Rational design microporous pillared-layer frameworks: syntheses, structures and gas sorption properties†
Chaoying
Gao
,
Shuxia
Liu
*,
Linhua
Xie
,
Chunyan
Sun
,
Jianfang
Cao
,
Yuanhang
Ren
,
Dan
Feng
and
Zhongming
Su
Key Laboratory of Polyoxometalates Science of Ministry of Education, College of Chemistry, Northeast Normal University, Changchun City, JiLin, P. R. China 130024. E-mail: liusx@nenu.edu.cn
First published on 3rd November 2008
Abstract
The solvothermal reactions of nickel nitrate hexahydrate, aromatic carboxylic acids, and either 4,4′-bipyridine (bipy) or pyrazine (pyz) produced three open-framework compounds, namely, Ni(HBTC)(pyz)·3DMF 2 (BTC = 1,3,5-benzentricarboxylate, DMF = N,N′-dimethylformamide), Ni2(BTEC)(bipy)3·3DMF 3 (BTEC = 1,2,4,5-benzenetetracarboxylate), together with previously reported Ni(HBTC)(bipy)·3DMF 1. Compound 2 is the product when pyz pillars replace bipy pillars in the reaction of 1, and it is a 3D porous network where honeycomb grid layers constructed by Ni-BTC groups are interlinked by pyz pillars. When the BTEC ligand takes the place of BTC ligand in the system, compound 3 is obtained and it is a 3D connectivity with 1D channels where Ni-BTEC chains are interlinked by bipy ligands. The three compounds feature permanent porosity, which was confirmed by N2 adsorption isotherm measurement, and the architectural features of 1–3 can be ascribed to pillared-layer motifs. Furthermore, hydrogen adsorption measured at 77 K for 1 shows an excess uptake of 3.03 wt% at 20 bar. Details of the synthesis conditions, crystal structures and property measurements of each new compound are described.
Introduction
The design and construction of metal–organic frameworks (MOFs) are of current interest due to a virtually endless number of possible structural topologies with variable properties1–3 and potential applications.4–8 The key to this success is in designing extraordinary permanent porosity even in the absence of guest molecules, to afford the desired robust frameworks.9–14 It has been proven that metal/multi-carboxylate species are sufficiently strong to stabilize networks against collapse;15–17 however, there is usually a lack of predicting the exact skeleton of the product. On the other hand, metal/pyridine frameworks require counterionic guests in the cavities or channels of the solids and removal of the charge compensation normally leads to damaging the frameworks. It has been shown that both problems might be circumvented by incorporating O- and N-donor coordinations in a mixed-ligand framework system.18–21In previous studies, we mainly focused on the transition metal–BTC22 systems under solvothermal conditions. Unfortunately, the corresponding products did not possess permanent porosity. Taking this reality into account and with the above considerations in mind, we employed the ‘pillaring’ strategy23 by incorporating O- and N-donor coordinations in a mixed-ligand framework system and successfully obtained compound Ni(HBTC)(bipy)·3DMF 124 with expected connectivity, where the honeycomb grid layers constructed of nickel-BTC groups were linked by bipy pillars to form a 3D highly porous framework (Fig. 1a). The investigative result forcefully indicated that the system containing two robust bridging ligands is effective for predicting the architecture of the product and stable enough against channel collapse. As part of our ongoing research, we introduced the pyz instead of the bipy to serve as the ‘pillar’ and obtained the Ni(HBTC)(pyz)·3DMF 2 (Fig. 1b), which realized the length of the linear ligand, controlling the pore size and free accessible volume as we had forecasted. In order to investigate the influence of the multicarboxylic acid on the formation of architectures, we also applied the H4BTEC to replace the H3BTC in the mixed-ligand system and synthesized a new compound, Ni2(BTEC)(bipy)3·3DMF 3 (Fig. 1c), the architecture of which can also be described as a pillared-layer motif. Moreover, compounds 2 and 3 feature permanent porosity just as 1. Herein, the crystal structures, synthesis investigation, along with N2 sorption properties of compounds 2, 3 and H2 uptake measurement for compound 1 will be presented and discussed.
