In situ synthesis of 5-substituted-tetrazoles and metallosupramolecular co-ordination polymers†
Wenbin
Yang
,
Xiang
Lin
,
Alexander J.
Blake
,
Claire
Wilson
,
Peter
Hubberstey
,
Neil R.
Champness
* and
Martin
Schröder
*
School of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: Neil.Champness@nottingham.ac.uk; M.Schroder@nottingham.ac.uk
First published on 14th October 2008
Abstract
A series of metal–organic supramolecular co-ordination polymers [Zn(HPO4)(Hpytz)] (1), [Cd(Hpytz)2(H2PO4)2(H2O)2] (2), [Zn(pytz)2(H2O)4]·2H2O (3) [pytz− = 5-(4-pyridyl)tetrazolate], [M(tzpyo)2(H2O)4] [M = Zn (4), Ni (5)] [tzpyo− = 4-tetrazolate-pyridine N-oxide] incorporating tetrazolyl ligands have been synthesized via in situ [2 + 3] cycloaddition reactions of 4-cyanopyridine, 4-cyanopyridine N-oxide with sodium azide in the presence of zinc, cadmium or nickel salts under hydrothermal reaction conditions at 130–160 °C. Use of higher temperatures leads to hydrolysis of nitriles to carboxylates and subsequently to decarboxylation, resulting in two unexpected byproducts, [inaH]+(NO3−) (6) [ina = isonicotinic acid] and [(4,4′-bipyH2)(H2PO4)2(H2O)2] (7). Single crystal X-ray structure analyses of 1 and 2, prepared under hydrothermal conditions in the presence of H3PO4, reveal κ1-co-ordination of the N-donor at the 2-position of the tetrazolyl ligands binding in a TZ-mode. The pyridyl N-centres in 1 and 2 are protonated resulting in the formation of Npy–H⋯O hydrogen-bonds within the solid-state structures. In the absence of H3PO4 similar hydrothermal reactions afforded 3, in which the pyridyl group is directly bound to Zn(II) and tetrazolate-ring is unco-ordinated (PY-mode). Single crystal X-ray structures of 4 and 5 show a TZ-binding mode analogous to that for 1 and 2, with the pyridyl N-oxide moiety forming hydrogen-bonds with co-ordinated water molecules to give supramolecular pillar-layered frameworks. Complex 2 exhibits a distorted 6-connected α-Po net of 412·63 topology; 3 possesses an attractive 3D (4,5,10)-connected trinary (3·42·53)2(3·44·54·6)4(32·48·514.612·77·82) net, while the isostructural complexes 4 and 5 show 3D (4,8)-connected layer-pillared network structures with a unique (46)2(412·612·84) topology. In 6, all hydrogen atoms of inaH+ cations are involved in the formation of hydrogen-bonds with either nitrate or other inaH+ cations resulting in a 2D (4,6)-connected binary net with (32·42·52)(34·44·54·63) topology. In 7, the dihydrogenphosphate anions are arranged viahydrogen-bonds into an unusual (4,4) grid-type sheet, and these layers are further pillared by organobipyridine cations to form a 3D open framework with channels occupied by helical chains of water dimers. The thermal stability of these metal-tetrazolate complexes has been studied by TGA and powder X-ray diffraction, and the effect of proton source (H3PO4) on the co-ordination mode of these tetrazolyl ligands is discussed.
Introduction
The synthesis of metal co-ordination polymers is an area of chemistry that has received an increasing level of attention since the highly influential reports of Robson.1 Over recent years this research area has expanded rapidly, reflecting the variety of synthetic strategies that allow the preparation of a given framework structure with specific physico-chemical properties.2 Driven by potential applications in co-ordination chemistry as ligands,3–14 in medicinal chemistry as a metabolically stable surrogates for the carboxylic acid group,15 and in materials science as high density energy materials,16 tetrazoles have aroused particular interest, and the literature on synthetic routes to this useful functional group is expanding rapidly. Generally, 5-functionalized tetrazoles are prepared by the intra- or intermolecular [2 + 3] dipolar cycloadditions of azides to nitriles, thiocyanates, cyanates and cyanamides (Scheme 1).17 However, these routes are all somewhat dangerous and/or cause environmental problems due to the use of expensive and toxic metal–organic azide complexes such as tin or organo-silicon azides, and to the presence of hydrazoic acid, which is toxic, volatile and explosive. Sharpless et al. have developed18 a methodology which overcomes these disadvantages and by which 5-substituted-1H-tetrazoles can be obtained in rather high yield by reaction of azides and nitriles in water in the presence of Zn(II) salts as Lewis acid catalysts. Although the precise role of the Zn catalyst and the mechanistic pathway(s) in the formation of tetrazoles remain unclear, Sharpless’ method offers a facile, safe and environmentally-friendly synthetic route to tetrazoles and, therefore, makes exploration of the co-ordination chemistry of tetrazolate ligands more accessible and attractive than in the past.![Inter- and intramolecular [2 + 3] dipolar cycloaddition](/image/article/2009/CE/b808496c/b808496c-s1.gif) | ||
| Scheme 1 Inter- and intramolecular [2 + 3] dipolar cycloaddition | ||
One of the most chemically intriguing features of tetrazolates is their ability to use up to four electron-donating N-centres. As illustrated in Scheme 2, the pH-sensitive tetrazolate heterocycle can exhibit versatile co-ordination modes ranging from κ1 to κ4, some of which have been observed in the construction of metal–organic frameworks.3–14,19 Moreover, the utilization of asymmetric bridging tetrazolate N-donors can introduce electronic asymmetry and lead to novel solid-state ferroelectric20 and second harmonic generation (SHG) materials.21 In addition, tetrazolate complexes have the potential to produce unusual extended networks through hydrogen-bonding since 5-substituted-tetrazoles, which exist in two tautomeric forms (1H and 2H), have relatively low pKa values in the range 3–5, similar to carboxylic acids. Thus, tetrazoles are stable in strongly acidic and basic media, as well as to oxidizing and reducing conditions.18a
 | ||
| Scheme 2 Potential co-ordination modes of 5-substituted-tetrazolates. | ||
In comparison with more common bridging ligands such as pyrazine, 4,4′-bipy and their analogues,22 di-,23 tri-,24 tetra-25 and hexacarboxylates26 and pyridine multicarboxylates,27 and five-membered aromatic heterocycles such as imidazole, pyrazole, triazole and their derivatives,22a,28 which have been extensively used in the construction of interesting polymeric frameworks, the networking ability and the full potential of the donor properties of tetrazoles have been less studied. A range of tetrazolate complexes of Zn(II),7,10,11a, 11b, 13,14 Cd(II)4a, 11c and Mn(II)7–9 and several extended 2D and 3D metal–organic co-ordination polymers3,5,6,10,12,20,21 of 5-substituted-tetrazole ligands have been synthesized and structurally characterized; in some cases the bridging tetrazolate ligands are generated in situ under hydrothermal conditions.
The present study focuses on the preparation of metal–organic co-ordination polymers by hydrothermal reactions involving the in situ generation of 5-substituted tetrazolate ligands. We report herein the hydrothermal synthesis, solid-state structures, supramolecular topological analysis and preliminary properties of five metal–tetrazolate complexes (1–5) and two unexpected by-products (6 and 7) obtained under more severe reaction conditions.
