DOI:
10.1039/B812447G
(Paper)
CrystEngComm, 2009,
11, 168-176
Ligand isomerism-controlled structural diversity of cadmium(II) perchlorate coordination polymers containing dipyridyladipoamide ligands†
Received
21st July 2008
, Accepted 19th September 2008
First published on 17th October 2008
Abstract
The syntheses, structures and ligand conformations of three Cd(II) coordination polymers [Cd(L1)(ClO4)2]∞ [L1 = N,N′-di(2-pyridyl)adipoamide], 1, {[Cd(L2)2(CH3OH)2](ClO4)2·2CH3OH}∞ [L2 = N,N′-di(3-pyridyl)adipoamide], 2 and [Cd(L3)2(ClO4)2(CH3CH2OH)2]∞ [L3 = N,N′-di(4-pyridyl)adipoamide], 3, are reported. Their structures have been characterized by X-ray crystallography. Complex 1 forms 1D zigzag chains, while complex 2 forms 2D pleated sheets which are positioned in an ABAB manner. Complex 3 shows a rare 3D entanglement from parallel polycatenation of 3-fold interpenetrated square layers. While the L1 ligands in 1 adopt AAAtrans conformations and the L2 ligands in 2 adopt GAGtrans conformations, the L3 ligands in 3 adopt AAAtrans and AGAtrans conformations. The structural types of the Cd(II) coordination polymers can be directed by ligand isomerism.
Introduction
Crystal engineering of coordination polymeric frameworks has attracted great attention not only because of their potential properties as novel zeolite-like materials for separation, ion exchange and catalysis, but also for their intriguing structural topologies.1,2 The range and variety of self-assembling inorganic structures that can be constructed relies on the presence of suitable metal–ligand interactions and supramolecular contacts, i.e., hydrogen bondings and other weak interactions.3 Although many coordination polymers with interesting structures and properties have been reported, the control of structural dimensionality of coordination polymers is still a challenge.4 Moreover, the structural types of the resulting coordination polymers are also affected by factors such as counterion,5 metal-to-ligand ratio6 and solvent.7
While coordination self-assembly based upon rigid, linear bipyridyl ligands has been extensively investigated, the use of ligands showing conformational flexibility was less explored.8 The coordination networks of metal complexes containing flexible bidentate ligands are less predictable due to possible occurrence of supramolecular isomerism involving the adoption of different ligand conformations. It is well known that the ligand 1,2-bis(4-pyridyl)ethane can adopt either a gauche or anti geometry, whereas the flexible ligand 1,3-bis(4-pyridyl)propane can exist in anti–anti, gauche–anti and two different gauche–gauche (cis–trans) conformations.8(k) Our recent work9 has shown that the dipyridyladipoamide ligands can be arranged in anti–anti–anti (AAA), anti–anti–gauche (AAG), anti–gauche–anti (AGA), anti–gauche–gauche (AGG), gauche–anti–gauche (GAG) and gauche–gauche–gauche (GGG) conformations, and based on the relative orientation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (or N–H) groups, each conformation can adopts cis or trans arrangement. The A and G conformations are given when the C–C–C–C torsion angle (θ) is 180 ≥ θ > 90° and 0 ≤ θ ≤ 90°, respectively.9a The Ag(I) coordination polymers containing the N,N′-di(2-pyridyl)adipoamide show new ligand conformations of GAGtrans, AAAtrans, GGG cis, and AGAcis, which also differ in the dihedral angle between the two pyridyl rings.9a More recently, the reaction of copper sulfate with N,N′-di(4-pyridyl)adipoamide led to the formation of a twelvefold diamondoid network, and the ligands adopt the AAAtrans and GAGtrans conformations.9b
O (or N–H) groups, each conformation can adopts cis or trans arrangement. The A and G conformations are given when the C–C–C–C torsion angle (θ) is 180 ≥ θ > 90° and 0 ≤ θ ≤ 90°, respectively.9a The Ag(I) coordination polymers containing the N,N′-di(2-pyridyl)adipoamide show new ligand conformations of GAGtrans, AAAtrans, GGG cis, and AGAcis, which also differ in the dihedral angle between the two pyridyl rings.9a More recently, the reaction of copper sulfate with N,N′-di(4-pyridyl)adipoamide led to the formation of a twelvefold diamondoid network, and the ligands adopt the AAAtrans and GAGtrans conformations.9b
To investigate the effect of ligand isomerism and conformation on the structural diversity of a metal complex with octahedral geometry, we have prepared three isomeric ligands N,N′-di(2-pyridyl)adipoamide, L1N,N′-di(3-pyridyl)adipoamide), L2 and N,N′-di(4-pyridyl)adipoamide), L3, which have a N,N′-bridging site with different donor atom positions. In addition, these ligands bear the amide groups with both the NH hydrogen donor and CO hydrogen acceptor which may result in interesting hydrogen bonds in the solid state structure. The reactions of these isomeric ligands with Cd(ClO4)2·6H2O have resulted in 1D, 2D and 3D Cd(II) coordination polymers, respectively. The synthesis, structure and ligand conformations of [Cd(L1)(ClO4)2]∞, 1, {[Cd(L2)2 (CH3OH)2](ClO4)2 ⋯ 2CH3OH}∞, 2 and [Cd(L3)2(ClO4)2(CH3CH2OH)2]∞, 3, form the subject of this report.
Experimental
General procedures
All manipulations were carried out under dry, oxygen-free nitrogen by using Schlenk techniques, unless otherwise noted. Solvents were dried and deoxygenated by refluxing over the appropriate reagents before use. Elemental analyses were obtained from a PE 2400 series II CHNS/O analyzer or a HERAEUS VaruoEL analyzer. IR spectra (KBr disk) were recorded on a Jasco FT/IR-460 plus spectrometer. Emission spectra were obtained from Hitachi F-4500 spectrometer. Differential scanning calorimetry (DSC) measurements were carried on a Dupon DSC Q-10.
Materials
The reagent Cd(ClO4)2·6H2O was purchased from Acros Chemical Co. The ligands N,N′-di(2-pyridyl)adipoamide (L1)9a and N,N′-di(3-pyridyl)adipoamide (L3)9b were prepared according to published procedures.
Synthesis
N,N′-di(3-dipyridyl)adipoamide (L2).