![Perspective view of the frameworks normal to the [100] plane in (a) 1, (b) 2, and (c) 3. For clarity the hydrogen atoms have been omitted. Ni: green; N: blue; O: red; C: dark.](/image/article/2009/CE/b812097h/b812097h-f1.gif) | ||
| Fig. 1 Perspective view of the frameworks normal to the [100] plane in (a) 1, (b) 2, and (c) 3. For clarity the hydrogen atoms have been omitted. Ni: green; N: blue; O: red; C: dark. | ||
Experimental
General materials and methods
All the reagents and solvents were purchased from commercial sources and used as received without further purification. Elemental analyses (C, H, and N) were performed on a Perkin-Elmer 2400 CHN elemental analyzer, Ni was determined by a PLASMA-SPEC(I) ICP atomic emission spectrometer. IR spectra were recorded in the range 400–4000 cm−1 on an Alpha Centaurt FT/IR spectrophotometer using KBr pellets. Powder X-ray diffraction measurements were performed on a Rigaku D/MAX-3 instrument with Cu Kα radiation in the angular range 2θ = 3–60° at 293 K. Thermal gravimetric analyses (TGA) were performed on a Perkin-Elmer TGA7 instrument in N2 atmosphere with a heating rate of 10 °C min−1.Preparations
Single-crystal X-ray crystallography
Diffraction intensities for compounds 2–3 were collected on a Siemens Smart CCD diffractometer with Mo Kα monochromatic radiation (λ = 0.71073 Å) 183 and 293 K. The linear absorption coefficients, scattering factors for the atoms, and the anomalous dispersion corrections were taken from the International Tables for X-ray Crystallography. Empirical absorption corrections were applied. The structures were solved by the direct method and refined by the full-matrix least-squares method on F2 using the SHELXTL-97 crystallographic software package.25 All H atoms were placed geometrically. Owing to disorder, partial H atoms on the ligand BTC have not been added for compound 2. Anisotropic thermal parameters were used to refine all nonhydrogen atoms for compounds 2 and 3.In both cases, solvent molecules within channels were highly disordered and attempts to locate and refine the solvent peaks were unsuccessful. Contributions to scattering due to these solvent molecules were removed for 2 and 3 with the SQUEEZE routine within PLATON and refined further with the data generated;26 the disordered components in 2 and 3 were all included in the final structure-factor calculations. The numbers of disordered guest molecules for both compounds were determined according to elemental analyses and thermogravimetric analyses.
For the crystallographic data of compound 2, there is severe disorder because of the high crystallographically imposed symmetry (6/mmm). In the asymmetric unit, all atoms are disordered except for the two atoms located on the six-fold axis, Ni1 and N1. The other atoms on BTC and pyz ligands are disordered at six positions along the crystallographically six-fold axis.27 Owing to the disorder, the peak heights for these disordered atoms are very low and they became unstable in the subsequent refinements. Therefore, most of these atoms (O1, O2, O3, C1, C3, C4, C5) are finally refined with their coordinates fixed. This constraint gives rise to relatively higher or lower Ueq values for some of the atoms (O1, C1, C4). The severe disorder of the compound also gives a relatively low data/parameter ratio (6.05), which results from the increased number of atoms with partial occupancy factors in the asymmetric unit. An ORTEP view of the compound and the pertinent figures about the disorder are shown in Fig. S1 and S9.†
The detailed crystallographic data and structure refinement results for compounds 2 and 3 are summarized in Table 1, selected bond lengths and angles in Table S1.†
| 2 | 3 | |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR 2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. | ||
| Empirical formula | NiC22N5O9H29 | Ni2C49N9O11H47 |
| Formula weight | 566.19 | 1055.34 |
| Crystal system | Hexagonal | Triclinic |
| Space group | P6/mmm | P-1 |
| a/Å | 9.6103(3) | 8.793(2) |
| b/Å | — | 10.091(3) |
| c/Å | 6.9723(5) | 15.404(4) |
| α/° | — | 87.826(4) |
| β/° | — | 88.941(5) |
| γ/° | — | 73.318(4) |