[Zn(HPO4)(Hpytz)] (1)
[Cd(Hpytz)2(H2PO4)2(H2O)2] (2)
[Zn(pytz)2(H2O)4]·2H2O (3)
[M(tzpyo)2(H2O)4] [M = Zn (4), Ni (5)]
[inaH]+(NO3−) (6)
[(4,4′-bipyH2)(H2PO4)2(H2O)2] (7)
[pytz− = 5-(4-pyridyl)tetrazolate, tzpyo− = 4-tetrazolate-pyridine-N-oxide, ina = isonicotinic acid, 4,4′-bipy = 4,4′-bipyridine]
Experimental
General
All chemicals were purchased from commercial sources and were used without further purification. Elemental analyses of C, H and N were performed by the Elemental Analysis Service of the School of Chemistry, University of Nottingham. Infrared spectra were measured as KBr disks on a Nicolet Avatar 360 FT-IR system over the 400–4000 cm−1 range. Thermal gravimetric analysis was performed with a Rheometric Scientific STA 1500H thermal analyzer at a heating rate of 1 °C min−1. X-Ray powder diffraction data were collected on a Philips X'pert powder diffractometer with Cu Kα radiation (λ = 1.5418 Å) from samples mounted on a flat glass plate sample holder. Scans of approximately 90 min were run for each sample over the range 5° ≤ 2θ ≤ 60° with a step size of 0.02° in 2θ. Simulated powder patterns were generated from the final single crystal refinement model using MERCURY version 1.4.2.29A similar hydrothermal reaction using NiCl2·6H2O in place of the Zn(II) salt led to the formation of pale green crystals of 5 in 40% yield based on Ni. Anal. Calc. For C12H16N10NiO6: C, 31.68; H, 3.54; N, 30.78%. Found: C, 31.65; H, 3.56; N, 30.85%. IR(cm−1): 3355(s, vb), 1591(m), 1530(s), 1463(s), 1438(s), 1388(s), 1219(s), 1175(m), 1044(w), 852(s), 766(w), 654(s).
[inaH]+(NO3−) (6): A mixture of Fe(NO3)2·6H2O (0.2161 g, 0.75 mmol), 4-cyanopyridine (0.1042 g, 1.0 mmol)), NaN3 (0.0667 g, 1.0 mmol), and H3PO4 (0.4238 g, 85% wt) in H2O (10 ml) was stirred for 5 min sealed in a 23 cm3 Parr bomb and heated at 180 °C for 4 d to yield a pale green gel-like solid. Colourless crystals of 6 were obtained in ca. 45% yield (based on 4-cyanopyridine) after removal of the gel-like solid and upon evaporation of the solvent. Anal. Calc. For C6H6N2O5: C, 38.72; H, 3.25; N, 15.05%. Found: C, 38.75; H, 3.26; N, 15.11%. Selected IR (KBr, cm−1): 3470(s, br), 1700(s), 1595(s), 1548(s), 1225(w), 1047(w), 1016(w), 866(w), 848(m), 766(s), 700(m), 690(s), 556(w), 508(w).
X-Ray crystallographic study
Suitable crystals were mounted in films of perfluoropolyether (Fomblin YR-1800, Alfa Aesar) on glass fibres and transferred into the cold stream of an Oxford Cryosystems open-flow cryostat30 on a Bruker SMART CCD diffractometer equipped with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Diffraction data were collected at 150 K, and an empirical absorption correction by SADABS31 was applied to the intensity data. All calculations were performed with the SHELXTL32 suite of programs. All structures were solved by direct methods, developed by iterative cycles of least-squares refinement and difference Fourier synthesis, and refined by the full-matrix least-squares with anisotropic displacement parameters for all non-hydrogen atoms. Hydrogen atoms bound to oxygen and to pyridyl nitrogen atoms were located from difference Fourier syntheses and then refined with distance restraints to O—H or freely for pyridyl NH; other H atoms were placed in calculated positions and refined using a riding model with Uiso = 1.2Ueq(C). Crystal data and other experimental details are summarized in Table 1.| Formula | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| C6H6N5O4PZn | C12H18CdN10O10P2 | C12H20N10O6Zn | C12H16N10O6Zn | C12H20N10NiO6 | C6H6N2O5 | C10H18N2O10 P2 | |
| Formula weight | 308.50 | 636.70 | 465.75 | 461.72 | 455.06 | 186.13 | 388.20 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| space group | P 21/c | P-1 | P-1 | C2/c | C2/c | P21/n | P21/c |
| a/Å | 5.4076(10) | 6.9577(5) | 7.1480(12) | 21.546(6) | 21.403(3) | 6.6590(8) | 18.947(3) |
| b/Å | 8.536(2) | 9.1474(7) | 7.7330(13) | 6.928(2) | 6.9036(9) | 8.1864(10) | 9.0330(14) |
| c/Å | 21.321(4) | 9.4756(7) | 8.6938(15)) | 11.201(3) | 11.1612(14) | 14.140(2) | 9.4944(14) |
| α/° | 90 | 100.334(2) | 88.434(3) | 90 | 90 | 90 | 90 |
| β/° | 94.219(3) | 105.472(2) | 89.527(3) | 95.737(5) | 95.837(2) | 90.398(2) | 92.736(2) |
| γ/° | 90 | 107.665(2) | 80.241(2) | 90 | 90 | 90 | 90 |
| V/Å3 | 981.5(5) | 531.09(12) | 473.4(2) | 1663.6(8) | 1640.6(6) | 770.80(17) | 1623.1(7) |
| Z | 4 | 1 | 1 | 4 | 4 | 4 | 4 |
| D calc /g cm−3 | 2.088 | 1.991 | 1.634 | 1.843 | 1.842 | 1.604 | 1.589 |
| µ(Mo Kα)/mm−1 | 2.678 | 1.256 | 1.352 | 1.539 | 1.246 | 0.142 | 0.323 |
| hkl ranges | −6 ≤ h ≤ 6 | −8 ≤ h ≤ 8 | −9 ≤ h ≤ 9 | −23 ≤ h ≤ 27 | −27 ≤ h ≤ 27 | −8 ≤ h ≤ 4 | −24 ≤ h ≤ 24 |
| −9 ≤ k ≤ 11 | −11 ≤ k ≤ 9 | −10 ≤ k ≤ 10 | −7 ≤ k ≤ 9 | −8 ≤ k ≤ 8 | −10 ≤ k ≤ 10 | 11 ≤ k ≤ 11 | |
| −25 ≤ l ≤ 27 | −12 ≤ l ≤ 12 | −11 ≤ l ≤ 11 | −13 ≤ l ≤ 14 | −14 ≤ l ≤ 14 | −17 ≤ l ≤ 18 | −12 ≤ l ≤ 12 | |
| Goodness-of-fit on F2 | 1.09 | 1.06 | 1.12 | 1.05 | 1.05 | 1.00 | 1.04 |
| R 1 (wR2) | 0.0255 (0.0598) | 0.0174(0.0456) | 0.0247(0.0636) | 0.0313(0.0795) | 0.0302(0.0769) | 0.0313(0.0700) | 0.0453(0.118) |
| (Δ/σ)max in final cycle | 0.001 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.002 |
| Largest difference peak and hole/e Å−3 | 0.45 and −0.29 | 0.37 and −0.41 | 0.42 and −0.23 | 0.64 and −0.27 | 0.66 and −0.24 | 0.21 and −0.18 | 0.67 and −0.43 |
Results and discussion
Synthesis
Hydrothermal reaction (140–170 °C) of NaN3, 4-cyanopyridine, H3PO4 and Zn(OAc)2·4H2O (for 1), or CdCl2·5H2O (for 2) in water in different molar ratios yielded [Zn(HPO4)(Hpytz)] (1), [Cd(Hpytz)2(H2PO4)2(H2O)2] (2), while the corresponding reactions of 4-cyanopyridine with NaN3 and ZnCl2 in the absence of H3PO4 gave [Zn(pytz)2(H2O)4]·2H2O (3) [pytz− = 5-(4-pyridyl)tetrazolate]. The hydrothermal reaction of 4-cyanopyridine N-oxide with NaN3, ZnCl2 or NiCl2 resulted in the formation of [M(tzpyo)2(H2O)4] [M = Zn (4), Ni (5)] [tzpyo− = 4-tetrazolate-pyridine N-oxide], respectively. For each reaction the disappearance of the cyano peak in the 2100 cm−1 region of the IR spectrum is consistent with a [2 + 3] cycloaddition between the nitrile and the azide, with tetrazolate stretching frequencies observed near 1400 cm−1 consistent with those of other metal-tetrazolate complexes reported previously.3–14We also investigated the reactions as a function of temperature. Thus, hydrothermal reaction of NaN3, 4-cyanopyridine, ZnCl2/Zn(OAc)2·4H2O and H3PO4 below 120 °C always afforded the inorganic salt [Zn3(PO4)2(H2O)4], while hydrothermal reaction at 180 °C resulted in the formation of 4-carboxypyridinium nitrate [inaH+](NO3) (6, ina = isonicotinic acid) viahydrolysis of 4-cyanopyridine. Another unexpected hydrogen-bonded organic salt [(4,4′-bipyH2)(H2PO4)(H2O)2] was produced in low yield at 200 °C by replacing 4-cyanopyridine with 4,4′-bipyridine-2-carbonitrile in the above reaction. These synthetic studies confirm that 5-substituted tetrazoles can be readily formed and isolated within the appropriate temperature range. However, use of higher temperatures leads to hydrolysis of nitriles to carboxylates and subsequently to decarboxylation.