Adipoyl chloride (1.00 g, 5.47 mmol) was added slowly to a DMF solution of 3-aminopyridine (1.03 g, 10.94 mmol), which was stirred for 15 min and triethylamine (1.11 g, 10.94 mmol) was then added. The mixture was refluxed for 6 h and then the volume was reduced to 10 mL by vacuum evaporation. A light yellow solid was obtained from the solution after standing at room temperature for 24 h. The solid was filtered off and washed with excess of cold water to give a white powder. Yield: 1.32 g (83%). 1H NMR (DMSO-D6, ppm) 10.11 (2H, s, NH), 8.68 (2H, d, H2-py), 8.19 (2H, d, H4-py), 7.99 (2H, d, H6-py), 7.28 (2H, t, H5-py), 2.39 (4H, t, COCH2), 1.60 (4H, p, COCH2CH2). Anal Calcd for C16H18O2N4 (MW = 298.34): C, 64.41; H, 6.08; N, 18.78%. Found: C, 64.40; H, 6.09; N, 18.65%. IR (KBr disk, cm−1): 3305(w), 3178(br), 1692(s), 1599(br), 1582(s), 1552(s), 1423(s), 1286(s), 1161(w), 809(m), 704(w), 628(w).
[Cd(L1)(ClO4)2]∞, 1.
An ethanol solution of L1 (0.30 g, 1.0 mmol) was layered on top of an aqueous solution of Cd(ClO4)2·6H2O (0.42 g, 1.0 mmol). After one month, needle-like crystals were found at the interface. The crystals were collected, washed with diethyl ether and then dried under vacuum. Yield: 0.45 g (74%). Anal Calcd for C16H18CdCl2N4O10 (MW = 609.64): C, 31.52; H, 2.98; N, 9.19. Found: C, 31.50; H, 2.95; N, 9.05. IR (cm−1): 3565(w), 3133(m), 1662(m), 1575(s), 1432(s), 1372(s), 1339(s), 1238(w), 965(m), 855(w), 731(w), 551(w).
{[Cd(L2)2(CH3OH)2](ClO4)2·2CH3OH}∞, 2.
A methanol solution of L2 (0.30 g, 1.0 mmol) was layered on top of an aqueous solution of Cd(ClO4)2· 6H2O (0.42 g, 1.0 mmol). After one month, needle-like crystals were found at the interface. The crystals were collected, washed with diethyl ether and then dried under vacuum. Yield: 0.85 g (82%). Anal Calcd for C36H52CdCl2N8O16 (MW = 1036.16): C, 41.73; H, 5.06; N, 10.81. Found: C, 41.50; H, 5.05; N, 10.78. IR (cm−1): 3587(w), 3198(m), 1680(m), 1587(s), 1427(s), 1386(s), 1329(s), 1244, 958(m), 847(w), 728(w), 556(w).
[Cd(L3)2(ClO4)2(CH3CH2OH)2]∞, 3.
Prepared as described for 1, except that L3 (0.30 g, 1.0 mmol) was used. Yield: 0.75 g (75%). Anal Calcd for C36H48CdCl2N8O14 (MW = 1000.12): C, 43.23; H, 4.84; N, 11.20. Found: C, 43.05; H, 4.83; N, 11.12. IR (cm−1): 3337(w), 3088(s), 1696(s), 1594(m), 1467(s), 1384(s), 1309(s), 1254(s), 1109(s), 929(m), 839(w), 480(w), 462(w).
The diffraction data for L2 and complexes 1–3 were collected on a Bruker AXS diffractometer, which was equipped with a graphite-monochromated Mo Kα (λKα = 0.71073 Å) radiation. Data reduction was carried by standard methods with use of well-established computational procedures.10 The structure factors were obtained after Lorentz and polarization corrections. The positions of the heavy atoms, including the cadmium atoms, were located by the direct method. The remaining atoms were found in a series of alternating difference Fourier maps and least-square refinements,11 except the hydrogen atoms of 2 and 3 which were added by using the HADD program and refined using a riding model. The atoms C16 in 2 and C18 in 3 were found to be disordered such that two positions for each atom can be located. Basic information pertaining to crystal parameters and structure refinement is summarized in Table 1, while selected bond distances and angles for L2 and complexes 1–3 are given in Table 2.
| Compound |
L2
|
1
|
2
|
3
|
|
R
1 = S||Fo| – |Fc||/S|Fo|.
wR
2 = [Sw(Fo2 – Fc2)2/Sw(Fo2)2]1/2. w = 1/[σ2(Fo2) + (ap)2 + (bp)], p = [max(Fo2 or 0) + 2(Fc2)]/3. a = 0.0549, b = 0.0939, L2; a = 0.0344, b = 1.1440, 1; a = 0.0842, b = 1.8685, 2; a = 0.0826, b = 7.9674, 3.
Quality-of-fit = [Sw(|Fo2| – |Fc2|)2/Nobserved – Nparameters)]1/2.
|
| Formula |
C16H18N4O2 |
C16H18CdCl2N4O10 |
C36H52CdCl2N8O16 |
C36H48CdCl2N8O14 |
|
FW
|
298.34 |
609.64 |
1036.16 |
1000.12 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Orthorhombic |
| Space group |
P21/n |
P21/n |
Pī
|
Pcca |
|
a/Å |
9.4559(12) |
9.1730(11 |
9.6762(11) |
16.779(2) |
|
b/Å |
7.8465(10) |
11.3098(13) |
9.9743(11) |
12.1263(13) |
|
c/Å |
10.6879(17) |
11.0517(10) |
12.4337(14) |
21.766(3) |
|
α/° |
90 |
90 |
76.553(9) |
90 |
|
β/° |
108.137(11) |
107.793(8) |
78.663(9) |
90 |
|
γ/° |
90 |
90 |
79.367(9) |
90 |
|
V/Å3 |
753.60(18) |
1091.7(2) |
1132.2(2) |
4428.6(9) |
|
Z
|
2 |
2 |
1 |
4 |
|
d
calc/g cm−3 |
1.315 |
1.855 |
1.520 |
1.500 |
|
μ(Mo Ka)/mm−1 |
0.090 |
1.307 |
0.677 |
0.686 |
| Crystal size/mm |
0.6 × 0.3 × 0.1 |
0.4 × 0.4 × 0.2 |
0.4 × 0.3 × 0.2 |
0.6 × 0.4 × 0.2 |
| Range (2θ) for data |
5.04 ≤ 2θ ≤ 49.98 |
5.06 ≤ 2θ ≤ 49.98 |
4.24 ≤ 2θ ≤ 49.98 |
4.46 ≤ 2θ ≤ 50.02 |
| Collection/° |
|
|
|
|
| Reflections collected |
1801 |
2543 |
4673 |
4495 |
| Independent reflections |
1305 |
1917 |
3919 |
3864 |
| |
[R(int) = 0.0338] |
[R(int) = 0.0271] |
[R(int) = 0.0346] |
[R(int) = 0.0262] |
| Data/restraints/parameters |
1305/0/136 |
1917/0/187 |
3919/0/305 |
3864/0/300 |
| Quality-of-fit indicatorc |
1.032 |
1.031 |
1.026 |
1.035 |
| Final R indices |
R1 = 0.0500, |
R1 = 0.0329, |
R1 = 0.0617, |
R1 = 0.0523, |
| [I > 2σ(I)] a,b |
wR2 = 0.1048 |
wR2 = 0.0713 |
wR2 = 0.1467 |
wR2 = 0.1381 |
|
R indices (all data) |
R1 = 0.1010, |
R1 = 0.0511, |
R1 = 0.0808, |
R1 = 0.0763, |
| |
wR2 = 0.1272 |
wR2 = 0.0793 |
wR2 = 0.1605 |
wR2 = 0.1554 |
Table 2 Selected bond distances (Å) and angles (°) for L2 and complexes 1–3a
|
L2
|
|
(A) −x + 2, −y, −z; −x, y, −z + 3/2 for L2. (A) −x, −y, −z + 1 for 1. (A) −x − 1, −y + 1, −z + 1 for 2. (A) −x + 1/2, −y + 1, z for 3.
