOH molecules (M = C, Si, Ge, Sn, Pb) revealed for them different crystal packing motifs: the C-, Si- and Ge-17,41 derivatives represent hydrogen-bonded tetramers with the O-atoms in a flatten tetrahedral arrangement, while Sn- and Pb-containing molecules have structures consisting of zigzag chains of planar Ph3M (M = Sn, Pb) groups joined by OH groups giving trigonal bipyramidal geometry at M,42 whilst in Ph3CSH,43 a precise analogue of Ph3COH as well as in PhCNH2,37 being isoelectronic with Ph3COH, there are no intermolecular hydrogen bonding despite the availability of potential H-donors (SH, NH2groups) and H-acceptors (S, N-lone pair), and the structures consist of isolated molecules with no indication that the H-atoms take part in any hydrogen bonding. Concluding this section, the row of the relative crown-containing binary systems (Ph3COH)2.12C4 (VI), (Ph3CNH2)2.12C4 (VII) (present work), (Ph3SiOH)2.12C4,24 and (Ph3CSH)2.12C425 represents the isomorphous aggregates with close unit cell dimensions and a common structural motif. It permits to conclude the ordering and leveling function of the 12C4 molecule upon its inclusion.
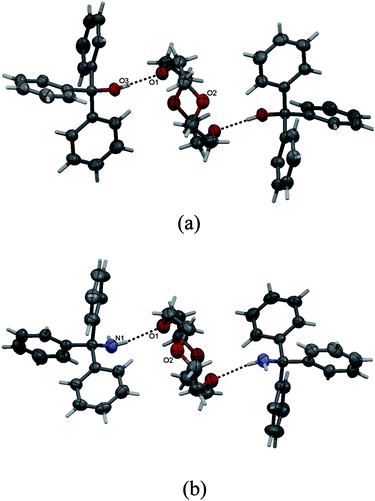
The view of complex VIII is shown in Fig. 3. The ternary salt-like adduct VIII crystallizes in the centrosymmetric space groupP21/c and, similar to VI and VII, the 12C4 molecule resides on an inversion center. The components in the crystal are held together via the diverse system of charge-assisted hydrogen bonds, NH+⋯O(crown), NH+⋯N−(NCS−) and NH+⋯S−(NCS−) (Fig. 3, Table 2).
 | ||
| Fig. 3 ORTEP drawing for VIII with a partial numbering scheme. Thermal ellipsoids are drawn at 50% probability level. | ||
The tetrahedral ammonia group provides one hydrogen for the NH+⋯O interaction with a crown molecule and two hydrogens for interionic NH+⋯N−(NCS−) and NH+⋯S(NCS−) interactions with two inversion-related NCS− anions. Just as in the two previous structures, these concerted interactions combine the components in the tape propagated along the c axis in the crystal (Fig. 4). The neighboring tapes are packed in an antiparallel arrangement to maximize the SPE. The crystal architecture of VIII is very close to structure organization in tritylammonium chloride 1,4-dioxane solvate, Ph3CNH3Cl.(CH2CH2O)2 (YAGSIJ refcode in CSD).44
 | ||
| Fig. 4 Fragment of tape in VIII. | ||
As was stressed above, incorporation of the amidosulfoxide group into the phenyl ring of IVa results in competition between the two donor groups (SO2NH2 and OH) of V for the crown's oxygens and following the Etter's rule45 in a preferable interaction of the stronger H-donor, SO2NH2group with the macrocyclic oxygens via two bifurcated NH⋯O hydrogen bonds with the formation of 1:2 molecular complex IX (Fig. 5, Table 2). IX crystallizes in triclinic space group P1 where the 18C6 molecule resides on an inversion center. The hydroxyl group seeking for the next H-acceptor is responsible for the association of these complexes into tapes viaOH⋯O(SO2) hydrogen bonds (Fig. 6, Table 2). Similar to the previous examples, the tape in IX is characterized by the hydrophilic interior saturated by hydrogen bonds and two external hydrophobic sides formed by the phenyl rings. Compound IX represents the first known example of the inclusion of spacious 18C6 molecule in the lattice of triphenylmethanol derivative. The common feature of the crystal packing for VI–IX is a ribbon motif with crown inclusion within the ribbon and the SPE formulated between the neighboring ribbons.
