Inclusion of a well resolved T4(2)6(2) water tape in a H-bonded, (4,7)-binodal 3D network†
George E.
Kostakis
,
Ghulam
Abbas
,
Christopher E.
Anson
and
Annie K.
Powell
*
Institut für Anorganische Chemie der Universität Karlsruhe, Engesserstr. 15, D-76131, Karlsruhe, Germany. E-mail: powell@aoc.uni-karlsruhe.de; Fax: +49 721 608 8142; Tel: +49 721 608 2135
First published on 30th October 2008
Abstract
The reaction of lanthanide nitrate with m-BDTH2 (1,3-benzeneditetrazol-5-yl) leads to a three dimensional (3D) hydrogen bonded framework formulated as {[Ln(H2O)8(m-BDTH)](m-BDT)·12(H2O)} (Ln: Ce,Nd). The topological description of the supramolecular architectures reveals a new binodal (4,7) net with a Schäfli symbol of (3.42.53)(32.42.54.65.72). A self-assembled T4(2)6(2) water tape is included and has been analyzed in comparison with known examples.
Introduction
The crystal engineering of supramolecular metal–organic architectures assembled through covalent bonds in addition to other weak cooperative interactions such as hydrogen bond, π–π stacking and electrostatic interactions has been rapidly developed and attracted considerable interest for designing and constructing new crystalline materials.1 Among these secondary interactions, the hydrogen bond is more important in the molecular assembly due to its suitable strength and directionality.2Hydrogen-bonded water clusters have been broadly studied both theoretically and experimentally as it is possible to provide useful information on the nature of cooperative association of a small collection of water molecules. So far, a variety of tetramer,3apentamer,3bhexamer,3coctamer3d and decamer3ewater clusters has been isolated in different solid crystalline hosts.3f–h Small water clusters (H2O)n (n = 4–6) are crucial building units for extended water morphologies including tapes4 and layers5 whose physical properties are closely associated with those of bulk water.65-Substituted-1H-tetrazoles are heterocyclic compounds that can be found in neutral, anionic or cationic form. They can act as ligands in metal complexes, form salts and can be both acceptors and donors for hydrogen bonding. The 1,3-benzeneditetrazol-5-yl (m-BDTH2) ligand is a useful organic spacer which has received less attention in the construction of coordinated metal–organic frameworks.7 In a recent publication we described the room temperature synthesis of a 3-D porous framework. In the structure, the [Ln(H2O)n]3+ ions act as pillars between π-stacked and H-bonded sheets of (m-BDTH)– organic anions in [Ln(H2O)n](m–BDTH)3·9(H2O) [Ln ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Pr, n = 9, Ln Gd, n = 8].3b
Pr, n = 9, Ln Gd, n = 8].3b
It is well known that the metal salt, temperature, solvent and the time of reaction affect the nature of the final product. Bearing this in mind, the next step in our systematic investigation was to modify these parameters and observe their influence on the structural diversity of the final products. Thus, the reaction of equimolar amounts of the corresponding hydrated lanthanide nitrate with m-BDTH2 in a H2O/CH3CN/EtOH solvent mixture under reflux for 2h in the presence of CH3CO2Na resulted after 3 weeks in the deposition of pale yellow or pink crystals of a compound formulated as [Ln(H2O)8(m-BDTH)](m-BDT)·12(H2O) [Ln: Ce(1), Nd (2)], respectively.