|
|
Distances
|
| O–C(3) |
1.217(3) |
N(1)–C(3) |
1.358(3) |
| N(1)–C(4) |
1.401(3) |
N(2)–C(7) |
1.333(3) |
| N(2)–C(8) |
1.343(3) |
C(1)–C(2) |
1.496(4) |
|
C(1)–C(1A) |
1.522(5) |
C(2)–C(3) |
1.508(4) |
| C(4)–C(8) |
1.375(3) |
C(4)–C(5) |
1.377(4) |
| C(5)–C(6) |
1.374(4) |
C(6)–C(7) |
1.363(4) |
|
Angles
|
| C(3)–N(1)–C(4) |
127.5(2) |
C(7)–N(2)–C(8) |
117.9(2) |
| C(2)–C(1)–C(1A) |
112.2(3) |
C(1)–C(2)–C(3) |
115.2(2) |
| O–C(3)–N(1) |
123.9(2) |
O–C(3)–C(2) |
123.0(2) |
| N(1)–C(3)–C(2) |
113.1(2) |
C(8)–C(4)–C(5) |
117.4(2) |
| C(8)–C(4)–N(1) |
123.5(2) |
C(5)–C(4)–N(1) |
119.1(2) |
| C(6)–C(5)–C(4) |
119.9(2) |
C(7)–C(6)–C(5) |
119.0(3) |
| N(2)–C(7)–C(6) |
122.5(3) |
N(2)–C(8)–C(4) |
123.2(3) |
| Complex 1 |
|
Distances
|
| Cd–O(1A) |
2.215(3) |
Cd–O(1) |
2.215(3) |
| Cd–N(1) |
2.267(3) |
Cd–N(1A) |
2.267(3) |
| Cd–O(2) |
2.409(4) |
Cd–O(2A) |
2.409(4) |
|
Angles
|
|
O(1A)–Cd–O(1) |
180.0 |
O(1A)–Cd–N(1) |
97.89(10) |
| O(1)–Cd–N(1) |
82.11(10) |
O(1A)–Cd–N(1A) |
82.11(10) |
| O(1)–Cd–N(1A) |
97.89(10) |
N(1)–Cd–N(1A) |
180.0 |
|
O(1A)–Cd–O(2) |
86.30(15) |
O(1)–Cd–O(2) |
93.70(15) |
| N(1)–Cd–O(2) |
85.53(12) |
N(1A)–Cd–O(2) |
94.47(12) |
|
O(1A)–Cd–O(2A) |
93.70(15) |
O(1)–Cd–O(2A) |
86.30(15) |
| N(1)–Cd–O(2A) |
94.47(12) |
N(1A)–Cd–O(2A) |
85.53(12) |
| O(2)–Cd–O(2A) |
180.00(11) |
|
|
| Complex 2 |
|
Distances
|
| Cd–O(3A) |
2.336(4) |
Cd–O(3) |
2.336(4) |
| Cd–N(3A) |
2.352(4) |
Cd–N(3) |
2.352(4) |
| Cd–N(1A) |
2.377(4) |
Cd–N(1) |
2.377(4) |
|
Angles
|
|
O(3A)–Cd–O(3) |
180.0 |
O(3A)–Cd–N(3A) |
89.22(16) |
| O(3)–Cd–N(3A) |
90.78(16) |
O(3A)–Cd–N(3) |
90.78(16) |
| O(3)–Cd–N(3) |
89.22(16) |
N(3A)–Cd–N(3) |
180.000(1) |
|
O(3A)–Cd–N(1A) |
88.94(16) |
O(3)–Cd–N(1A) |
91.06(16) |
|
N(3A)–Cd–N(1A) |
89.18(15) |
N(3)–Cd–N(1A) |
90.82(15) |
|
O(3A)–Cd–N(1) |
91.06(16) |
O(3)–Cd–N(1) |
88.94(16) |
|
N(3A)–Cd–N(1) |
90.80(15) |
N(3)–Cd–N(1) |
89.20(15) |
|
N(1A)–Cd–N(1) |
180.000(1) |
|
|
| Complex 3 |
|
Distances
|
| Cd–N(1A) |
2.336(3) |
Cd–N(1) |
2.336(3) |
| Cd–O(7) |
2.372(4) |
Cd–O(7A) |
2.372(4) |
| Cd–N(3) |
2.364(4) |
Cd–N(3A) |
2.364(4) |
|
Angles
|
|
N(1A)–Cd–N(1) |
179.71(19) |
N(1A)–Cd–O(7) |
89.94(14) |
| N(1)–Cd–O(7) |
89.85(14) |
N(1A)–Cd–O(7A) |
89.85(14) |
| N(1)–Cd–O(7A) |
89.94(14) |
O(7)–Cd–O(7A) |
86.5(2) |
|
N(1A)–Cd–N(3) |
94.90(12) |
N(1)–Cd–N(3) |
85.29(12) |
| O(7)–Cd–N(3) |
89.72(15) |
O(7A)–Cd–N(3) |
173.94(15) |
|
N(1A)–Cd–N(3A) |
85.29(12) |
N(1)–Cd–N(3A) |
94.90(12) |
| O(7)–Cd–N(3A) |
173.94(15) |
O(7A)–Cd–N(3A) |
89.72(15) |
| N(3)–Cd–N(3A) |
94.41(18) |
|
|
Results and discussions
Structure of L2
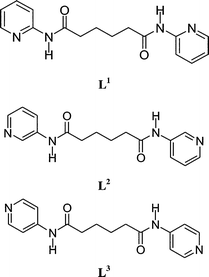
The structure of L2 was solved in the space groupP21/n with two molecules in a unit cell. The molecule of L2 lies about an inversion center. Fig. 1(a) shows the molecular structure of L2, which is planar with the dihedral angle between the two pyridyl rings of 0°. Fig. 1(b) shows the packing diagram for L2, and Fig. 1(c) depicts a side view showing the undulation. The molecules of L2 are linked by N–H---N interactions (H⋯N = 2.052 Å, N⋯N = 2.971 Å; ∠N–H⋯N 174.9°) to form 2D pleated grids of hcb topology (hexagonal layers) that stacks as ABAB along [1,0,−1].