Structural descriptions
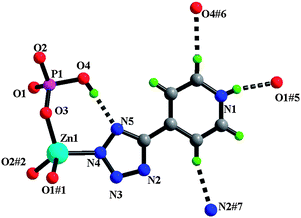 | ||
| Fig. 1 View of asymmetric unit and unique hydrogen-bonds for 1 (symmetry codes are defined in Table 2). | ||
In 1 three O3ZnN tetrahedra are bridged by three phosphate groups to form a six-membered ring of corner-sharing tetrahedra. A series of such six-ring units is fused to form an extended two-dimensional zinc oxo-phosphate network structure (Fig. 2a) in which 5-(4-pyridyl)tetrazolate ligands bind to the metal centre through the tetrazolate N-donor in the 2-position and protrude into the inter-lamellar regions. The stacking of adjacent layers in an ABAB sequence along the crystallographic a direction reveals the organic groups oriented perpendicular to the plane formed by the zinc oxophosphate layers. The 5-(4-pyridyl)tetrazole rings in adjacent layers are parallel, considerably overlapped, and separated by ∼3.4 Å suggesting potential π–π stacking interactions (Fig. 2b). One intra-layer hydrogen-bond is observed between the tetrazolate nitrogen at the 1-position and P–O(4)H, characterized by a O(4)⋯N(5) distance of 2.763(2) Å [H⋯N 1.939(11) Å, ∠O–H⋯N 172(3)°] (Table 2). An important structural feature of 1 is the observed protonation of the pyridyl ring rather than the tetrazolate group, which is indicated by the location in a difference Fourier map of H1 in a suitable position near N1. The presence of strong interlayer N–H⋯O hydrogen-bonds between the pyridinium nitrogen proton N(1) and P=O(1)#5 groups [N⋯O 2.768(3) Å, H⋯O 1.98(3) Å and ∠NHO 166(3)°; #5 = x, −y + 3/2, z + 1/2] confirms the presence of an NH group in this position. Three N–H⋯O hydrogen-bonds are complemented by a series of C–H⋯O and C–H⋯N interactions between adjacent layers (Table 2). Complex 1 represents the first example of a metal phosphate with co-ordinated tetrazolate ligands.
| 1 a | |||||
| Zn(1)-O(1)#1 | 1.9412(14) | N(1)-C(6) | 1.339(3) | O(2)#2–Zn(1)-N(2) | 113.98(7) |
| Zn(1)-O(2)#2 | 1.9168(15) | N(2)-N(3) | 1.328(3) | O(3)-Zn(1)-N(4) | 107.23(7) |
| Zn(1)-O(3) | 1.9304(15) | N(2)-C(7) | 1.347(3) | O(2)-P(1)-O(1) | 112.15(9) |
| Zn(1)-N(4) | 2.0102(18) | N(3)-N(4) | 1.326(3) | O(2)-P(1)-O(3) | 113.01(9) |
| P(1)-O(1) | 1.5330(15) | N(4)-N(5) | 1.344(2) | P(1)-O(3)-Zn(1) | 133.50(9) |
| P(1)-O(2) | 1.5163(15) | N(5)-C(7) | 1.336(3) | ||
| P(1)-O(3) | 1.5188(15) | ||||
| P(1)-O(4) | 1.5891(16) | ||||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
| N(1)-H(1)⋯O(1)#5 | 0.80(3) | 1.98(3) | 2.768(3) | 166(3) | |
| O(4)-H(4)⋯N(5) | 0.83(3) | 1.94(3) | 2.763(2) | 172(3) | |
| C(2)-H(2A)⋯O(4)#6 | 0.95 | 2.33 | 3.278(3) | 173 | |
| C(5)-H(5A)⋯N(2)#7 | 0.95 | 2.33 | 3.243(3) | 162 | |
| 2 b | |||||
|---|---|---|---|---|---|
| Cd(1)-O(1) | 2.2735(12) | N(1)-C(1) | 1.342(2) | O(1)-Cd(1)-N(3) | 86.99(5) |
| Cd(1)-N(3) | 2.3300(14) | N(2)-N(3) | 1.338(2) | O(1)-Cd(1)-O(5) | 91.93(4) |
| Cd(1)-O(5) | 2.3151(12) | N(3)-N(4) | 1.324(2) | O(5)-Cd(1)-N(3) | 89.06(5) |
| P(1)-O(1) | 1.4960(13) | N(5)-C(6) | 1.340(2) | O(1)-P(1)-O(2) | 117.60(7) |
| P(1)-O(2) | 1.5082(13) | C(3)-C(6) | 1.469(2) | O(1)-P(1)-O(4) | 106.09(7) |
| P(1)-O(3) | 1.5716(12) | C(4)-C(5) | 1.378(3) | O(2)-P(1)-O(4) | 109.10(7) |
| P(1)-O(4) | 1.5828(12) | ||||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
| O(5)-H(5C)⋯O(3)#1 | 0.85(3) | 1.99(3) | 2.8212(18) | 166(2) | |
| O(5)-H(5B)⋯O(2)#2 | 0.85(3) | 1.87(3) | 2.7069(17) | 166(3) | |
| N(1)-H(1)⋯O(4)#3 | 0.76(3) | 1.99(3) | 2.746(2) | 180(3) | |
| O(3)-H(3)⋯O(2)#4 | 0.85(3) | 1.68(3) | 2.5199(17) | 172(3) | |
| O(4)-H(4)⋯N(5)#5 | 0.85(3) | 1.81(3) | 2.6309(19) | 162(3) | |
| 5 c | |||||
|---|---|---|---|---|---|
| a Symmetry codes: #1 x −1, y, z; #2 −x + 1, y−1/2, −z + 1/2; #3 x + 1, y, z; #4 −x + 1, y + 1/2, −z + 1/2; #5 x, −y + 3/2, z + 1/2; #6 −x + 2, −y + 2, −z + 1; #7 −x, −y + 1, −z + 1. b Symmetry codes: #1 −x−1, −y + 1, −z; #2 −x, −y + 1, −z; #3 −x, −y + 2, −z + 1; #4 −x − 1, −y, −z; #5 −x, −y + 1, −z + 1. c Symmetry codes: #1 −x + 1, y, −z + 1/2; #2 x + 1/2, y − 1/2, z; #3 −x + 1/2, −y + 1/2, −z; #4 x + 1/2, y + 1/2, z; #5 x, −y, z − 1/2. | |||||
| Ni(1)-O(2) | 2.0706(13) | N(3)-N(4) | 1.332(2) | O(3)#1-Ni(1)-O(3) | 81.60(8) |
| Ni(1)-O(3) | 2.0580(14) | N(4)-N(5) | 1.323(2) | O(3)-Ni(1)-O(2) | 96.57(6) |
| Ni(1)-N(4) | 2.0724(16) | N(5)-C(6) | 1.341(2) | O(3)-Ni(1)-O(2)#1 | 177.79(5) |
| O(1)-N(1) | 1.334(2) | C(1)-C(2) | 1.369(3) | O(3)-Ni(1)-N(4) | 89.40(6) |
| N(1)-C(5) | 1.352(2) | C(2)-C(3) | 1.400(3) | O(2)-Ni(1)-N(4) | 88.90(6) |
| N(1)-C(1) | 1.352(2) | C(3)-C(4) | 1.399(3) | O(2)-Ni(1)-N(4)#1 | 89.44(6) |
| N(2)-N(3) | 1.328(2) | C(3)-C(6) | 1.462(3) | O(2)#1-Ni(1)-N(4)#1 | 88.90(6) |
| N(2)-C(6) | 1.351(2) | C(4)-C(5) | 1.372(3) | N(4)-Ni(1)-N(4)#1 | 177.74(8) |
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