| V/Å3 | 557.67(5) | 1308.3(6) |
| Z | 1 | 1 |
| Dc/g cm−3 | 1.686 | 1.340 |
| µ(Mo Kα)/mm−1 | 0.938 | 0.785 |
| N o. unique data | 260 | 5052 |
| θ/° | 2.45−25.87 | 1.32−26.09 |
| F(000) | 296 | 548 |
| Limiting indices | −11 ≤ h ≤ 8 | −7 ≤ h ≤ 10 |
| −9 ≤ k ≤ 11 | −12 ≤ k ≤ 12 | |
| −8 ≤ l ≤ 8 | −17 ≤ l ≤ 19 | |
| Data/restraints/parameters | 260/31/43 | 5052/0/253 |
| GOF (F2) | 1.167 | 0.923 |
| R 1 a(I > 2σ(I)) | 0.0453 | 0.0546 |
| wR 2 b(all data) | 0.1237 | 0.1271 |
Gas sorption measurements
A Micromeritics ASAP 2020 Surface Area and Porosity Analyzer was used to measure N2 sorption. The hydrogen adsorption isotherm was recorded using a RUBOTHERM magnetic suspension balance (Ankersmid B.V., Netherlands). In order to remove guest solvent molecules in the frameworks, the as-synthesized crystals 1–3 were heated under vacuum at 150 °C overnight. Before the measurement, the samples were vacuumed again by using the ‘outgas’ function of the surface area analyzer for 10 h at 150 °C. A sample of 0.108 g for 2, 0.092 g for 3 and 0.114 g for 1 was used for the gas sorption measurement and was maintained at 77 K with liquid nitrogen.Results and discussion
Syntheses of the compounds
The solvothermal reaction is a relatively complex process, and the final products under a given set of conditions are often unpredictable. For the system discussed here, the stoichiometry of the starting materials is crucial for the formation of targeted networks. Throughout a systematic work carried out under similar solvothermal conditions, it was found that compounds 2 and 3 could be obtained only in the specifical conditions, in which the amount of diamine ligands (pyz or bipy) should be more than that of aromatic carboxylic acid ligands (H3BTC or H4BTEC); however, the stoichiometry of Ni(NO3)2 to carboxylic acid was not rigorous for the isolation of the target products. Specifically, if the amount of pyz was less than that of H3BTC ligands in the reaction of 2, light-green crystals Ni(HBTC)(DMF)2·(guest)24 were isolated; if the amount of bipy was less than that of H4BTEC ligands in reaction system 3, amorphous powder but not crystals was obtained. It is also noteworthy to mention that if the color of the crystals produced is light-green not dark-green, suggesting that the diamine ligands did not take part in the coordination: for example, in the reaction of 2 if the crystals produced are light-green, we can estimate that the product is Ni(HBTC)(DMF)2·(guest);24 if the crystals produced are dark-green, we can estimate that the product is the target product compound 2.Reaction systems 2 and 3 were cooled manually at a rate of 10 °C h−1 to obtain crystals. And if the reaction systems were cooled unaided, no suitable crystals could be isolated. In addition, the increasing reaction time was beneficial for improving the crystal yields. Both compounds are stable upon exposure to the atmosphere and insoluble in water and common organic solvents such as ethanol, acetonitrile, chlorform and acetone. The phase purities of the bulk samples were identified by X-ray powder diffraction (Fig. S7 and S8).†
Structure description of 2
An X-ray single-crystal diffraction study was performed on the as-synthesized materials, revealing that 2 is a 3D microporous pillared-layer framework. The coordinative environment around the metal centre is presented in Fig. 2a, where the nickel (II) centre is bounded by three BTC units and two pyz units. One of the BTC units is completely deprotonated and coordinates to three metal centers in a bidentate fashion, whereas each of the other two units coordinates to three metal centers in a monodentate fashion; the remaining free carbonyls hydrogen-bond strongly to adjacent free carbonyls. In such a mode, the connectivity of the BTC ligand and Ni atoms results in a 2D layer parallel to the ab plane (Fig. 2c). The polar positions on the nickel(II) centre are occupied by pyz molecules, which disorder at two positions (Fig. S9†). The pyz ligands pillar the 2D layers generating the 3D framework of 2 (Fig. 1b). There are two distinct types of channels in network 2: the openings of the