 | ||
| Fig. 5 ORTEP drawing for IX with a partial numbering scheme. Thermal ellipsoids are drawn at 50% probability level. | ||
 | ||
| Fig. 6 Packing diagram for IX (a), fragment of tape (b), fragments of adjacent tapes with SPE between two V molecules. | ||
Compound X of the 1:1:1 stoichiometry is formed as a result of the interaction of IVd with a linear 2-methoxyetanol molecule. X crystallizes in monoclinic space groupC2/c. The content of the asymmetric unit is shown in Fig. 7. Similar to VIII it is a ternary complex where two hydrogens of the same ammonia group are involved in the NH+⋯O(COO−) and NH+⋯O(2-methoxyethanol) contacts (Table 2). Furthermore, inside the complex the terminal hydroxyl group of the 2-methoxyethanol molecule participates in OH⋯O−(COO−) interaction with the second carboxyl oxygen. These three hydrogen bonds close the eleven-membered ring, R33(11) using a graph set notation.45
 | ||
| Fig. 7 ORTEP drawing for X with the partial numbering scheme. Thermal ellipsoids are drawn at 50% probability level. | ||
Contrary to VIII where each ammonia group interacts with two anions and one neutral crown molecule, in X the third ammonia hydrogen is involved into NH+⋯O interaction with the second 2-methoxyethanol molecule related by the two-fold axis with the basic one. These concerted interactions combine two complexes into a six-membered calix-like capsule stabilized by 8 charge-assisted hydrogen bonds within the hydrophilic core that is covered by the hydrophobic exterior formed by six phenyl rings of two tritylammonia cations and two terminal methyl groups of 2-methoxyethanol molecules (Fig. 8). The CH⋯F interactions22,23,46 between triflate anion and 2-methoxyethanol molecule are responsible for the association of the 6-membered globules into tape running along the b direction in the crystal.
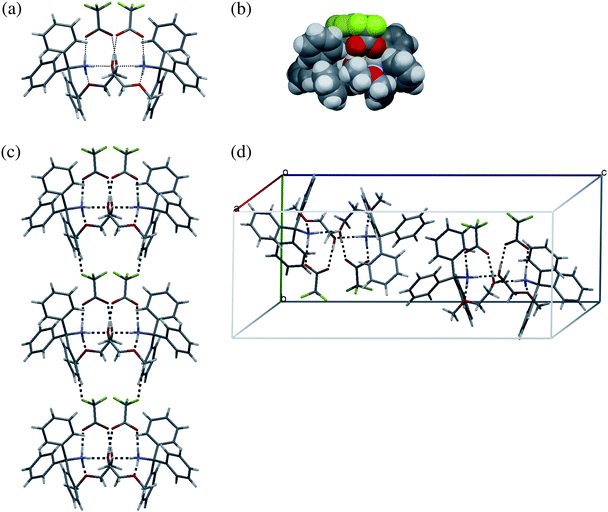 | ||
| Fig. 8 Packing diagram for X: (a) network of hydrogen bonds shown by dashed lines within the six-membered cluster, (b) space-filling representation of the cluster, (c) column of clusters sustained by CH⋯F interactions, (d) the SPE between adjacent clusters. | ||
Thus, the present study amplifies the scanty family of trytilammonia salts by two novel representatives. Contrary to the trytile sulfonates studied so far21 all of which represent binary adducts and formulate the well-defined [4 + 4] ion-pair clusters, VIII and X alongside the tritylammonium chloride acetone and tritylammonium chloride dioxane solvates [19,44] belong to the ternary compounds with the inclusion of the different O-containing neutral molecules (acetone, dioxane, 12C4, 2-methoxyethanol) in the ionic crystal lattice of the parent salt. The size and topology of these guests appear to be crucial for the final aggregates: linear acetone and 2-methoxyethanol molecules facilitate the formation of the isolated 6-membered clusters while the cyclic double-face 1,4-dioxane and 12C4 molecules provide extended 1D structures. In any closest environment [three anions,21 two anions and one neutral molecule19,44 or one anion and two neutral molecules (present work)] all three ammonium hydrogens are involved in the hydrogen bonding system.
Conclusions
For the first time the approaches of supramolecular chemistry were used to estimate the preferable in size crown ethers (in the order of 12–18-membered CEs) for interaction with triphenylmethane derivatives. The 12C4 molecule has proven to be the best candidate as it is selectively included in the crystal lattice of neutral Ph3COH and Ph3CNH2 in the 1:2 ratio and in the ionic network of Ph3CNH3NCS in the 1:1:1 ratio. Contrary to the pure forms of Ph3COH, Ph3SiOH, Ph3CSH, and Ph3CNH2 which differ essentially by the crystal packing, the corresponding binary aggregates with 12C4 appear to be isomorphous and ordered with each macrocyclic molecule entrapping by four bulky host molecules. The functionalization of one of the phenyl rings in the Ph3COH molecule by the H2NSO2-group provides its involvement in the interaction with 18C6 in a traditional manner and excludes the hydroxyl group from the host–guest interactions. The supramolecular architecture of the studied tritylammonium salts is dictated by the concerted effect of the topology of the anion and the included neutral molecule, being 1D structure for Ph3CNH3·NCS assimilating cyclic 12C4 molecule and the isolated six-membered capsule upon inclusion of linear 2-methoxyethanol molecules in the ionic lattice of Ph3NH3·CF3COO. In all cases the common packing motif maximizes the SPE arrangement of the host molecules.