Results and discussion
The full structures of 1 and 2 were determined by single-crystal X-ray diffraction. These two compounds are isomorphous, crystallising in the monoclinic space groupP21/n with Z = 4 and consist of one LnIII, one singly deprotonated (m-BDTH)− ligand, one doubly deprotonated (m-BDT)2− organic dianion, eight aqua ligands and twelve lattice water molecules in the asymmetric unit.In compound 1Ce(1) is ligated by eight water molecules and one nitrogen atom N(3), forming a capped square antiprism, the eight Ce–O distances within the antiprism lie in the range 2.446(3)–2.588(3) Å and the distance to the capping nitrogen is 2.862(4) Å. BVS analysis reveals that Ce(1) is in the trivalent oxidation state. There are two crystallographically independent organic moieties. The first is partly deprotonated and ligated to the Ce(1) with N(3) from the deprotonated tetrazole ring forming in this way a [Ce(H2O)8(m-BDTH)]2+ cation (Fig. 1), while the second is completely deprotonated equalizing the positive charge, but not coordinated to the lanthanide centre. The hydrogens of all the water ligands, the N–H hydrogen of the organic molecule and the hydrogens of eleven out of twelve lattice waters could be located and refined, allowing us to study the hydrogen-bonding in the compound.
 | ||
| Fig. 1 The asymmetric unit of 1. Ce green, O red, N blue, C grey. The lattice water molecules are omitted for clarity. | ||
Twenty water molecules are included in the asymmetric unit, therefore a variety of O–H⋯O, O–H⋯N and N–H⋯O hydrogen bonding interactions (Tables S1 and S2†) are observed in both crystal structures. Both organic moieties can act as hydrogen bond donors and acceptors from the ligated water molecules. The ligated monoanion is hydrogen bonded to two [Ce(H2O)8(m-BDTH)]2+ forming a zig–zag 1D cationic chain perpendicular to the b axis, (Fig. S1).† Generally, the [Ce(H2O)8(m-BDTH)]2+ cation is hydrogen bonded to the three aforementioned cations and to four (m-BDT)2− organic moieties (Fig. 2). At the same time, the non-coordinated organic moiety forms five hydrogen bonds to ligated water molecules of four [Ce(H2O)8(m-BDTH)]2+ cations generating a 3D supramolecular architecture (Fig. 2). For the topological description of the 3D supramolecular structure, each (m-BDT)2− organic moiety and each [Ce(H2O)8(m-BDTH)]2+ cation can be considered as a 4- and 7-connecting node, respectively. (Fig. 2) Thus, the 3D hydrogen bonded framework of 1 can be simplified to a new binodal (4,7) net with a Schäfli symbol of (3.42.53)(32.42.54.65.72).8 The long notation of the net topology can be described as 3.4.3.5.4.52 for the four connected node and 3.3.3.3.4.4.4.5.5.5.5.5.6.6.62.62.62.62.63.63.75 for the seven-connected node. (Fig. 3) Furthermore, the ligated organic monoanion and the non-coordinated organic moiety are stacked parallel to the (0 1 1) plane through strong π–π interactions with a mean interplane distance of only 3.24 Å further stabilizing the overall 3D supramolecular framework of 1.
![Hydrogen bonds formed in compound 1 between the 4-connected node (m-BDT)2− organic moiety (blue) and four [Ce(H2O)8(m-BDTH)]2+ cations (green) (upper). Hydrogen bonds formed between the emphasized 7-connected node [Ce(H2O)8(m-BDTH)]2+ (red) and three different [Ce(H2O)8(m-BDTH)]2+ (green) and four (m-BDT)2− organic moieties (blue) (lower).](/image/article/2009/CE/b811376a/b811376a-f2.gif) | ||
| Fig. 2 Hydrogen bonds formed in compound 1 between the 4-connected node (m-BDT)2− organic moiety (blue) and four [Ce(H2O)8(m-BDTH)]2+ cations (green) (upper). Hydrogen bonds formed between the emphasized 7-connected node [Ce(H2O)8(m-BDTH)]2+ (red) and three different [Ce(H2O)8(m-BDTH)]2+ (green) and four (m-BDT)2− organic moieties (blue) (lower). | ||