| O(2)-H(2B)⋯O(1)#2 | 0.836(10) | 1.903(11) | 2.7231(19) | 167(2) | |
| O(3)-H(3C)⋯O(1)#3 | 0.838(10) | 1.848(12) | 2.668(2) | 165(3) | |
| O(3)-H(3B)⋯O(1)#4 | 0.838(10) | 1.899(11) | 2.7254(19) | 169(2) | |
| O(2)-H(2C)⋯N(3)#5 | 0.833(10) | 2.050(12) | 2.858(2) | 163(2) | |
![View of 1; (a) two-dimensional layers viewed along the c axis; (b) three-dimensional structure formed by hydrogen-bonds and π–π stacking interactions (viewed along [100] direction, Zn green, P purple, O red, N blue, C deep gray, H light gray).](/image/article/2009/CE/b808496c/b808496c-f2.gif) | ||
| Fig. 2 View of 1; (a) two-dimensional layers viewed along the c axis; (b) three-dimensional structure formed by hydrogen-bonds and π–π stacking interactions (viewed along [100] direction, Zn green, P purple, O red, N blue, C deep gray, H light gray). | ||
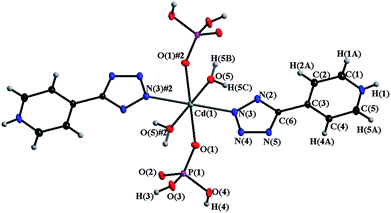 | ||
| Fig. 3 View of local co-ordination geometry at the Cd(II) centre in 2 (50% ellipsoid probability). | ||
Interestingly, each [(H2O)2Cd(H2PO4)2] unit is connected via moderately short OW-H⋯O–P [O⋯O 2.8212(18) Å, H⋯O 1.99(3) Å, ∠OHO, 166(2)°] and short linear P–O–H⋯O–P hydrogen-bonds [O⋯O 2.5199(17) Å, H⋯O 1.68(3) Å, ∠OHO 172(3)°] into sheets with three kinds of hydrogen-bonded rings characterized by graph set notations R22(8), R22(12) and R44(12). To clearly understand the 3D supramolecular framework in 2, we first consider each [(H2O)2Cd(H2PO4)2] unit as a four-connected node, taking double P–O–H⋯O–P or OW–H⋯O–P bonds as a single inter-node connection. A two-dimensional (4,4) layer is then obtained, with grid lengths of 6.96 and 9.67 Å and a grid angle of 64.37°, which can be clearly seen in Fig. 4a. Individual sheets are corrugated and interconnected through Hpytz ligands acting as pillars into a 3-D supramolecular network (Fig. 4b). Each pillaring Hpytz ligand is co-ordinated to a Cd(II) centre within a (4,4) layer with a Npy–H⋯Ophosphate interaction [N⋯O 2.746(2) Å, H⋯O 1.99(3) Å, ∠NHO 180(3)°] through the pyridinium hydrogen-bond to a phosphate oxygen acceptor, O(4), of the adjacent layer. These N–H⋯O hydrogen-bonds are supported by further Ophosphate-H⋯Ntz contacts [O⋯N 2.6309(19) Å, H⋯O 1.81(3) Å, ∠OHN, 162(3)°, Table 2] and π–π interactions between neighbouring, parallel Hpytz pillars separated by a distance of 3.36 Å.
 | ||
| Fig. 4 Views of (a) hydrogen-bonded sheets in 2 (viewed along the c axis); (b) and (c) 3D pillar-layered supramolecular structure (viewed close to a and b axis, respectively). H atoms on carbons are omitted for clarity (Cd green, P purple, O red, N blue, H light cyan). | ||
[M(tzpyo)2(H2O)4] [M = Zn (4), Ni (5)]
Compound 5 is isomorphous with 4, which has been reported recently.13b Both crystallize in the space groupC2/c and exhibit 3-D supramolecular arrays formed by π–π stacking interactions and hydrogen-bonds. In 5, each Ni(II) ion resides in an octahedral co-ordination geometry [(NiN2(OH2)4] of which the equatorial plane consists of four co-ordinated H2O molecules, while the apical positions are occupied by two N-donors at the 2-position of the tetrazolate. The metal sits on a two-fold axis resulting in only three unique co-ordination bonds [Ni1–O2 2.0706(13), Ni1–O3 2.0580(14), Ni1–N4 2.0725(16) Å] (Fig. 5a).![a) View of molecular structure of 5 (symmetry codes: #1, (−x, y, −z + 1/2); #2, (x + 1/2, y − 1/2, z); #3, (−x + 1/2, −y + 1/2, −z); #4, (x + 1/2, y + 1/2, z); #5, (x, −y, z − 1/2); #6, (−0.5 + x, 0.5 + y, z); #7, (0.5 − x, 0.5 − y, −z); and #8, (−0.5 + x, −0.5 + y, z). b) View of supramolecular hydrogen-bonded layer formed by [Ni(OH2)4]2+ and the oxygen atoms of tzpyo− ligands (viewed along the a axis). (c) and (d) 3D framework of 5 (viewed along the b and c axis, respectively).](/image/article/2009/CE/b808496c/b808496c-f5.gif) | ||
| Fig. 5 a) View of molecular structure of 5 (symmetry codes: #1, (−x, y, −z + 1/2); #2, (x + 1/2, y − 1/2, z); #3, (−x + 1/2, −y + 1/2, −z); #4, (x + 1/2, y + 1/2, z); #5, (x, −y, z − 1/2); #6, (−0.5 + x, 0.5 + y, z); #7, (0.5 − x, 0.5 − y, −z); and #8, (−0.5 + x, −0.5 + y, z). b) View of supramolecular hydrogen-bonded layer formed by [Ni(OH2)4]2+ and the oxygen atoms of tzpyo− ligands (viewed along the a axis). (c) and (d) 3D framework of 5 (viewed along the b and c axis, respectively). | ||
The ability of pyridine N-oxides to form hydrogen-bonds with water molecules is well established in supramolecular chemistry and crystal engineering.41 Here, each oxygen of the tzpyo− ligands is hydrogen-bonded to three co-ordinated water molecules from three [Ni(tzpyo)2(H2O)4] units, resulting in the formation of 2D ordered hydrogen-bonded {ONi(H2O)4}n layers (Fig. 5b). These hydrogen-bonds are rather short with Oligand–Ow distances in the range 2.668(2)–2.858(2) Å, and Oligand–H–Ow angles of 163(2)–169(2)° (see Table 2).