Acknowledgements
E.V.G. and W.J.W. are indebted to the National Science Council of Taiwan 94-2811-M-032 project for financial support.References
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017 CrossRef CAS.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021 CrossRef.
- Inclusion Compounds. Structural Aspects of Inclusion Compounds formed by Organic Host Lattices, ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Academic Press, London, Orlando, San Diego, San Francisco, New York, Toronto, Montreal, Sydney, Tokyo, San Paulo, 1984, vol. 2, 499 pp Search PubMed.
- Host Guest Complex Chemistry. Macrocycles. Synthesis, Structure, Applications, ed. A. Vogtle and E. Weber,Springer, Berlin, 1985, 511 pp Search PubMed.
- J.-M. Lehn, Supramolecular Chemistry/ Concepts and Perspectives, VCH Verlagsgesellschaft mbH, Weinheim; New York; Basel; Cambridge; Tokyo, 1995, 334 p Search PubMed.
- G. R. Desiraju, in Comprehensive Supramolecular Chemistry, ed. J-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Wogtle, Pergamon, Oxford, New York, Tokyo, 1996, vol. 6, pp 1–22 Search PubMed.
- P. Dastidar andI. Goldberg, in Comprehensive Supramolecular Chemistry, ed. J-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Wogtle, Pergamon, Oxford, New York, Tokyo, 1996, vol. 6, pp 305–350 Search PubMed.
- (a) F. Toda, in Comprehensive Supramolecular Chemistry, ed. J-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Wogtle, Pergamon, Oxford, New York, Tokyo, 1996, vol. 6, pp 465–516 Search PubMed; (b) E. Weber, N. Dörpinghaus and I. Goldberg, J. Chem. Soc., Chem. Commun., 1988, 1566 RSC; (c) S. A. Bourne, L. R. Nassimbeni, M. L. Niven, E. Weber and A. Wierig, J. Chem. Soc., Perkin Trans., 1994, 2, 1215 Search PubMed; (d) M. R. Caira, L. R. Nassimbeni, M. L. Niven, W.-D. Schubert, E. Weber and N. Dörpinghaus, J. Chem. Soc., Perkin Trans., 1990, 2, 2129 Search PubMed; (e) A. Jacobs, N. L. Z. Masuku, L. R. Nassimbeni and J. H. Taljaard, CrystEngComm, 2008, 10, 322 RSC.
- E. Weber, in Comprehensive Supramolecular Chemistry, ed. J-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Wogtle, Pergamon, Oxford, New York, Tokyo, 1996, vol. 6, pp 535–592 Search PubMed.
- M. R. Caira andL. R. Nassimbeni, in Comprehensive Supramolecular Chemistry, ed. J-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Wogtle, Pergamon, Oxford, New York, Tokyo, 1996, vol. 6, pp 825–850 Search PubMed.
- A. Reichert,H. Ringsdorf andP. Schuhmacher, in Comprehensive Supramolecular Chemistry, ed. J-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Wogtle, Pergamon, Oxford, New York, Tokyo, 1996, vol. 9, pp 313–350 Search PubMed.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723 CrossRef CAS.
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. Panunto, J. Am. Chem. Soc., 1990, 112, 8415 CrossRef CAS.
- (a) K.-K. D. Ng and H. Hart, Tetrahedron, 1995, 51, 7883 CrossRef CAS; (b) E. Weber, N. Dörpinghaus and I. Csöregh., J. Chem. Soc., Perkin Trans. 2, 1990, 2167 RSC; (c) K. Eckardt, H. Paulus, H. Fuess, N. Onoda-Yamamuro and R. Ikeda, J. Incl. Phenom., 1999, 35(3), 431 Search PubMed.
- E. Weber, K. Skobridis and I. Goldberg, J. Chem. Soc., Chem. Commun., 1989, 1195 RSC.
- R. West, R. H. Baney and D. L. Powell, J. Am. Chem. Soc., 1960, 82, 6269 CrossRef CAS.
- K. F. Bowes, C. Glidewell and J. N. Low, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o409 CrossRef.
- S. A. Bourne, L. Johnson, C. Marais, L. R. Nassimbeni, E. Weber, K. Skobridis and F. Toda, J. Chem. Soc., Perkin Trans. 2, 1991, 1707 RSC.