![A topological presentation of the resulting new (4,7)-connected 3D H-bonding binodal net of 1 and 2. Colour code: [Ln(H2O)8(m-BDTH)]2+ green node; m-BTD2− blue node (down).](/image/article/2009/CE/b811376a/b811376a-f3.gif) | ||
| Fig. 3 A topological presentation of the resulting new (4,7)-connected 3D H-bonding binodal net of 1 and 2. Colour code: [Ln(H2O)8(m-BDTH)]2+ green node; m-BTD2− blue node (down). | ||
Interestingly, one coordinated (O3) and five lattice water molecules (O11, O12, O14, O15 and O19) form a cyclic water hexamer through hydrogen bonds (Fig. 4). In the structure of 2, the hexamer is slightly different compared to that in 1. Within the hexamer the O⋯O⋯O angles are in the range of 107.5–134.8°, (106.77–135.20° for 2) while the six O⋯O⋯O⋯O torsional angles in the ring are 0.8, −3.0, 4.7, −4.3, 2.9 and −0.9° (0.9, −2.9, 4.7, −4.4, 3.4 and −1.4° for 2). The hexamer in both structures thus approximates to a planar hexagon. The O⋯O distance within the hexamer ranges from 2.673 to 2.778 Å in 1 and from 2.676 to 2.776 Å in 2 with an average value of 2.728 Å and 2.731 Å for 1 and 2, respectively, which can be compared with the O⋯O distance in ice Ih (2.759 Å at 200 K).9 Within the hexamer, O3 and O12 act as double donors, O11 and O14 act as double acceptors, while O15 and O19 act as single donor and single acceptor. Furthermore, each hexamer is self-assembled to the symmetry related oxygen atoms through hydrogen bonds into fused four- and six-membered water rings forming in this way an infinite 1D ‘ladder-like’ water tape entitled T4(2)6(2), parallel to the (0 1 1) plane (Fig. 4). The O⋯O distance within the tetramer ranges from 2.673 to 2.852 Å in 1 and from 2.676 to 2.861 Å in 2, with an average value of 2.764 Å in 1 and 2.765 Å in 2, while the plane deviation between the four- and six-membered water rings is defined at 53.79 and 53.84° in 1 and 53.58 and 53.64° in 2. Compared to our recent work,3b the increase of the temperature in the reaction system results in the formation of a 6-member planar water cluster, while at room temperature 5-memberd planar water clusters are formed. Moreover, it is worth mentioning that in both cases the formed water cluster is developed in the region of one ligated water molecule.
 | ||
| Fig. 4 A planar-shaped hexamer water cluster, the T4(2)6(2) ‘ladder-like’ water tape formed by the water hexamers (upper) and the resulting 3D supramolecular structure of 1 projected onto the (0 1 1). (lower). (Symmetry transformations used to generate equivalent atoms: * = 1/2 + x, 1/2 − y, 1/2 − z, ** = x − 1/2, 1/2 − y, 1/2 + z, *** = x − 1/2, 1/2 − y, z − 1/2). | ||
Up to now, the T4(2)6(2) motif has rarely been reported in the literature. Mascal et. al reported an investigation on the H-bonding water clusters with a T4(2)6(2) motif based on a CSD research.3h As shown in Table 1, there are 22 compounds which form defined 1D water tapes with a T4(2)6(2) motif through a symmetry operation. Among them, in the fourteen examples of water chain the hexamerwater cluster possesses a chair conformation. The remaining eight compounds exhibit a hexamerwater cluster which approximates to a planar hexagon. The water chain inlaid in 1 represents the first example of a planar-shaped T4(2)6(2) motif assembled from six crystallographically independent water molecules. Compared to other planar ladder-like tapes, the average O⋯O distance 2.728 Å in the “tread” of 1 is shorter than any previously reported (Table 2). The ‘riser’ in 1 is defined at 2.764 Å close to the values calculated from other examples of 2.751 Å10p and 2.776,10i while the deviation between ‘riser’ and ‘tread’ is defined at 53.82°, similar to the calculated values of 51.07°10l and 50.85°10q (Scheme 1).