 | ||
| Fig. 6 (a) View of structure of 6 and crystallographically independent hydrogen-bonds (symmetry codes are defined in Table 3). (b) View of 2-D supramolecular sheet formed by inaH+ cations and nitrate anions via strong O–H⋯O and N–H⋯O and weak C–H⋯O hydrogen-bonds. (c) View of 2-D (4,6)-connected net for 6. | ||
The tzpyo− ligands serve as pillars that connect two-dimensional hydrogen-bonded sheets to create a 3D network (Fig. 5c and 5d). The oxygen of the tzpyo− ligand is part of a hydrogen-bonded sheet, while the tetrazolate 2-N centres of the same ligand bind to a Ni(II) cation from the adjacent sheet. In addition, Ow-H⋯Ntzhydrogen-bonds [O⋯N 2.858(2) Å, H⋯N 2.050(12), ∠OHN, 163(2)°] are adopted between adjacent layers (see Table 2). In 5 adjacent tetrazolate and pyridine N-oxide moieties are almost parallel (dihedral angle: 3.06°) and stack in an ABAB fashion along the direction parallel to the crystallographic b axis (Fig. 5c). The spacing between adjacent tetrazole and pyridine N-oxide moieties (ca. 3.52 Å, centroid–centroid) indicates effective π–π stacking interactions which can be presumed to contribute to the adopted structural arrangement.
| 6 a | ||||
|---|---|---|---|---|
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) |
| N(1)-H(1)⋯O(4)#1 | 0.965(17) | 1.770(18) | 2.7346(15) | 178.1(15) |
| O(2)-H(2)⋯O(4) | 0.881(17) | 1.710(17) | 2.5807(12) | 169.6(15) |
| C(1)-H(1A)⋯O(3)#1 | 0.95 | 2.50 | 3.1944(17) | 130 |
| C(2)-H(2A)⋯O(5)#2 | 0.95 | 2.46 | 3.3756(16) | 161 |
| C(4)-H(4A)⋯O(3)#3 | 0.95 | 2.34 | 3.1761(16) | 147 |
| C(5)-H(5A)⋯O(1)#1 | 0.95 | 2.43 | 3.1515(17) | 132 |
| 7 [ b ] | ||||
|---|---|---|---|---|
| a Symmetry codes: #1 −x + 1, y, −z + 1/2; #2 x + 1/2, y − 1/2, z; #3 −x + 1/2, −y + 1/2, −z; #4 x + 1/2, y + 1/2, z; #5 x, −y, z − 1/2. b Symmetry codes: #1 x, −y + 3/2, z − 1/2; #2 −x + 2, y − 1/2, −z + 9/2; #3 x, −y + 3/2, z + 1/2; #4 −x + 1, y + 1/2, −z + 5/2 #5 x − 1, −y + 3/2, z − 3/2; #6 x + 1, y, z + 1; #7 −x + 2, y + 1/2, −z + 9/2; #8 −x + 2, −y + 1, −z + 4; #9 −x + 1, −y + 1, −z + 3. | ||||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) |
| O(3)-H(3B)⋯O(1)#1 | 0.834(10) | 1.770(12) | 2.5876(19) | 166(3) |
| O(4)-H(4B)⋯O(2)#2 | 0.839(10) | 1.696(10) | 2.5328(19) | 174(3) |
| O(7)-H(7B)⋯O(5)#3 | 0.830(10) | 1.760(12) | 2.580(2) | 169(3) |
| O(8)-H(8B)⋯O(6)#4 | 0.842(10) | 1.639(11) | 2.478(2) | 173(3) |
| O(9)-H(9C)⋯O(10)#5 | 0.842(10) | 1.885(13) | 2.712(3) | 167(3) |
| O(9)-H(9B)⋯O(5) | 0.843(10) | 2.027(14) | 2.844(2) | 163(3) |
| O(10)-H(10C)⋯O(9)#6 | 0.840(10) | 1.870(11) | 2.706(3) | 174(3) |
| O(10)-H(10B)⋯O(1) | 0.840(10) | 1.942(10) | 2.780(2) | 176(3) |
| N(1)-H(1)⋯O(2) | 0.95(3) | 1.67(3) | 2.616(2) | 173(2) |
| N(2)-H(2)⋯O(6) | 0.95(3) | 1.65(3) | 2.592(2) | 173(3) |
| C(1)-H(1A)⋯O(1)#7 | 0.95 | 2.48 | 3.373(2) | 157 |
| C(5)-H(5A)⋯O(3)#8 | 0.95 | 2.56 | 3.258(3) | 13 |
| C(6)-H(6A)⋯O(8) | 0.95 | 2.46 | 3.159(3) | 130 |
| C(9)-H(9A)⋯O(9)#9 | 0.95 | 2.40 | 3.319(3) | 162 |
The crystal structure of 6 is rather simple but interesting. As shown in Fig. 6b, all hydrogen atoms in each 4-carboxypyridinium cation (inaH+) are involved in hydrogen-bonding interactions with either nitrate anions or adjacent inaH+ moieties (Table 3). Through these extensive hydrogen-bonds, each nitrate anion connects four 4-carboxypyridinium cations to form a supramolecular sheet, in which each 4-carboxypyridinium cation is surrounded by four nitrate anions, and hydrogen-bonded to two symmetry-related cations. Weak π–π stacking interactions are observed between neighbouring supramolecular sheets.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2. As depicted in Fig. 7b, the dihydrophosphate moieties interact via O–H···O hydrogen-bonds into an unusual 2-D square grid-type sheet with P⋯P separations ranging from 4.83 to 4.85 Å. All hydrogen-bonds within the layered morphology have O⋯O separations of less than 2.6 Å and near-linear ∠O–H⋯O angles in the range of 167–176° (Table 3). Although several 1D hydrogen-bonded (H2PO4−)n chains have been crystallographically characterized in recent years,42 to the best of our knowledge, there is only one previously reported example of extended hydrogen-bonded {H2PO4−}n layers.43 In 7, the organobipyridine cations serve as pillars linking the dihydrogenphosphate layers via linear strong N–H⋯O hydrogen-bonds and weak C–H⋯O contacts (Table 3) to form a 3-D supramolecular open-framework with 1D channels of dimensions of ca. 5.1 × 6.1 Å running parallel to the c axis (Fig.7c). Each 1D channel accommodates a helical chain of water dimers. As shown in Fig.7d, two crystallographically distinct water molecules (O9 and O10) are hydrogen-bonded within the structure,44 and these are assembled along the crystallographic 21-screw axis by linear O10–H10C⋯O9 hydrogen-bonds into extended helical chains in a 1:1 ratio of left- and right-handedness. They are further anchored to the organic phosphoric host framework via O9–H9B⋯O5 and O10–H10B⋯O1 and weak C55–H55A⋯O9 hydrogen-bonding. As a result, the two crystallographically distinct oxygen atoms of the water molecules have different coordination numbers The centre O9 is 4-co-ordinate (to two hydrogen atoms acting as acceptors with two lone pairs as donors) in an approximate tetrahedral arrangement, while O10 shows a 2 + 1 triangular coordination (Fig. 7d).