 |
| | Fig. 1 (a) A molecular structure of L2. (b) The molecules of L2 are interlinked through extensive N–H⋯N interactions to form 2D pleated grids. (c) A side view showing the undulation. | |
Structure of [Cd(L1)(ClO4)2]∞, 1
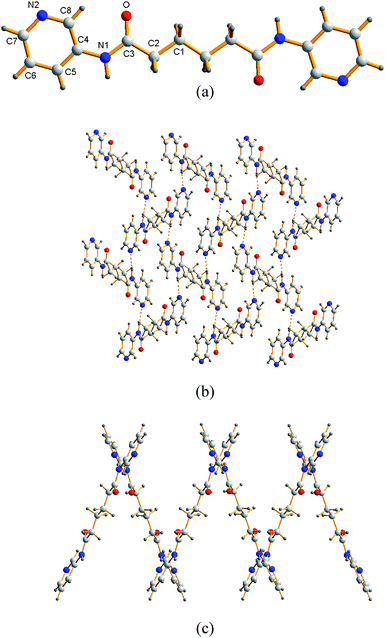
The crystals of complex 1 conform to the space groupP21/n. A molecular structure showing the arrangement about the Cd(II) metal center is shown in Fig. 2(a). The Cd(II) atom lies on an inversion center and the L1 ligand lies about another independent inversion center. The Cd(II) metal center, which is coordinated by two pyridyl nitrogen atoms [Cd–N = 2.267(3) Å] and two carbonyl oxygen atoms [Cd–O = 2.215(3) Å] of two L1 ligands and two oxygen atoms of two trans ClO4− anions [Cd–O = 2.409(4) Å] adopts a distorted octahedral geometry. The L1 ligands are coordinated to the Cd(II) metal centers in a tetradentate bonding mode through two pyridyl nitrogen atoms and two carbonyl oxygen atoms, resulting in 1D zigzag chains. The 1D zigzag chains are interlinked through extensive N–H⋯O (H⋯O = 2.203 Å, N⋯O = 3.009 Å, ∠N–H⋯O = 164.1°) interactions to the oxygen atoms of the coordinated ClO4− anions, Fig. 2(b). The chains are linked to the 4 nearest neighbor parallel chains with a Cd⋯Cd distance of 8.25 Å, Fig. 2(c), resulting in an overall 3D net of simple cubic topology pcu, as expected from rod packing of this kind.12 This tetradentate bonding mode is in marked contrast to those found in the Ag(I) complexes of L1 ligands, where the L1 ligands are coordinated to the Ag(I) metal centers in a bidentate mode through only the two pyridyl nitrogen atoms.9a
 |
| | Fig. 2 (a) A molecular structure of 1 showing the environment about the Cd(II) atom. Symmetry transformations used to generate equivalent atoms: A, −x, −y, −z + 1. (b) The zigzag chains are interlinked by the N–H⋯O interactions. (c) The chains are linked to the 4 nearest neighbor parallel chains. | |
Structure of {[Cd(L2)2 (CH3OH)2](ClO4)2·2CH3OH}∞, 2
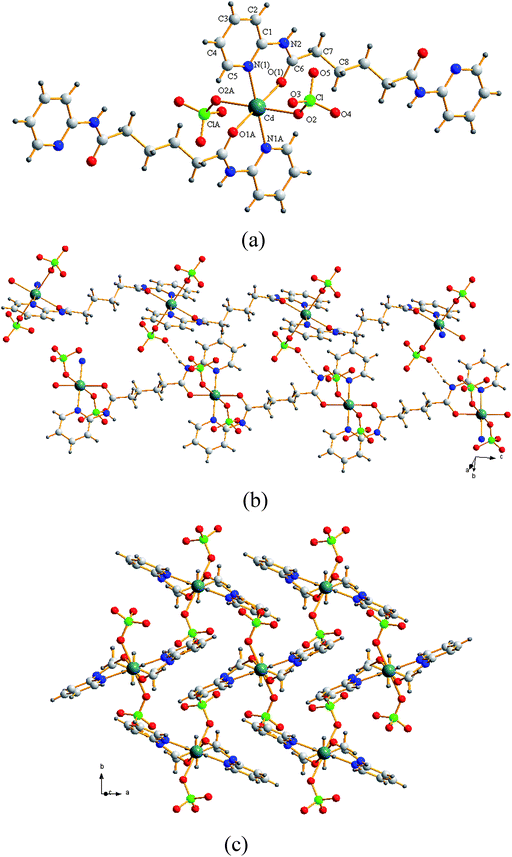
The crystal structure of 2 was solved in the space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the building block is shown in Fig. 3(a). The Cd(II) atom lies on an inversion center and the two independent L2 ligands lie about other independent inversion centers. The Cd(II) metal center which exhibits an octahedral arrangements is coordinated by four pyridyl nitrogen atoms [Cd–N = 2.351(4) and 2.379(4) Å] of four L2 ligands and two oxygen atoms [Cd–O = 2.336(4) Å] of two cis methanol molecules. The extended structure contains layers of 2D pleated nets based on the sql topology with rectangular grid-like corrugated sheets showing dimensions of 17.19 × 17.96 Å2, Fig. 3(b), which are positioned in an ABAB manner, Fig. 3(c). All the 2D nets are equivalent and are composed of 60-membered metallocycles. The distortion from a perfect square is due to the presence of two independent L2 ligands which adopt the same GAGtrans conformation, but different in torsional angles. The sql nets are interlinked by the ClO4− anions through N–H⋯O (H⋯O = 2.044 Å, N⋯O = 2.898 Å, ∠N–H⋯O = 172.0°) and weaker C–H⋯O (H⋯O = 2.719 Å, C⋯O = 3.467 Å, ∠C–H⋯O = 138.1°; H⋯O = 2.802 Å, C⋯O = 3.654 Å, ∠C–H⋯O = 152.8°) interactions, Fig. 3(d).