| 1 | 2 | |
|---|---|---|
| Formula | C16H49CeN16O20 | C16H49N16NdO20 |
| M r | 925.83 | 929.95 |
| Crystal size/mm | 0.05 × 0.17 × 0.22 | 0.04 × 0.34 × 0.42 |
| Color | Pale yellow | Pink |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| T/K | 100 | 100 |
| a/Å | 11.2155(5) | 11.2104(17) |
| b/Å | 28.9919(13) | 28.980(4) |
| c/Å | 11.9052(5) | 11.9110(18) |
| β/° | 111.216(1) | 111.184(2) |
| V/Å3 | 3608.7(3) | 3608.2(9) |
| Z | 4 | 4 |
| ρ calcd/g cm−3 | 1.704 | 1.712 |
| µ(Mo-Kα)/mm−1 | 1.361 | 1.538 |
| F(000) | 1900 | 1908 |
| Reflns collected | 18231 | 24695 |
| Unique reflns | 6787 | 8155 |
| Reflns with I > 2σ(I) | 6042 | 6949 |
| Parameters/restraints | 594/45 | 594/51 |
| GoF on F2 | 1.071 | 1.131 |
| R 1 [I > 2σ(I)] | 0.0418 | 0.0478 |
| wR 2 (all data) | 0.0902 | 0.1205 |
| Configuration of the hexamer in the T4(2)6(2) motif | Coordinated water | Calculated average distance between O atoms | Dimension of the host | ||
|---|---|---|---|---|---|
| 6 member | 4 member | ||||
| ANAPHS10a | chair– shaped | no | 2.792 | 2.826 | 0D |
| CELGEF 10b | chair– shaped | no | 2.842 | 2.877 | organic |
| CISPOJ 10c | chair– shaped | no | 2.842 | 2.898 | organic |
| FEDSOW 10d | chair– shaped | no | 2.880 | 2.857 | 1D |
| GOMGIY 10e | planar | no | 2.808 | 2.803 | organic |
| MEPYRZ 10f | chair– shaped | no | 2.760 | 2.849 | organic |
| QIPPUA 10g | chair– shaped | no | 2.776 | 2.843 | organic |
| UDEDAI 10h | chair– shaped | no | 2.780 | 2.801 | organic |
| UMEZUH 10i | planar | no | 2.782 | 2.776 | 2D |
| VOBXIT 10j | chair– shaped | no | 2.880 | 2.884 | organic |
| WEKCIY 10k | chair– shaped | no | 2.726 | 2.903 | organic |
| XEVHIP 10l | planar | no | 2.770 | 2.816 | 0 D |
| XIPGUY 10m | chair– shaped | no | 2.821 | 2.822 | 0 D |
| XOPSIE 10n | chair– shaped | no | 2.885 | 2.939 | 1 D |
| YABBOS10o | planar | no | 3.024 | 3.000 | 1 D |
| YIKWUK 10p | planar | no | 2.783 | 2.751 | 2 D |
| YUJHAM 10q | planar | no | 2.805 | 2.844 | organic |
| ZOGZAW 10r | planar | no | 2.799 | 2.838 | 1 D |
| ZUHMIY 10s | chair– shaped | no | 2.827 | 2.839 | organic |
| 10t | planar | no | 2.858 | 2.829 | 1D |
| 10u | chair – shaped | no | 2.744 | 2.775 | 0D |
| 10v | chair– shaped | no | 3.019 | 2.878 | 1D |
| this work | planar | yes | 2.728 | 2.764 | 0D |
 | ||
| Scheme 1 Schematic representation of ‘riser’ and ‘tread’ in the ladder. | ||
Thermal studies
The thermogravimetric analysis (TGA) of compound 1 was performed between 25 and 700 °C at a heating rate of 5 °C min−1 under nitrogen atmosphere (Fig. 5). The weight loss begins at the start of the experiment. The removal of the lattice–water molecules takes place in two subsequent endothermic steps at 105 °C. The calculated value for ten lattice–water molecules (19%) corresponds to the experimental ones (19.50%). Then, the dehydrated compound is stable up to the abstraction of the ligated water molecule which is accomplished at 190 °C. The calculated value for the remaining ten water molecules (19.50%) is not in agreement with the experimental value (15.60%). This indicates that two water molecules remain ligated to the cerium metal centres. Upon temperature increase, the partially dehydrated compound begins to decompose taking place in two discrete steps. First, an endothermic step is completed at 300 °C followed by a second exothermic step at 580 °C. The weight loss which is observed in these two steps corresponds to the total evacuation of N2 gas and the sublimation of the carbon (theoretical 45.03%, experimental 44.90%). The final residue corresponds to the predicted Ce2O3 (theoretical 17.90%, experimental 18.50%).·12H2O 1.](/image/article/2009/CE/b811376a/b811376a-f5.gif)