:2:2. As depicted in Fig. 7b, the dihydrophosphate moieties interact via O–H···O hydrogen-bonds into an unusual 2-D square grid-type sheet with P⋯P separations ranging from 4.83 to 4.85 Å. All hydrogen-bonds within the layered morphology have O⋯O separations of less than 2.6 Å and near-linear ∠O–H⋯O angles in the range of 167–176° (Table 3). Although several 1D hydrogen-bonded (H2PO4−)n chains have been crystallographically characterized in recent years,42 to the best of our knowledge, there is only one previously reported example of extended hydrogen-bonded {H2PO4−}n layers.43 In 7, the organobipyridine cations serve as pillars linking the dihydrogenphosphate layers via linear strong N–H⋯O hydrogen-bonds and weak C–H⋯O contacts (Table 3) to form a 3-D supramolecular open-framework with 1D channels of dimensions of ca. 5.1 × 6.1 Å running parallel to the c axis (Fig.7c). Each 1D channel accommodates a helical chain of water dimers. As shown in Fig.7d, two crystallographically distinct water molecules (O9 and O10) are hydrogen-bonded within the structure,44 and these are assembled along the crystallographic 21-screw axis by linear O10–H10C⋯O9 hydrogen-bonds into extended helical chains in a 1:1 ratio of left- and right-handedness. They are further anchored to the organic phosphoric host framework via O9–H9B⋯O5 and O10–H10B⋯O1 and weak C55–H55A⋯O9 hydrogen-bonding. As a result, the two crystallographically distinct oxygen atoms of the water molecules have different coordination numbers The centre O9 is 4-co-ordinate (to two hydrogen atoms acting as acceptors with two lone pairs as donors) in an approximate tetrahedral arrangement, while O10 shows a 2 + 1 triangular coordination (Fig. 7d).
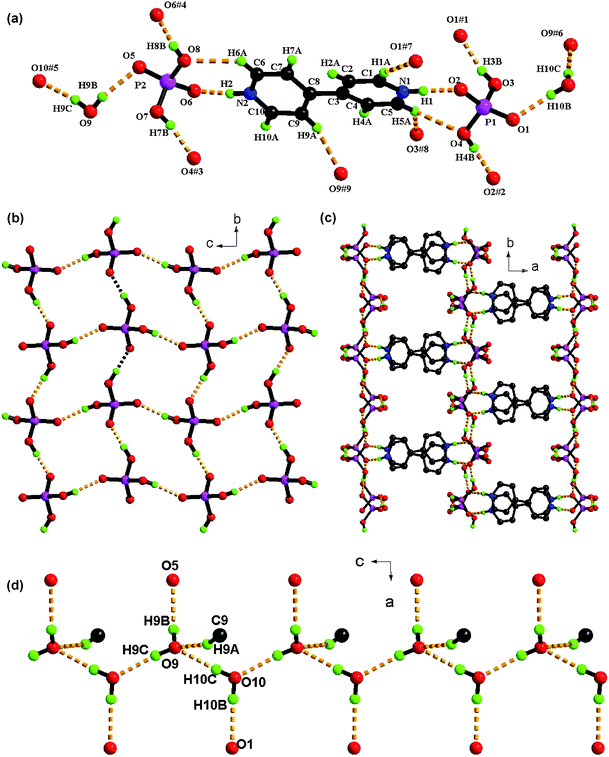 | ||
| Fig. 7 (a) View of structure of 7 and crystallographically independent H-bonding arrangements (symmetry codes are defined in Table 3). (b) An unusual square grid-type dihydrogenphosphonate layer formed through O–H⋯O links; (c) the organobipyridine cations connecting the dihydrogenphosphonate layers into a 3D supramolecular network with 1D channels along c axis. (d) Perspective view of the 1D helical chains which are anchored to the host framework via O–H··O and C–H⋯O contacts. | ||
Discussion
Coordination modes of 5-substituted-tetrazoles
5-Substituted-tetrazoles can be formed readily under appropriate hydrothermal conditions, and are stable in acidic, neutral and basic media. The 5-(4-pyridyl)tetrazole (Hpytz) ligand can exhibit a range of forms (Scheme 3) and these can bind to metal ions by different modes, (i)–(v): through the tetrazolate N-centre (‘TZ-mode’), the pyridyl (‘Py-mode’) or both pyridyl and tetrazolate N-donors (‘Py-TZ-mode’). From the structures of complexes 1–3 prepared under hydrothermal conditions in the presence or absence of H3PO4, it can be seen that the degree of acidification of the aqueous reaction solution influences the observed co-ordination modes of the functional groups of 5-functionalized-tetrazoles. Under the acidic conditions employed in the synthesis of complexes 1 and 2, the ligand pyridyl nitrogen is protonated in preference to any of the tetrazolate nitrogens and the 5-(4-pyridyl)tetrazole ligand, therefore, binds the metal centre only through the 2-N-donor of the tetrazolate [‘TZ-mode’, (i) in Scheme 3] in the isolated product. In solution, however, a range of protonation and complexation forms (i)-(v) are possible (Scheme 3). | ||
| Scheme 3 Co-ordination and protonation modes of substituted tetrazoles. | ||
The ‘Py-TZ’ bridging mode (V) (Scheme 3) of the 5-(4-pyridyl)tetrazolate ligand has been observed previously in co-ordination polymers of Zn(II),10,11a, 12a Cd(II),4aCu(I),12a and Cu(II),3 where the pyridyl N-atom and various N-centres of the tetrazolate ring are involved in co-ordination to the metal centres. Here, we report the first observation of the pure ‘TZ-mode’ in the metal complexes 1 and 2, in which only the N-centre at the 2-position of the tetrazolate is co-ordinated to a metal centre, while the pyridyl group is protonated allowing the formation of complex hydrogen-bonded nets. In the structures of 4 and 5, the 4-tetrazolato-pyridine N-oxide (tzpyo−) ligand exhibits a similar terminal ‘TZ-mode’, with the tetrazolate moiety bound to the metal centre, leaving the pyridyl N-oxide group to form three hydrogen-bonds with water molecules.
Supramolecular topologies of 2–7
The self-assembly of isolated molecules into valence bonded polymeric structures has been the subject of a number of studies,45 and it is useful to analyse the topologies of the resultant networks 2–7 in the present work.In 2 and 7, the [Cd(H2PO4)(H2O)2] (2) or the dihydrogenphosphate moieties (7) form a (4,4) net via O–H⋯O hydrogen-bonds (Fig. 4 and Fig. 7), while the Hpytz ligands (2) or the organobipyridine cations (7) act as linear pillars that link adjacent layers to form 6-connected distorted α-Po 41263 net (Fig. 8).46
 | ||
| Fig. 8 View of 6-connected contorted α-Po net for 2 and 7. | ||
During the preparation of this manuscript, the crystal structure of 3, composed of an un-coordinated lattice water molecule and a Zn(II) cation co-ordinated by two pytz− ligands and four H2O molecules, appeared in the literature.9a A highly related Mn(II) complex, [Mn(pytz)2(H2O)4]·2H2O, has also been described recently and this exhibits an isostructural metal complex and a similar supramolecular arrangement viahydrogen-bonding interactions.9b No topological analysis, however, was provided by these authors.9a Compound 3 comprises a (4,5,10)-connected trinary net comprising 4-connected lattice H2O nodes, 5-connected tetrazole nodes and 10-connected [Zn(H2O)4]2+ nodes (Fig. 9a-f). The interconnection of these different nodes viahydrogen-bonds, as well as valence bonds between [Zn(H2O)4]2+ and tetrazoles, results in a highly complex 3-D (4,5,10)-connected net (Fig. 9g) with (3·42·53)2(3·44·54·6)4(32·48·514.612·77·82) topology. Thus each 4-connected H2O node is surrounded by two tetrazolate ligands and two [Zn(H2O)4]2+ nodes, each 5-connected tetrazole node is linked to three [Zn(H2O)4]2+ nodes and two H2O nodes, and each [Zn(H2O)4]2+ node is connected to six tetrazoles and four H2O nodes (Fig. 9).