and the building block is shown in Fig. 3(a). The Cd(II) atom lies on an inversion center and the two independent L2 ligands lie about other independent inversion centers. The Cd(II) metal center which exhibits an octahedral arrangements is coordinated by four pyridyl nitrogen atoms [Cd–N = 2.351(4) and 2.379(4) Å] of four L2 ligands and two oxygen atoms [Cd–O = 2.336(4) Å] of two cis methanol molecules. The extended structure contains layers of 2D pleated nets based on the sql topology with rectangular grid-like corrugated sheets showing dimensions of 17.19 × 17.96 Å2, Fig. 3(b), which are positioned in an ABAB manner, Fig. 3(c). All the 2D nets are equivalent and are composed of 60-membered metallocycles. The distortion from a perfect square is due to the presence of two independent L2 ligands which adopt the same GAGtrans conformation, but different in torsional angles. The sql nets are interlinked by the ClO4− anions through N–H⋯O (H⋯O = 2.044 Å, N⋯O = 2.898 Å, ∠N–H⋯O = 172.0°) and weaker C–H⋯O (H⋯O = 2.719 Å, C⋯O = 3.467 Å, ∠C–H⋯O = 138.1°; H⋯O = 2.802 Å, C⋯O = 3.654 Å, ∠C–H⋯O = 152.8°) interactions, Fig. 3(d).
 |
| | Fig. 3 (a) A molecular structure of 2 showing the environment about the Cd(II) atom. (b) The structure of 2 contains layers of 2D pleated nets based on the sql topology, which are positioned in an ABAB manner, (c). (d) The 2D nets are interlinked through N–H⋯O and C–H⋯O interactions. | |
Structure of [Cd(L3)2(ClO4)2(CH3CH2OH)2]∞, 3
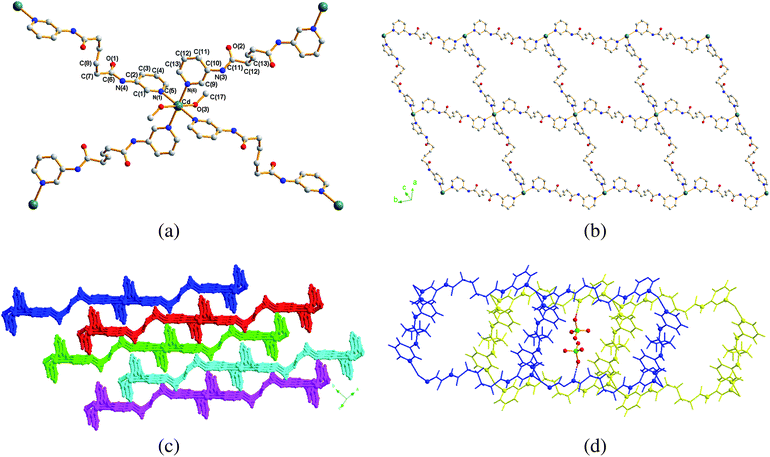
The crystal structure of 3 was solved in the space groupPcca and the building block is shown in Fig. 4(a). The Cd(II) metal center, which lies on a twofold axis bisecting the N(1)–Cd–N(1A), N(3)–Cd–N(3A) and O(7)–Cd–O(7A) angles, exhibits an octahedral arrangement. Each Cd(II) metal atom is coordinated by four nitrogen atoms [Cd–N = 2.337(4) and 2.367(4) Å] of four L3 ligands and two oxygen atoms [Cd–O = 2.358(4) Å] of two cis ethanol molecules. One of the L3 ligands lies on an independent twofold axis and the other one lies about an inversion center. The structure that is a complex 3D entanglement13 may be described in two steps: there are 3-fold interpenetrated sql layers that stack as ABAB, but with a distance between the 3-fold thick layers short enough to produce polycatenation among them. The two independent L3 ligands adopt two different conformations of AAAtrans and AGAtrans, resulting in rectangular grid-like corrugated sheets showing dimensions of 20.98 (AGAtrans) × 22.70 (AAAtrans) Å2. All sql are equivalent and are composed of 68-membered metallocycles, Fig. 4(b). Three sql interpenetrate parallelly (i.e. with the same average plane), forming a 2D layer with a thickness of approximately 14.92 Å (2D → 2D interpenetration), Fig. 4(c). Fig. 4(d) shows a schematic view of the three-fold parallel interpenetration. In addition, layers of the three-fold parallel interpenetration sheets polycatenated with other parallel but offset sql layers, giving the final 3-D entanglement, Fig. 4(e). Each sql thus is catenated with six others. While two are coplanar, two are “above” at different heights and the other two are “below” at different heights, resulting in a Doc = 6 and Is = 3.13e,f The mean planes of the two sets of three-fold parallel interpenetrating sheets which are positioned “above” and “below” are coincident, i.e. these average planes are in an ABAB fashion. Fig. 4(f) shows that each window of the three-fold interpenetrating sheets has six others passing through it. Another peculiar feature of this kind of entanglement is that only two sql from each set of the 3-fold layers “above” and “below” catenate with any given sql and not with all three (hence Density of catenation Doc = 2 + 2 + 2 and not 2 + 3 + 3). This is due to the undulation of the layers: one sql is left out from the polycatenation. Such a strange and very rare feature has been previously observed for polycatenation of the 2-fold hcb (hexagonal) layer with Doc = 3 = 1 + 1 + 1 (and not 1 + 2 + 2).13(f) No other examples are known.