Reversible solvent uptake is a widespread property for coordination or hydrogen bonded frameworks either in the form of lattice molecules11a or acting as bridges.11b On heating the crystals of compound 1 at 60 °C in vacuum for 120 min, the dehydrated product has an amorphous phase, indicating a collapse of the initial structure. Rehydration of the heated sample affords a different product from the initial material (Fig. 6). It will thus be of interest to investigate the use of alternative methods such as hydrothermal synthesis to gauge whether the water tapes can be modified or disrupted. Initial experiments show that polycrystalline materials are accessible with different IR spectra and X-ray powder patterns from those of the compounds reported here (see ESI, Fig. S2).†
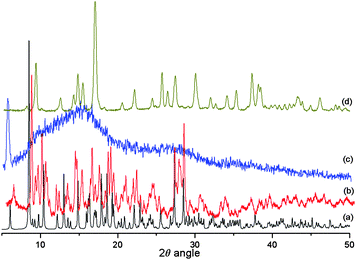 | ||
| Fig. 6 (a) Theoretical powder pattern of 1, (b) experimental powder pattern of 1, (c) heated sample at 60 °C in vacuum after 2h, (d) rehydrated sample. | ||
IR spectroscopy
The IR spectra of compounds 1 and 2 show a broad peak centred at 3375 cm−1, which can be assigned to the O–H stretching vibration of the water molecules of the tape. These bands occur at higher energies than those reported for ice (3220 cm−1),9 and are similar to those observed for other water chains.10,12Conclusions
In conclusion, we have synthesized and characterized an H-bonded and π-stacked 3D supramolecular architecture {[Ln(H2O)8(m-BDTH)](m-BDT)·12(H2O)} (Ln = Ce, Nd) possessing a new (4,7)-connected binodal net with a Schläfli symbol of (3.42.53)(32.42.54.65.72). In both crystal structures a well-resolved water 1D ‘ladder-like’ chain possessing a T4(2)6(2) motif is observed. The present finding provides new structural data, which could enhance the understanding of the structural aspects of water. This work represents a part of our systematic efforts to determine new synthetic pathways in order to synthesize multidimensional networks, possessing special structural motifs and/or magnetic properties.Experimental
General information
All chemicals and solvents used for the synthesis were obtained from commercial sources and were used as received, without further purification. All reactions were carried out in aerobic conditions. The elemental analysis (C, H, N) were carried out at the Institut für Anorganische Chemie at the Universität Karlsruhe (TH) using an Elementar Vario EL analyzer. Fourier transform IR spectra were measured on a Perkin-Elmer Spectrum One spectrometer with samples prepared as KBr discs. X-Ray powder diffraction patterns for all compounds were measured at room temperature using a Stoe STADI-P diffractometer with a Cu-Kα radiation.Synthesis