![(a) to (c) Perspective views of the 4, 5 and 10-connectivity, respectively, in 3 (Zn green, C dark grey, H cyan, N blue, O red). (d) to (f) Topological representation of the 4, 5 and 10-connectivity, respectively, in 3 (10-connected [Zn(H2O)4]2+ nodes, 5-connected tetrazole nodes, and 4-connected non-coordinated H2O nodes are shown as green, blue and red balls, respectively). (g) View of 3D (4,5,10)-connected trinary net with (3·42·53)2(3·44·54·6)4(32·48·514.612·77·82) topology for 3 (viewed close to [100] direction).](/image/article/2009/CE/b808496c/b808496c-f9.gif) | ||
| Fig. 9 (a) to (c) Perspective views of the 4, 5 and 10-connectivity, respectively, in 3 (Zn green, C dark grey, H cyan, N blue, O red). (d) to (f) Topological representation of the 4, 5 and 10-connectivity, respectively, in 3 (10-connected [Zn(H2O)4]2+ nodes, 5-connected tetrazole nodes, and 4-connected non-coordinated H2O nodes are shown as green, blue and red balls, respectively). (g) View of 3D (4,5,10)-connected trinary net with (3·42·53)2(3·44·54·6)4(32·48·514.612·77·82) topology for 3 (viewed close to [100] direction). | ||
Topologically, the supramolecular frameworks of 4 and 5 exhibit an attractive 3-D (4,8)-connected binary net with (46)2(412·612·84) topology, in which the O-centre of each tzpyo− ligand and the [M(H2O)4]2+ moiety can be envisaged as 4-connected and 8-connected nodes, respectively. This net consists of two-dimensional (3,6)-connected [OM(H2O)4] layers further pillared by the 4-tetrazolate-pyridine moiety of tzpyo− ligands (Fig. 10).
 | ||
| Fig. 10 (a) View of the 3-D (4,8)-connected binary (46)2(412·612·84) net for 4 or 5 and (b) of a selected (3,6)-connected layer. | ||
The structure of 6 exhibits a 2D (4,6)-connected binary net of (32·42·52)(34·44·54·63) topology (Fig. 6c), in which the nitrate anion and the inaH+ cation can be regarded as 4- and 6-connected nodes, respectively. Each nitrate node links to four inaH+ nodes which in turn link to four nitrate nodes and two inaH+ nodes.
Thermal stability and powder XRD analysis
To examine the thermal stability of these metal–tetrazolate complexes and their structural variation as a function of temperature, thermal gravimetric analysis (TGA) (Table 4) and powder X-ray diffraction (PXRD) were performed on single-phase polycrystalline samples 1–5. A representative example is given by 2, where the loss of six water molecules (four co-ordinated and two presumably formed by OH− from phosphate moieties and H+ on pyridyl nitrogens) is observed between 205 and 230 °C (obsd. weight loss 12.69%, calcd. 12.61%). Subsequently, the loss of the 5-substituted-tetrazolate ligands is observed above 240 °C to form an unidentified product. Similar thermal behaviour was observed for 1, but with a slightly higher temperature for the release of water molecules, 215-250 °C for 1 vs. 205–230 °C for 2, and for the decomposition of the framework (260 °C for 1vs. 240 °C for 2). PXRD measurements on the dehydrated forms of 1 and 2 revealed that they are amorphous.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a Temperature range for the loss of lattice H2O, co-ordinated H2O and/or those formed by OH− from phosphate moieties and H+ on ligands for 1–5. b Temperature for the initiation of release of tetrazolate ligand. | |||||
| T range/°Ca | 215–250 | 205–230 | 80–150 | 50–175 | 50–180 |
| Weight-loss (obsd)/% | 6.77 | 12.69 | 21.45 | 14.33 | 16.93 |
| Weight-loss (calcd)/% | 5.83 | 12.61 | 23.19 | 15.59 | 15.84 |
| State (before loss of solvent molecules) | Crystalline | Crystalline | Crystalline | Crystalline | Crystalline |
| State (after loss of solvent molecules) | Amorphous | Amorphous | Crystalline | Crystalline | Amorphous |
| Temperature (°C)[b] | 260 | 240 | 250 | 210 | 210 |
The thermal behaviour of 3, as revealed by TGA, is quite distinct from that displayed by 1 and 2. Firstly, a weight loss of 21.45% is observed between 80 and 150 °C, which can be assigned to the release of two lattice and four co-ordinated water molecules per formula unit (calcd. weight loss 23.19%). The compound then reaches a plateau with no further weight loss up to ∼250 °C. With further increases of temperature, the ligand molecules (pytz−) are released forming an unidentified product (Fig. 11).
 | ||
| Fig. 11 TGA analysis of compounds 1–5. | ||
The structures of 3 and 4 both contain metal cations each co-ordinated by four water molecules and two transnitrogen donors from pyridyl (3) or tetrazolate rings (4), leaving an un-co-ordinated tetrazolate ring or pyridyl N-oxide moiety, respectively. The dehydrated forms of 3 and 4 were studied by powder X-ray diffraction. Complex 3 was heated at 170 °C for one day and then exposed to air. The PXRD data for the final product were found to be consistent with that of the crystal structure of 3, indicating that the dehydrated form of 3 rapidly absorbs water from the atmosphere and so reverts to its original form.
However, the powder X-ray diffraction profile of the solid obtained by dehydrating 4 at 170 °C under N2 flow to form 4a is significantly different from that of the starting material 4 (Fig. 12). The observation of sharp reflections indicates that 4a retains crystallinity, demonstrating that thermal treatment of 4 gives rise to a solid-state crystalline phase conversion, probably involving a change of co-ordination environment around the Zn(II) metal centres. A single crystal X-ray diffraction pattern of the new crystalline phase was obtained, but the data were not of sufficient quality to allow structure solution. Moreover, the dehydrated form 4a can effectively adsorb water molecules from air to give [Zn(tzpyo)2]·mH2O. The PXRD pattern profile of the sample obtained by exposure of 4a to air for one (4b) or two days (4c) is identical to that of 4a, while its TGA behaviour is significantly different from that of 4 (see the inset diagram in Fig. 12). From TGA data, about 2.5 water molecules per formula unit are adsorbed by 4a after exposure to air for two days. The adsorption properties of 4a with other guest molecules (e.g., methanol and benzene) or gases will form the basis of further investigations.