- M. Canle L, W. Clegg, I. Demirtas, M. R. J. Elsegood and H. Maskill, J. Chem. Soc., Perkin Trans. 2, 2000, 85 RSC.
- I. Csöregh, S. Hirano, S. Toyota, P. Bombisz and F. Toda, CrystEngComm, 2004, 6, 60 RSC.
- N. Tohnai, Y. Mizobe, M. Doi, S. Sukata, T. Hinoue, T. Yuge, I. Hisaki, Y. Matsukawa and M. Miyata, Angew. Chem., Int. Ed., 2007, 46, 2220 CrossRef CAS.
- N. Hayashi, T. Mori and K. Matsumoto, Chem. Commun., 1998, 1905 RSC.
- D. Schollmeyer, O. V. Shishkin, T. Rühl and M. O. Vysotsky, CrystEngComm, 2008, 10, 715 RSC.
- E. A. Babaian, M. Huff, F. A. Tibbals and D. C. Hrncir, J. Chem. Soc., Chem. Commun., 1990, 306 RSC.
- M. S. Fonari, E. V. Ganin and W.-J. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o431 CrossRef.
- K. Wijaya, O. Moers, P. G. Jones and A. Blaschette, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 818 CrossRef.
- D. Henschel, A. Blaschette and P. G. Jones, Z. Naturforsch, 1995, 50b, 128 Search PubMed.
- D. Henschel, A. Blaschette and P. G. Jones, Z. Naturforsch., 1995, 50b, 229 Search PubMed.
- D. Henschel, M. Naveke, T. Hamann, A. Blaschette and P. G. Jones, Z. Naturforsch., 1995, 50b, 913 Search PubMed.
- J. Dale and K. J. Daasvatn, J. Chem. Soc., Chem. Commun., 1976, 295 RSC.
- E. V. Ganin, V. F. Makarov, S. A. Kotlyar, N. G. Lukyanenko, M. S. Fonari, A. A. Dvorkin and A. YuSimonov, Russ. Khim. Geterotsikl. Soedin., 1988, 1190 Search PubMed.
- A. A. Dvorkin, M. S. Fonari, S. T. Malinovskii, E. V. Ganin, A. YuSimonov, V. F. Makarov, S. A. Kotlyar and N. G. Lukyanenko, Russ. Zh. Strukt. Khim., 1989, 30, 96 Search PubMed.
- Z. Otwinowski andW. Minor, in Methods in Enzymology, Macromolecular Crystallography, Part A, ed. C. W. Carter and R. M. Sweet, Academic Press, London, 1996, vol. 276, p 307 Search PubMed.
- G. M. Sheldrick, SADABS, 1997,Bruker AXS Inc., Madison, WI-53719, USA Search PubMed.
- G. M. Sheldrick: SHELXS97 and SHELXL97, Programs for Crystal Structure Solution and Refinement.University of Göttingen, Germany, 1997 Search PubMed.
- G. Ferguson, J. F. Gallagher, C. Glidewell, J. N. Low and S. N. Scrimgeour, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1272 CrossRef.
- C. Glidewell and G. Ferguson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, C50, 924 CrossRef CAS.
- A. Michalides, D. Henschel, A. Blaschette and P. G. Jones, Z. Naturforsch., 1995, 50b, 1018 Search PubMed.
- (a) A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed; (b) I. Dance and M. Scudder, J. Chem. Soc., Chem. Commun., 1995, 1039 RSC; (c) R. Thaimattam, F. Xue, J. A. R. P. Sarma, C. W. Mak and G. R. Desiraju, J. Am. Chem. Soc., 2001, 123, 4432 CrossRef CAS; (d) J. J. Perry, G. J. McManus and M. J. Zaworotko, Chem. Commun., 2004, 2534 RSC.
- F. N. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- (a) H. Puff, K. Braun and H. Reuter, J. Organomet. Chem., 1991, 409, 119 CrossRef CAS; (b) G. Ferguson, J. F. Gallagher, D. Murphy, T. R. Spalding, C. Glidewell and H. D. Holden, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1228 CrossRef.
- C. Glidewell and D. C. Liles, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 129 CrossRef.
- G. Bernardinelli, M. Geoffroy and R. Franzi, Z. Kristallogr., 1991, 195, 147 CAS.
- S. Parsons, K. Borisenko, D. Rankin and P. Wood, private communication, 2004 Search PubMed.
- (a) M. Etter, J. Phys. Chem., 1991, 95, 4601 CrossRef CAS; (b) M. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- E. D'Oria and J. J. Novoa, CrystEngComm, 2008, 10, 423 RSC.
Footnote |
| † CCDC reference numbers 690773–690777. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b810076d |
| This journal is © The Royal Society of Chemistry 2009 |