Ni(HBTC) layers are aligned to generate honeycomb channels (their dimensions are approximately 8 Å at the widest and 5 Å at the narrowest spacing considering the van der Waals radii of O atoms in the framework) running along the c-axis (Fig. S10†); normal to [100] and [010], the pyz pillars give further rectangle channels (the cross section is around 3.3 Å in height and 6 Å in width, taking into account the van der Waals radii of the C atoms in the framework) parallel to the layers extending throughout the ab-plane (Fig. 1b), The effective free volume of 2 was calculated by PLATON analysis as 46.3% of the crystal volume (3094.5 Å3 out of the 6679.4 Å3 unit cell volume).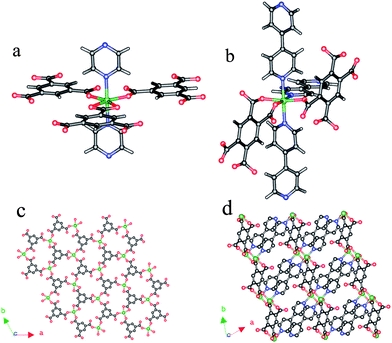 | ||
| Fig. 2 The coordinative environments around the metal centres in (a) compound 2 and (b) compound 3. A fragment of the 2D layer (c) included in compound 2 and (d) compound 3. Ni: green; N: blue; O: red; C: dark, H: white. | ||
Previously, we reported an isoreticular framework 1. With a different linear N-donor pillar, the lengths of these linear ligands control the pore sizes and free accessible volumes of the isoreticular frameworks featuring tunable structural properties.28
Rosseinsky and co-workers29 have reported a compound [Ni3(BTC)2(4,4′-bipy)3(C2H6O2)3(H2O)3·(guest)] synthesized by the diffusion method with the same metal cation and ligands. However, the compound becomes noncrystalline upon removal of the guest molecules. With a dissimilar synthetical route and connectivity, compounds 1 and 2 show their predominance of avoiding collapse in the absence of guest molecules (vide infra), the ability of which lies in the core of determining its suitability for applications.
In addition, Kwon and co-works reported four compounds based on a BTC-bipy mixed-ligand.30 The authors discussed in detail the diversification of the hydrothermal reaction products induced by naphthalene molecules. However further investigation of host–guest properties was not mentioned. Besides, Lee and co-workers31 have also reported a one-dimensional copper coordination polymer based on the BTC-bipy mixed-ligand. In summary, though many compounds constructed by BTC-bipy mixed-ligands have been reported, only the pillared-layer compounds 1 and 2 have been proven to be porous.
Structure description of 3
X-Ray crystallography reveals that 3 is a 3D neutral porous framework. The coordination environment of the metal centre for 3 is presented in Fig. 2b, where the nickel(II) centre is bound by three oxygen atoms from two different BTEC ligands and three nitrogen atoms from three bipy molecules. The Ni atom is related with a neighboring equivalent Ni atom through the crystallographic inversion center. Each BTEC linker is coordinated to four nickel centers in di-monodentate and monodentate fashions, and this connection leads Ni-BTEC to an infinite ladder chain (Fig. S11a).† The adjacent chains are further interlinked by one type of bipy (associated with N1 and N2) ligands to form a 2D layer (Fig. 2d). Such layers are pillared by the other type of bipy, giving rise to the 3D framework of 3 (Fig. 1c). There are two types of channels along the [100] and [010] directions (Fig. S11c and d).† The channel along the [100] direction is much more open. The windows offering access to these channels are defined by the van der Waals surfaces of the bipy molecules, and are identical in size, 7 × 7 Å2, at their closest edge to edge contact. Compared to the structure of 2, the architecture of 3 can also be considered as a pillar-layer motif, except that the layers are much thicker, which might be owing to more coordination sites for the ligand H4BTEC compared to H3BTC, resulting in a more complicated connectivity of the layers. The effective free volume of 3 was calculated by PLATON analysis as 42.4% of the crystal volume (555.3 Å3 out of the 1368.3 Å3 unit cell volume).It should be noted that several compounds based on BTEC-bipy have been reported previously.32,33 However, in comparison with 3, they are isolated under hydrothermal conditions and part coordination sites of metal centers are occupied by water molecules.