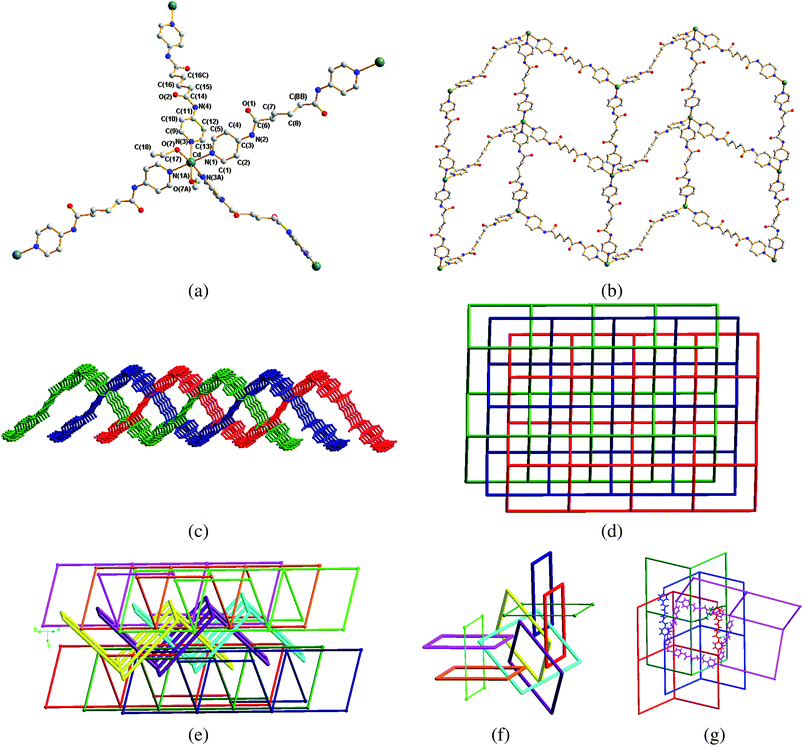 |
| | Fig. 4 (a) The building block of 3, showing octahedral environment about the Cd(II) metal center. Symmetry transformations used to generate equivalent atoms: A, 0.5 − x, 1 − y, z; B, −0.5 − x, 2 − y, z; C, −x, −y, −z. (b) A drawing showing the undulating nature of a single sql layer. (c) A side view of the three-fold 2D → 2D parallel interpenetrating sql sheets (view down the a axis). (d) A schematic view of the three-fold sql interpenetration. (e) A drawing showing the parallel polycatenation of 3-fold thick layers to give a 3D entanglement. From the figure it is evident that the removal of 3 layers will disconnect the entanglement (Index of Separation Is = 3). (f) The central square window (cyan) of the three-fold interpenetrating sheets has six others passing through it, (Density of catenation Doc = 6). Note that the thinner green squares are not catenated with the central one. (g) A drawing showing the N–H⋯O interactions that interlink the sql nets. | |
The resulting 3D entanglement contains 1D channels which are occupied by the ClO4− anions, interacting with the cations through N–H⋯O (H⋯O = 2.381 Å, N⋯O = 3.174 Å, ∠N–H⋯O = 153.7°) and weaker C–H⋯O (H⋯O = 2.568 Å, C⋯O = 3.419 Å, ∠C–H⋯O = 150.6°) hydrogen bonds. The framework is also supported by the N–H⋯O (H⋯O = 2.092 Å, N⋯O = 2.946 Å, ∠N–H⋯O = 172.2°) and weaker C–H⋯O (H⋯O = 2.541 Å, C⋯O = 3.400 Å, ∠C–H⋯O = 153.8° for pyridyl hydrogen atoms; H⋯O = 2.502 Å, C⋯O = 3.331 Å, ∠C–H⋯O = 143.3° for methylene hydrogen atoms) interactions resulting form the adjacent L3 ligands. The weak H-bonds produce one single 3D complex net. Fig. 4(g) depicts a molecular structure showing the N–H⋯O interactions that interlinked the sql nets.
Conformations of the ligands
Table 3 lists the ligand conformations found for the L2 ligand and complexes 1–3. The L1 ligands in complex 1 adopt an AAAtrans conformation and the L2 ligands in complex 2 adopt a GAGtrans conformation for the formation of 1D chains and 2D sql pleated sheets, while the L3 ligands adopt both the AAAtrans and AGAtrans conformations for complex 3 to exhibit a 3D polycatenated entangled framework. The conformation of the L1 ligand in complex 1 which adopts the tetradentate bonding mode is in marked contrast to those found in the Ag(I) complexes of L1 ligands, where the L1 ligands are coordinated to the Ag(I) metal centers in a bidentate mode through only the two pyridyl nitrogen atoms and adopt AAAtrans, GGGcis and AGAcis conformations.9(a) It is also noted that for the two independent L2 ligands in complex 2, which adopt the same GAGtrans conformation, a difference in the torsional angles is observed. In complex 3, the L3 ligand with AGAtrans conformation shows a dihedral angle of 18.1° between the two pyridyl rings while the two pyridyl rings of the ligand with an AAAtrans conformation are coplanar. For comparison, it is noted that in the polymeric complex [CuSO4(L3)(H2O)2]∞, which shows a twelvefold diamondoid network, the L3 ligands adopt the AAAtrans and GAGtrans conformations which also differ in the dihedral angle between the two pyridyl rings.9(b)
Table 3 Ligand conformations and corresponding angles for L2 and complexes 1–3
| Compounds |
Diagram |
Torsion angle/° |
Conformation |
Dihedral angle/° |
|
Due to the disorder of the C(16) atom, there are two sets of torsion angles.
|
|
L2
|

|
−178.6, 180.0, 178.6 |
AAA
trans |
0 |
|
1
|
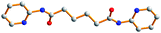
|
−179.4, 180, 179.4 |
AAA
trans |
0 |
|
2
|
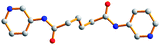
|
52.2, 180, −52.2 |
GAG
trans |
0 |
| |
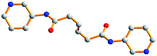
|
68.2, 180, −68.2a
−59.7, 180, 59.7
|
GAG
trans |
0 |
|
3
|
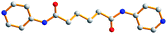
|
−179.0, −180, 179.0 |
AAA
trans |
0 |
| |
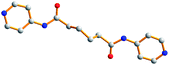
|
169.7, 60.3, 169.7 |
AGA
trans |
18.1 |
Luminescent properties
The photoluminescent properties of the frameworks ligands 1, 2 and 3 were investigated in the solid state at room temperature. Complexes 1, 2 and 3 emit at λmax = 392, 422 and 408 nm, respectively, upon excitation at 246, 246 and 244 nm, while the free L1, L2 and L3 ligands exhibit emission at 385, 397 and 394 nm, respectively, upon excitation at 250, 246 and 247, respectively. Since there is no significant energy shift observed upon coordinating ligands to the Cd atoms, the emission bands in 1, 2 and 3 may be tentatively assigned as intraligand (IL) π → π* transitions.