m-BDTH2 was prepared as described previously.3b The same procedure was employed to prepare compounds 1 and 2 and hence only the compound [Ce(H2O)8(m-BDTH)](m-BDT)·12(H2O) 1 is described here in detail. A solution of m-BDTH2 (0.021 g, 0.1 mmol) in CH3CN/EtOH (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, total volume 10 ml) was added dropwise to a stirred solution of Ce(NO3)3·6(H2O) (0.044 g, 0.1 mmol) and CH3CO2Na (0.014 g, 0.1 mmol) in water (10 mL). The resulting colourless solution was refluxed for 2 h, filtered and left to evaporate slowly. Pale yellow crystals in 80% yield (based on Ce) of 1 were isolated after 1 week, washed with ethanol and dried in air. Analysis (%) Found (calc. for C16H47N16O20Ce): C, 20.38 (20.80); H, 5.07 (5.13); N, 24.13 (24.27). IR (KBr), ν/cm−1 3375 (br, vs), 2414 (m), 1669 (m), 1650 (m), 1567 (m), 1449 (m), 1418 (w) 1384 (w), 1331 (w), 1253 (w), 1222 (w), 1170 (m), 1079 (m), 1040 (m), 994 (m), 903 (w), 762 (m), 737 (m), 681 (w). Synthesis of [Nd(H2O)8(m-BDTH)](m-BDT)·12(H2O) (2). Yield: 75% based on Nd. Analysis (%) Found (calc. for C16H47N16NdO20): C, 20.75 (20.85); H, 5.12 (5.17); N, 24.22 (24.27). IR (KBr), ν/cm−1 3375 (br, vs), 2414 (m), 1669 (m), 1650 (m), 1567 (m), 1449 (m), 1418 (w) 1384 (w), 1331 (w), 1253 (w), 1222 (w), 1170 (m), 1079 (m), 1040 (m), 994 (m), 903 (w), 762 (m), 737 (m), 681 (w).
:1, total volume 10 ml) was added dropwise to a stirred solution of Ce(NO3)3·6(H2O) (0.044 g, 0.1 mmol) and CH3CO2Na (0.014 g, 0.1 mmol) in water (10 mL). The resulting colourless solution was refluxed for 2 h, filtered and left to evaporate slowly. Pale yellow crystals in 80% yield (based on Ce) of 1 were isolated after 1 week, washed with ethanol and dried in air. Analysis (%) Found (calc. for C16H47N16O20Ce): C, 20.38 (20.80); H, 5.07 (5.13); N, 24.13 (24.27). IR (KBr), ν/cm−1 3375 (br, vs), 2414 (m), 1669 (m), 1650 (m), 1567 (m), 1449 (m), 1418 (w) 1384 (w), 1331 (w), 1253 (w), 1222 (w), 1170 (m), 1079 (m), 1040 (m), 994 (m), 903 (w), 762 (m), 737 (m), 681 (w). Synthesis of [Nd(H2O)8(m-BDTH)](m-BDT)·12(H2O) (2). Yield: 75% based on Nd. Analysis (%) Found (calc. for C16H47N16NdO20): C, 20.75 (20.85); H, 5.12 (5.17); N, 24.22 (24.27). IR (KBr), ν/cm−1 3375 (br, vs), 2414 (m), 1669 (m), 1650 (m), 1567 (m), 1449 (m), 1418 (w) 1384 (w), 1331 (w), 1253 (w), 1222 (w), 1170 (m), 1079 (m), 1040 (m), 994 (m), 903 (w), 762 (m), 737 (m), 681 (w).
X-Ray crystallography
Single-crystal X-ray crystallographic data were collected at 100 K on a Bruker SMART Apex CCD diffractometer using graphite-monochromated Mo-Kα radiation. Crystallographic data and details of measurement and refinement are summarized in Table 1. Semi-empirical absorption corrections were made using SADABS.13a The structures were solved using direct methods, followed by full-matrix least-squares refinement against F2 (all data) using SHELXTL.13b Anisotropic refinement was used for all non-H atoms; organic H atoms were placed in calculated positions, while the coordinates of the hydroxo H atoms were refined. The O–H bond lengths and, where necessary, the H–O–H angles were restrained. The crystallographic data and refinement parameters are listed in Table 1.Acknowledgements
This work is dedicated to Prof. Nick Hadjiliadis on the occasion of his retirement. Financial support from the DFG Center for Functional Nanostructures is gratefully acknowledged.References
- (a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef; (b) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS; (c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (d) J. M. Lehn, in Concepts and Perspectives in Supramolecular Chemistry, ed. U. Anton, VCH, Verlagsgesellschaft mbH. D-69451 Weinheim, 1995 Search PubMed; (e) M. W. Hosseini, Coord. Chem. Rev., 2003, 240, 157 CAS.