Thermogravimetric analyses
The thermal stabilities of 2 and 3 were investigated between ambient temperature and ca. 600 °C (Fig. S3 and Fig. S4).† The TGA curves reveal that there are plateaus before the frameworks collapse at a high temperature range.The release of the DMF guest molecules for compound 2 is observed from ambient temperature to 300 °C, leading to a weight loss of 40.14%, which corresponds the loss of three DMF molecules per Ni centre (calculated 38.73%). The plateau appears between 300 and 350 °C, and the framework structure begins to collapse at higher temperature.
Compound 3 loses its solvent molecules from room temperature to 215 °C and the number of guest molecules per formula unit was also calculated to be three. No weight loss is observed between 215 and 320 °C, and a higher temperature leads to the decomposition of the framework.
Infrared spectroscopy
The infrared spectra of 2 and 3 (Fig. S5 and Fig. S6)† are clearly informative regarding the presence of the primary building blocks of the complexes, showing the characteristic bands of the O-donor and N-donor ligands, hence fully supporting the presence of organic components.In the infrared spectra, the absorptions for the antisymmetric stretching vibrations vas(–CO2−) of the carboxyl appear between 1674 and 1544 cm−1 for 2, 1675 and 1542 cm−1 for 3; symmetric stretching vibrations vs(–CO2−) are observed in the range of 1448–1378 cm−1 for 2, 1483–1367 cm−1 for 3. The carboxyl frequencies for the aforementioned stretches appear to be shifted to lower values compared to those of free aromatic acid, indicating changes in the vibrational status of the ligand upon coordination to the metal ion.34,35 The bands located around 1610 and 1450 cm−1, evident in both spectra, strongly support the existence of the coordinated N-donor ligands arising from the symmetric stretching vibrations vs(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N).36
N).36
X-Ray powder diffraction (XRPD)
The plateaus in both TGA curves are informative to some degree, suggesting that removal of the included guest molecules may not lead to the decomposition of the channel structure and inspiring us to carry out the desolvation experiments to confirm this supposition. Freshly prepared samples of 2 and 3 were placed in a high vacuum oven at 150 °C for 10 h with 39.12% for 2 (calculated 38.73%) and 20.26% for 3 (calculated 20.78%) weight loss to get the evacuated solids. The structures of these phases were studied by measuring the XRPD patterns. Fig. 3 shows the observed XRPD patterns compared to those of as-synthesized solids 2 and 3. The agreement of the peaks in all diagrams demonstrates that the frameworks of 2 and 3 are basically retained upon complete removal of the guest molecules, though some of the peaks are broadened after vacuation.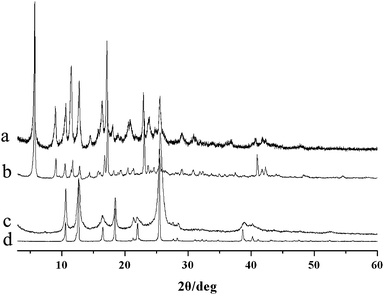 | ||
| Fig. 3 XRPD patterns of the evacuated samples (a for 3, c for 2) and as-synthesized samples (b for 3, d for 2). | ||
N2 sorption properties
The porosity and specific surface areas of framework materials 2 and 3 were estimated by measuring nitrogen gas sorption isotherms at 77 K. The isotherms are shown in Fig. 4. In both cases, the sorption of nitrogen reaches near-saturation at low relative pressures (p/p0 < 0.05) and thereafter increases very slowly up to 1 atm. This behavior is type I isotherms typical for a microporous material. There is no significant hysteresis between soption and desorption traces. The total amount uptake is 140 cm3 g−1 for 2 and 66 cm3 g−1 for 3. By applying the Langmuir equations, the Langmuir surface area is estimated to be 464 m2 g−1 for 2 and 240 m2 g−1 for 3.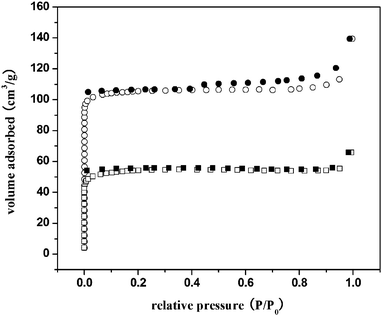 | ||
| Fig. 4 N2 gas sorption isotherms (77K) of 2 (circles) and 3 (squares) measured at 150 °C. Open symbols, sorption; filled symbols, desorption; Po = 1 atm. | ||