Thermal analyses
Thermal gravimetric analyses (TGA) were carried out to examine the thermal stabilities of 1–3. The samples were heated up in nitrogen gas at a pressure of 1 atm with a heating rate of 10 °C min−1. The TGA curves, Fig. 5, show that 1 is stable up to 200 °C. A total weight loss of 16.4% occurred in the temperature range 200–270 °C, due to the removal of one of the bonded ClO4− anions per formula unit (calcd 16.3%). The second weight loss of 25.6% occurred in the temperature range 300–350 °C, corresponding to the decomposition of the 1-D chains resulting from the removal of the methylene groups of the L1 ligands and another bonded ClO4− anion per formula unit (calcd 25.5%). The pyridyl and amide groups of the L1 ligands were removed in the temperature range 350–600 °C. The final residue percentage of 21.8% is consistent with the formation of CdO (calcd 21.1%) in 1 in the temperature range of 600–700 °C.
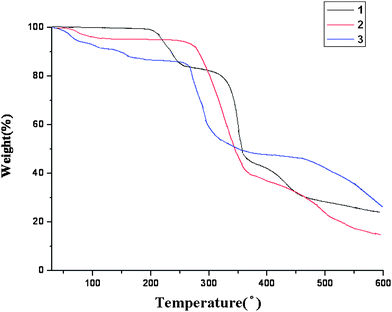 |
| | Fig. 5
TGA curves for complexes 1–3. | |
Complex 2 is stable up to 80 °C. A total weight loss of 6.2% occurred in the temperature range 80–132 °C, presumably due to the removal of two cocrystallized CH3OH molecules per formula unit (calcd 6.2%). The second weight loss of 45.50% occurred in the temperature range 250–360 °C, which can be ascribed to the decomposition of the 2-D nets, due to the removal of two bonded CH3OH molecules, the methylene groups of the L2 ligands and the uncoordinated ClO4− anions per formula unit (calcd 45.51%). The pyridyl and amide groups of the L2 ligands were removed in the temperature range 357–600 °C. The final residue percentage of 12.3% is consistent with the formation of CdO (calcd 12.4%) in 2. Complex 3 is stable up to 78 °C. A total weight loss of 9.1% occurred in the temperature range 78–140 °C, due to the removal the two bonded ethanol groups (calcd 9.2%). The second weight loss of 9.8% occurred in the temperature range 140–270 °C, due to the decomposition of one of the uncoordinated ClO4− anions per formula unit (calcd 9.9%). The third weight loss of 31.5% occurred in the temperature range 270–326 °C, which can be ascribed to the removal the methylene groups of the L3 ligand and the uncoordinated ClO4− anion per formula unit (calcd 31.6%). The pyridyl and amide groups of the L3 ligands were removed in the temperature range 326–700 °C. The final residue percentage of 12.7% is consistent with the formation of CdO (calcd 12.8%) in 3. From the TGA data obtained, it seems that the formation of 3D entanglement in 3 does not increase its thermal stability as compared with the other 1D and 2D complexes.
Conclusions
In this study, we have successfully prepared three Cd(II) coordination polymers showing 1D zigzag chains, 2D pleated sql sheets and a rare polycatenated interpenetrated 3D network, respectively, by using three isomeric ligands. All the Cd(II) centers possess octahedral geometry. A new tetradentate bonding mode of the L1 ligand was observed in complex 1. The results show that the structural types of the Cd(II) coordination polymers can be directed by ligand isomerism. Obviously, these three isomeric ligands are sufficiently flexible to adjust to the stereochemical requirements for the formation of complexes 1–3, which adopt conformations that maximize their intra- and intermolecular forces.
Acknowledgements
We are grateful to the National Science Council of the Republic of China for support. This research was also supported by the project of the specific research fields in Chung Yuan Christian University, Taiwan, R. O. C. under grant CYCU-95-CR-CH.
References
-
(a)
E. R. T. Tiekink and J. J. Vittal, Frontiers in Crystal Engineering, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed
 ;
(b)
R. Robson, B. E. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins and J. Lieu, Supramolecular Architecture, ACS Publications, Washington, DC, 1992 Search PubMed ;
(c) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef CAS ;
(d) H. Gudbjartson, K. Biradha, K. M. Poirier and M. J. Zaworotko, J. Am. Chem. Soc., 1999, 121, 2599 CrossRef CAS ;
(e) C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 397 RSC ;
(f) M. Fujita and K. Ogura, Coord. Chem. Rev., 1996, 148, 249 CrossRef CAS ;
(g) S.-I. Noro, R. Kitauta, M. Kondo, S. Kitagawa, T. Ishii, H. Matauzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568 CrossRef CAS ;
(h) S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739 CAS ;
(i) M. J. Zaworotko, Chem. Soc. Rev., 1994, 23, 283 RSC ;
(j) D. M. L. Goodgame, D. A. Grachvogel and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 153 CrossRef CAS ;
(k) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, Chem.–Eur. J., 2002, 8, 1520 CrossRef ;
(l) D. S. Reddy, K. E. Dewa and Y. Aoyama, Angew. Chem., Int. Ed., 2000, 39, 4266 CrossRef CAS .
;
(b)
R. Robson, B. E. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins and J. Lieu, Supramolecular Architecture, ACS Publications, Washington, DC, 1992 Search PubMed ;
(c) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef CAS ;
(d) H. Gudbjartson, K. Biradha, K. M. Poirier and M. J. Zaworotko, J. Am. Chem. Soc., 1999, 121, 2599 CrossRef CAS ;
(e) C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 397 RSC ;
(f) M. Fujita and K. Ogura, Coord. Chem. Rev., 1996, 148, 249 CrossRef CAS ;
(g) S.-I. Noro, R. Kitauta, M. Kondo, S. Kitagawa, T. Ishii, H. Matauzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568 CrossRef CAS ;
(h) S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739 CAS ;
(i) M. J. Zaworotko, Chem. Soc. Rev., 1994, 23, 283 RSC ;
(j) D. M. L. Goodgame, D. A. Grachvogel and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 153 CrossRef CAS ;
(k) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, Chem.–Eur. J., 2002, 8, 1520 CrossRef ;
(l) D. S. Reddy, K. E. Dewa and Y. Aoyama, Angew. Chem., Int. Ed., 2000, 39, 4266 CrossRef CAS .