- A. Ballbh, R. T. Darshak, P. Dastidar and E. Suresh, CrystEngComm, 2002, 4, 135 RSC.
- (a) S. Supriya, S. Manikumari, P. Raghavaiah and S. K. Das, New J. Chem., 2003, 27, 218 RSC; (b) G. E. Kostakis, G. Abbas, C. E. Anson and A. K. Powell, CrystEngComm, 2008, 10, 1117 RSC; (c) H.-H. Song and B.-Q. Ma, CrystEngComm, 2007, 9, 625 RSC; (d) J. L. Atwood, L. J. Barbour, T. J. Ness, C. L. Raston and P. L. Raston, J. Am. Chem. Soc., 2001, 123, 7192 CrossRef CAS; (e) L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 2000, 859 RSC; (f) L. Infantes and S. Motherwell, CrystEngComm, 2002, 4, 454 RSC; (g) L. Infantes, J. Chisholm and S. Motherwell, CrystEngComm, 2003, 5, 480 RSC; (h) M. Mascal, L. Infantes and J. Chisholm, Angew. Chem., Int. Ed., 2006, 45, 32 CrossRef.
- (a) B. Y. Saha and A. Nangia, Chem. Commun., 2006, 1825 RSC; (b) R. Luna–Carcia, B. M. Damian– Murillo, V. Barba, H. Hopfl, H. I. Beltran and L. S. Zamudio–Rivera, Chem. Commun., 2005, 5527 RSC; (c) J. P. Naskar, M. G. B. Drew, A. Hulme, D. A. Tocher and D. Datta, CrystEngComm, 2005, 7, 67 RSC; (d) J.-M. Zheng, S. R. Batten and M. Du, Inorg. Chem., 2005, 44, 3371 CrossRef CAS; (e) X. Mei and C. Wolf, CrystEngComm, 2006, 8, 377 RSC; (f) B.-Q. Ma, H.-L. Sun and S. Gao, Chem. Commun., 2004, 2220 RSC.
- (a) K. Raghuraman, K. K. Katti, L. J. Barbour, N. Pillarsetty, C. L. Barnes and K. V. Katti, J. Am. Chem. Soc., 2003, 125, 6955 CrossRef CAS; (b) C. Janiak and T. G. Scharman, J. Am. Chem. Soc., 2002, 124, 14010 CrossRef CAS; (c) P. S. Lakshminarayanan, E. Suresh and P. Ghosh, J. Am. Chem. Soc., 2005, 127, 13132 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- C. Jiang, Z. Yu, C. Jiao, S. Wang, J. Li, Z. Wang and Y. Cui, Eur. J. Inorg. Chem., 2004, 4669 CrossRef CAS.
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- K. D. Eisenberg and W. Kauzmann, The Structure and Properties of Water, Oxford University Press, Oxford, 1969 Search PubMed.