Compared with the architecture 1 reported previously, the pillars in 2 are shorter and the layers in 3 are closed. The differences in the structures of 1–3 lead to a great disparity in the numerical value of the surface area28 as well as the amount of gas adsorption (Fig. S12).† It is believed that the shape and size of channels are responsible for this trend.29
H2 sorption property
As the automobile and fuel cell industries look for an efficient method to store and transport hydrogen, metal–organic porous materials have been closely examined for their gas sorption properties. With a higher surface area and considerable extra-framework volume,24 compound 1 is intriguing for inspecting its hydrogen sorption capacity. The high-pressure hydrogen storage capability of compound 1 was evaluated at 77 K. As shown in Fig. 5, the sorption isotherm of compound 1 shows a type I profile which reveals an excess uptake of 3.03 wt% at 20 bar, which corresponds to a H2storage capacity of 337.8 mL g−1 and is equivalent to the adsorption of about 6.4 H2 molecules per formula unit. Such a high hydrogen uptake at moderate pressures is attributed to the full usage of the pores in compound 1, revealing the potential use of pillared-layer MOF materials for hydrogen storage capacity.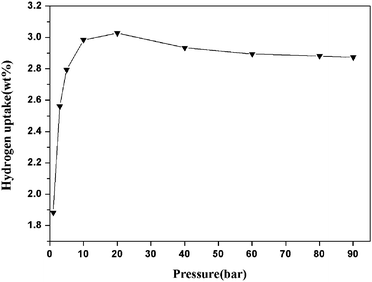 | ||
| Fig. 5 High-pressure hydrogen adsorption isotherm (excess adsorption) for 1 at 77 K. Note that the downturn in the excess adsorption isotherm at higher pressure is a well-understood phenomenon.37 | ||
Conclusions
In summary, we have successfully synthesized and fully characterized a family of 3D metal–organic frameworks based on the pillared-layer motif. The metal-carboxylate-diamine system allows a systematic modulation of the linker or pillar to tune the shape and size of the channels. Gas sorption studies reveal that closely related materials possess a permanent porosity in terms of reverisble gas sorption and MOF. 1 shows adsorption behavior that is favorable towards substantial hydrogen uptake at moderate pressure, revealing the potential use of pillared-layer MOF materials for hydrogen storage capacity. Work is in progress, focused on the mixed-ligand reaction systems to realize rational design and construction of a series of tunable microporous MOFs for soption of gas molecules.Acknowledgements
This work was supported by the National Science Foundation of China (Grant No. 20571014), the Program for New Century Excellent Talents at University (NCET-07-0169), the Program for Changjiang Scholars and Innovative Research Team in University and the Analysis and Testing Foundation of the Northeast Normal University. We thank Mr Yongcun Zou for his experimental support at gas adsorption isotherm measurement.References
- Y. Z. Zheng, M. L. Tong, W. Xue, W. X. Zhang, X. M. Chen, F. Grandjean and G. J. Long, Angew. Chem., Int. Ed., 2007, 46, 6076 CrossRef CAS.
- Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2007, 46, 6638 CrossRef CAS.
- X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, Y. G. Li and L. Xu, Angew. Chem., Int. Ed., 2006, 45, 7411 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwing, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS.
- B. Moulton and M. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS.
- X. S. Wang, S. Ma, D. Sun, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 16474 CrossRef CAS.
- S. Q. Ma, D. F. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2007, 129, 1585.
- B. L. Chen, S. Q. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H. C. Zhou, Inorg. Chem., 2007, 46, 1233 CrossRef CAS.
- L. Pan, D. H. Olson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616 CrossRef CAS.
- C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136 CrossRef CAS.
- D. Tanaka, S. Horike, S. Kitagawa, M. Ohba, M. Hasegawa, Y. Ozawa and K. Toriumi, Chem. Commun., 2001, 1496 RSC.