-
(a) In this paper we adopt the three-letter symbol for nets, proposed by M. O'Keeffe, that can be retrieved from the RCSR database (Reticular Chemistry Structure Resource, http://rcsr.anu.edu.au/). sql = square layer, pcu = primitive cubic. See also: O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 22 Search PubMed ;
(b) O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 515 CrossRef ;
(c) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS ;
(d) O. Delgado-Friedrichs, M. D. Foster, M. O'Keeffe, D. M. Proserpio, M. M. J. Treacy and O. M. Yaghi, J. Solid State Chem., 2005, 178, 2533 CrossRef CAS ;
(e) O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Phys. Chem. Chem. Phys., 2007, 9, 1035 RSC .
-
(a) R. P. Sijbesma and E. W. Meijer, Curr. Opin. Colloid Interface Sci., 1999, 4, 24 CrossRef CAS ;
(b) S. C. Zimmerman and P. S. Corbin, Struct. Bonding, 2000, 96, 63 CAS ;
(c) D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83 RSC ;
(d) D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229 CrossRef CAS .
-
(a) Y.-N. Chi, F.-Y. Cui, Y.-Q. Xu and C.-W. Hu, Inorg. Chem. Commun., 2007, 10, 695 CrossRef CAS ;
(b) Z. He, Z.-M. Wang and C.-H. Yan, CrystEngComm, 2005, 7, 143 RSC ;
(c) J. Seo, M. R. Song, J.-E. Lee, S. Y. Lee, I. Yoon, K.-M. Park, J. Kim, J. H. Jung, S. B. Park and S. S. Lee, Inorg. Chem., 2006, 45, 952 CrossRef CAS .
-
(a) H.-H. Li, Z.-R. Chen, J.-Q. Li, C.-C. Huang, Y. F. Zhang and G.-X. Jia, Cryst. Growth Des., 2006, 6, 1813 CrossRef CAS ;
(b) M. O. Awaleh, A. Badia and F. Brisse, Cryst. Growth Des., 2006, 6, 2674 CrossRef CAS ;
(c) S. Noro, R. Kitaura, M. Kondo, S. Kitagawa, T. Ishii, H. Matsuzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568 CrossRef CAS ;
(d) Y.-H. Wang, K.-L. Chu, H.-C. Chen, C.-W. Yeh, Z.-K. Chan, M.-C. Suen and J.-D. Chen, CrystEngComm, 2006, 8, 84 RSC ;
(e) C.-Y. Lin, Z.-K. Chan, C.-W. Yeh, C.-J. Wu, J.-D. Chen and J.-C. Wang, CrystEngComm, 2006, 8, 841 RSC .
- X. H. Bu, W. Chen, W. F. Hou, R. H. Zhang and F. Brisse, Inorg. Chem., 2002, 41, 3477 CrossRef CAS .
- A. J. Blake, N. R. Champness, P. A. Cooke, J. E. B. Nicolson and C. J. Wilson, J. Chem. Soc., Dalton Trans., 2000, 3811 RSC .
-
(a) M. B. Zaman, M. D. Smith and H.-C. z. Loye, Chem. Commun., 2001, 2256 RSC ;
(b) M.-L. Tong, Y.-M. Wu, J. Ru, X.-M. Chen, H.-C. Chang and S. Kitagawa, Inorg. Chem., 2002, 41, 4846 CrossRef CAS ;
(c) M. J. Plater, M. R. St. J. Foreman, T. Gelbrich and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2000, 1995 RSC ;
(d) M. J. Plater, M. R. St. J. Foreman, T. Gelbrich, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2000, 3065 RSC ;
(e) M. Fujita, Y. J. Kwon, M. Miyazawa and K. Ogure, J. Chem. Soc., Chem. Commun., 1994, 1977 RSC ;
(f) M. Fujita, Y. J. Kwon, O. Sasaki, K. Yamaguchi and K. Ogura, J. Am. Chem. Soc., 1995, 117, 7287 CrossRef CAS ;
(g) M. O. Awaleh, A. Badia and F. Brisse, Inorg. Chem., 2005, 44, 7833 CrossRef CAS ;
(h) M. O. Awaleh, A. Badia and F. Brisse, Cryst. Growth Des., 2005, 5, 1897 CrossRef CAS ;
(i) M. Du, C.-P. Li and X.-J. Zhao, Cryst. Growth Des., 2006, 6, 335 CrossRef CAS ;
(j) S. Muthu, J. H. K. Yip and J. J. Vittal, J. Chem. Soc., Dalton Trans., 2002, 4561 RSC ;
(k) See, for example: L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, CrystEngComm, 2002, 4, 121 Search PubMed .
-
(a) H.-C. Chen, H.-L. Hu, Z.-K. Chan, C.-W. Yeh, H.-W. Jia, C.-P. Wu, J.-D. Chen and J.-C. Wang, Cryst. Growth Des., 2007, 7, 698 CrossRef CAS ;
(b) Y. F. Hsu, C.-H. Lin, J.-D. Chen and J.-C. Wang, Cryst. Growth Des., 2008, 8, 1094 CrossRef CAS .
-
(a)
XSCANS, Release, 2.1, Siemens Energy & Automation, Inc, Madison, Wisconsin, USA, 1995 Search PubMed .
-
SHELXTL 5.10. Bruker Analytical X-ray Instruments Inc., Karlsruche, Germany, 1997 Search PubMed .
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504 CrossRef CAS .
-
(a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1461 CrossRef ;
(b)
S. R. Batten and R. Robson, in Molecular Catenanes, Rotaxanes and Knots, A Journey Through the World of Molecular Topology, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999, pp 77–105 Search PubMed ;
(c) S. R. Batten, CrystEngComm, 2001, 3, 67 RSC ;
(d) S. R. Batten, Curr. Opin. Solid State Mater. Sci., 2001, 5, 107 CrossRef CAS ;
(e) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247 CrossRef CAS ;
(f) S. Banfi, L. Carlucci, E. Caruso, G. Ciani and D. M. Proserpio, Cryst. Growth Des., 2004, 4, 29 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.