- (a) V. Cody and J. Hazel, Acta Crystallogr., Sect. B, 1977, 33, 3180 CrossRef; (b) N. U. Kamath and K. Venkatesan, Acta Crystallogr., Sect. C, 1984, 40, 559 CrossRef; (c) D. S. S. Gowda, Puttaraja and R. Rudman, J. Crystallogr. Spectrosc. Res., 1984, 14, 359 CrossRef CAS; (d) J. M. Harrowfield, R. P. Sharma, B. W. Skelton, P. Venugopalam and A. H. White, Aust. J. Chem., 1998, 51, 775 CAS; (e) A. J. Dobson and R. E. Gerkin, Acta Crystallogr., Sect. C, 1999, 55, 1192 CrossRef; (f) A. W. M. Braam, J. C. Eikelenboom, G. van Dijk and A. Vos, Acta Crystallogr., Sect. B, , 37, 259 Search PubMed; (g) R. Custelcean, C. Afloroaei, M. Vlassa and M. Polverejan, Angew. Chem., Int. Ed., 2000, 39, 3094 CrossRef CAS; (h) M. Mascal, M. Lera, A. J. Blake, M. Czaja, A. Kozak, M. Makowski and L. Chmurzynski, Angew. Chem., Int. Ed., 2001, 40, 3696 CrossRef CAS; (i) Y.-Q. Jiang, Z.-X. Xie and S. W. Ng, Acta Crystallogr., Sect. E, 2003, 59, m947 CrossRef; (j) A. Vichet, J. P. Galy, A. Baldy, J. Barbe, J. Feneau-Dupont and J.-P. Declercq, Acta Crystallogr., Sect. C, 1991, 47, 2508 CrossRef; (k) D. Korbonits, E. Tobias-Heja, K. Simon and P. Kolonits, Liebigs Ann., 1994, 19 Search PubMed; (l) P. C. McGowan, T. J. Podesta and M. Thornton-Pett, Inorg. Chem., 2001, 40, 1445 CrossRef CAS; (m) I. S. De la Cueva-Torregrosa, J. M. Gonzalez-Perez, A. G. Sicilia-Zafra, E. Bugella-Altamirano, J. Niclos-Gutierrez and A. Castineiras-Campos, J. Coord. Chem., 2000, 51, 235 CrossRef CAS; (n) J. C. Kim, A. J. Lough and H. Jo, Inorg. Chem. Commun., 2002, 5, 616 CrossRef CAS; (o) S. -B. Teo, C. -H. Ng, S. -G. Teoh and H. -K. Fun, J. Coord. Chem., 1991, 24, 151 CAS; (p) G. De Munno, R. Ruiz, F. Lloret, J. Faus, R. Sessoli and M. Julve, Inorg. Chem., 1995, 34, 408 CrossRef CAS; (q) B. M. Kariuki and W. Jones, Acta Crystallogr., Sect. C, 1995, 51, 1234 CrossRef; (r) K. Miyoshi, J. Wang and T. Mizuta, Chem. Lett., 1995, 721 CrossRef CAS; (s) G. Mendoza-Diaz, W. L. Driessen and J. Reedijk, Acta Crystallogr., Sect. C, 1996, 52, 960 CrossRef; (t) F. Li, T. -H. Li, W. Su, S. -Y. Gao and R. Cao, Eur. J. Inorg. Chem., 2006, 1582 CrossRef CAS; (u) M. Li, S. Chen, J. Xiang, H. He, L. Yuan and J. Sun, Cryst. Growth Des., 2006, 6, 1250 CrossRef CAS; (v) X.-Y. Song, L.-C. Li, D.-Z. Liao, Z.-H. Jiang and S.-P. Yan, Cryst. Growth Des., 2007, 7, 1220 CrossRef CAS.
- (a) C.–C. Wang, W.–Z. Lin, W.–T. Huang, M.–J. Ko, G.–H. Lee, M.–L. Ho, C.–W. Lin, C.–W. Shih and P.–T. Chou, Chem. Commun., 2008, 1299 RSC; (b) G. E. Kostakis, L. Casella, N. Hadjiliadis, E. Monzani, N. Kourkoumelis and J. C. Plakatouras, Chem. Commun., 2005, 3859 RSC.
- B. Sreenivasulu and J. J. Vittal, Angew. Chem., Int. Ed., 2004, 43, 5769 CrossRef CAS.
- (a) G. M. Sheldrick, SADABS (the Siemens Area Detector Absorption Correction), University of Göttingen, 1996 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details (Fig. S1, Tables S1 and S2). CCDC reference numbers 693449 and 702192. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b811376a |
| This journal is © The Royal Society of Chemistry 2009 |