- Q. R. Fang, G. S. Zhu, Z. Jin, M. Xue, X. Wei, D. J. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2006, 45, 6126 CrossRef CAS.
- C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS.
- L. Z. Zhang, W. Gu, B. Li, X. Liu and D. Z. Liao, Inorg. Chem., 2007, 46, 622 CrossRef CAS.
- B. Q. Ma, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2005, 44, 4912 CrossRef CAS.
- (a) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS; (b) H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS.
- (a) R. Kitaura, F. Iwahori, R. Matsuda, S. Kitagawa, Y. Kubota, M. Takata and T. C. Kobayashi, Inorg. Chem., 2004, 43, 6522 CrossRef CAS; (b) T. K. Maji, K. Uemura, H. C. Chang, R. Matsuda and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 3269 CrossRef CAS.
- D. N. Dybtsev, M. P. Yutkin, E. V. Peresypkina, A. V. Virovets, C. Serre, G. Férey and V. P. Fedin, Inorg. Chem., 2007, 46, 6843 CrossRef CAS.
- (a) L. H. Xie, S. X. Liu, B. Gao, C. D. Zhang, C. Y. Sun, D. H. Li and Z. M. Su, Chem. Commun., 2005, 2402 RSC; (b) L. H. Xie, S. X. Liu, C. Y. Gao, R. G. Cao, J. F. Cao, C. Y. Sun and Z. M. Su, Inorg. Chem., 2007, 46, 7782 CrossRef CAS.
- G. Férey, Chem. Mater., 2001, 13, 3084 CrossRef CAS.
- C. Y. Gao, S. X. Liu, L. H. Xie, Y. H. Ren, J. F. Cao and C. Y. Sun, CrystEngComm, 2007, 9, 545 RSC.
- G. M. Sheldrick, SHELX-97, Program for Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- Being isoreticular frameworks, 1 and 2 had been presumed to be in the same space group. But attempts to solve the metal organic framework structure of 2 in space groupP-62m just as 1 were unsuccessful. The subtle distinction in the space group may be caused by disorders in different directions from the pillars in 1 and 2.
- The calculated guest accessible free volumes for frameworks 1 and 2 were found to be 54.8 and 46.3%, respectively, which is in the perfect correlation with the linker length decrease from bipy (7.05 Å) to pyz (2.70 Å). In the same way, the free sizes of the intersecting channels vary from 7.0 × 6 Å for 1 and 3.3 × 6 Å for 2. And the Langmuir surface area is estimated to be 1282 m2 g−1 for 1 and 464 m2 g−1 for 2 by measuring nitrogen gas sorption isotherms.
- T. J. Prior, D. Bradshaw, S. J. Teat and M. J. Rosseinsky, Chem. Commun., 2003, 500 RSC.
- (a) E. Y. Choi and Y.-UK. Kwon, Inorg. Chem. Commun., 2004, 7, 942 CrossRef CAS; (b) E. Y. Choi and Y.-UK., Inorg. Chem., 2005, 44, 538 CrossRef CAS.
- H. S. Huh, D. W. Min, Y. K. Lee and S. W. Lee, Bull. Korean Chem. Soc., 2002, 23, 619 CAS.
- X. L. Wang, C. Qin and E. B. Wang, Cryst. Growth Des., 2006, 6, 438.
- C. D. Wu, C. Z. Lu, S. F. Lu, H. H. Zhuang and J. S. Huang, Inorg. Chem. Commun., 2002, 5, 171 CrossRef CAS.
- C. Djordjevic, M. Lee and E. Sinn, Inorg. Chem., 1989, 28, 719 CrossRef CAS.
- G. B. Deacon and R. J. Philips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- J. R. Bao, Y. L. Zhao and X. W. Zhu, Spectrosc. Spectral Anal., 2007, 27, 539 CAS.
- P. G. Menon, Chem. Rev., 1968, 68, 277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of 2 and 3 in cif format; TG curves, XRPD patterns, and additional figures relative to the compounds 2 and 3 (PDF). CCDC reference numbers 665814–700659. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b812097h |
| This journal is © The Royal Society of Chemistry 2009 |