![PXRD patterns for 4: (I) simulated from single crystal diffraction data; (II) as-synthesized crystals of 4; (III): new crystalline phase [Zn(tzpyo)2] (4a) obtained by thermal treatment of 4 at 170 °C for 12 h under N2 flow (60 ml min−1); (IV) and (V) samples of [Zn(tzpyo)2]·mH2O obtained by exposure of 4a to air for one (4b) and two days (4c), respectively; (VI) 4a obtained by reheating 4c at 170 °C for 24 h. The inset shows significant differences in TGA behaviour for 4 and 4c.](/image/article/2009/CE/b808496c/b808496c-f12.gif) | ||
| Fig. 12 PXRD patterns for 4: (I) simulated from single crystal diffraction data; (II) as-synthesized crystals of 4; (III): new crystalline phase [Zn(tzpyo)2] (4a) obtained by thermal treatment of 4 at 170 °C for 12 h under N2 flow (60 ml min−1); (IV) and (V) samples of [Zn(tzpyo)2]·mH2O obtained by exposure of 4a to air for one (4b) and two days (4c), respectively; (VI) 4a obtained by reheating 4c at 170 °C for 24 h. The inset shows significant differences in TGA behaviour for 4 and 4c. | ||
The dehydrated solid 4a has broadened peaks with shoulders, and indexing was therefore unreliable. However, the sample 4c obtained by exposure of 4a to air for two days can be indexed and gives a monoclinic P unit cell with a = 21.66, b = 8.72, c = 14.08 Å and β = 133.5°, significantly different from the original monoclinic C unit cell of 4 [a = 21.546(6), b = 6.928(2), c = 11.201(3) Å and β = 95.737(5)°]. The sample 4b obtained by exposure of 4a to air for one day is very similar to 4c. However, 4b has two additional small peaks that can not be indexed, although it appears that the bulk of the sample of 4b is same as 4c.
In contrast, although 5 is structurally isomorphous to 4, the dehydrated phase (5a) obtained by thermal treatment of 5 at 180 °C under nitrogen flow (60 mL min−1) is amorphous. However, when 5a is exposed to air for several days, the resultant solid (5b) shows a low degree of crystallinity with weak diffraction peaks observed (Fig. 13). Some of these peaks may be assigned to the partial re-generation of 5. More conclusively, 5b shows a similar TGA behaviour to that of 5 with no significant weight loss observed before 110 °C and complete loss of water around 180 °C. Thus, although 4 and 5 are isomorphous, thermal treatment leads to the formation of a microporous crystalline phase 4a, which can effectively re-absorb water from the atmosphere to give [Zn(tzpyo)2]·mH2O (4b and 4c). In contrast amorphous phase 5a, which can partially re-coordinate water molecules from air to give [Ni(tzpyo)2(H2O)m] (5b). Such differences may be attributed to the greater lability and different stereochemistry for the Zn(II) centre in 4 compared with Ni(II) in 5.
![PXRD patterns for 5: (I) simulated from single crystal diffraction data; (II) as-synthesized crystals of 5; (III): an amorphous phase [Zn(tzpyo)2] (5a) obtained by thermal treatment of 5 at 180 °C overnight under N2 flow (60 ml min−1); (IV) a new sample obtained by exposure of 5a to air for several days (5b). The inserted diagram shows the similar TGA behaviour of 5 and 5b.](/image/article/2009/CE/b808496c/b808496c-f13.gif) | ||
| Fig. 13 PXRD patterns for 5: (I) simulated from single crystal diffraction data; (II) as-synthesized crystals of 5; (III): an amorphous phase [Zn(tzpyo)2] (5a) obtained by thermal treatment of 5 at 180 °C overnight under N2 flow (60 ml min−1); (IV) a new sample obtained by exposure of 5a to air for several days (5b). The inserted diagram shows the similar TGA behaviour of 5 and 5b. | ||
Conclusions
5-Substituted-tetrazoles can be readily formed by reaction of NaN3 with aromatic nitriles under appropriate hydrothermal conditions, and are stable in acidic, neutral and basic media. However, hydrothermal reactions at higher temperatures may result in hydrolysis of nitriles, followed by decarboxylation. Multidentate 5-functionalized-tetrazolate ligands have been generated in situ under hydrothermal conditions, and bind to Zn(II), Cd(II) or Ni(II) salts to afford diverse supramolecular network structures ranging from 2D grids to 3D supramolecular pillared layers. Significantly, this work confirms that the co-ordination mode of the functional groups of 5-functionalized-tetrazoles is tunable by controlling the pH of the reaction conditions. In addition, both 5-functionalized tetrazolate ligands, pytz− and tzpyo−, have rigid backbones and show co-ordinative versatility due to their multiple donor sites.Acknowledgements
This work was supported by Engineering and Physical Science Research Council (EPSRC), the University of Nottingham and the CVCP (ORS award to WY). M.S. gratefully acknowledges receipt of a Royal Society Wolfson Merit Award and of a Leverhulme Trust Senior Research Fellowship.References
- (a) B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546 CrossRef CAS; (b) R. Robson, B. F. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins and J. P. Liu, ACS Symp. Ser., 1992, 499, 256 CAS.
- (a) N. R. Champness, Dalton Trans., 2006, 877 RSC; (b) N. R. Champness and M. Schröder, Curr. Opin. Sol. State Mater. Sci., 1998, 3, 419 Search PubMed; (c) H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS; (d) A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Cote, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110 CrossRef CAS; (e) D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1 RSC; (f) X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358 CrossRef; (g) X. Lin, A. J. Blake, C. Wilson, X. Sun, N. R. Champness, M. W. George, P. Hubberstey, R. Mokaya and M. Schröder, J. Am. Chem. Soc., 2006, 128, 10745 CrossRef CAS; (h) Y. Kubota, M. Takata, R. Matsuda, R. Kitaura, S. Kitagawa and T. C. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 4932 CrossRef CAS; (i) S. Horike, R. Matsuda, D. Tanaka, S. Matsubara, M. Mizuno, K. Endo and S. Kitagawa, Angew. Chem., Int. Ed., 2006, 43, 7226 CrossRef; (j) J. Jia, X. Lin, C. Wilson, A. J. Blake, N. R. Champness, P. Hubberstey, G. Walker, E. J. Cussen and M. Schröder, Chem. Commun., 2007, 840 RSC; (k) A. J. Blake, N. R. Champness, M. Crew, L. R. Hanton, S. Parsons and M. Schröder, J. Chem. Soc., Dalton Trans., 1998, 1533 RSC; (l) A. J. Blake, N. R. Brooks, N. R. Champness, M. Crew, L. R. Hanton, P. Hubberstey, S. Parsons and M. Schröder, J. Chem. Soc., Dalton Trans., 1999, 2813 RSC; (m) M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey, A. L. Realf and M. Schröder, J. Chem. Soc., Dalton Trans., 2000, 3261 RSC; (n) A. N. Khlobystov, M. T. Brett, A. J. Blake, N. R. Champness, P. M. W. Gill, D. P. O'Neill, S. J. Teat, C. Wilson and M. Schröder, J. Am. Chem. Soc., 2003, 125, 6753 CrossRef CAS.
- (a) T. T. Luo, H. L. Tsai, S. L. Yang, Y. H. Liu, R. D. Yadav, C. C. Su, C. H. Ueng, L. G. Lin and K. L. Lu, Angew. Chem., Int. Ed., 2005, 44, 6063 CrossRef CAS; (b) H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC.
- (a) X. Xue, X. S. Wang, L. Z. Wang, R. G. Xiong, B. F. Abraham, X. Z. You, Z. L. Xe and C. M. Che, Inorg. Chem., 2002, 41, 6544 CrossRef CAS; (b) T. Wu, B. H. Yi and D. Li, Inorg. Chem., 2005, 44, 4130 CrossRef CAS.
- J. Tao, Z. J. Ma, R. B. Huang and L. S. Zheng, Inorg. Chem., 2004, 43, 6133 CrossRef CAS.
- L. Carlucci, G. Ciani and D. M. Proserpio, Angew. Chem., Int. Ed., 1999, 38, 3488 CrossRef CAS.
- F. A. Mautner, C. Gspan, K. Gatterer, M. A. S. Goher, M. A. M. Abu-Youssef, E. Bucher and W. Sitte, Polyhedron, 2004, 23, 1217 CrossRef CAS.
- (a) X. S. Wang, Y. Z. Tang, X. F. Huang, Z. R. Qou, C. M. Che, P. W. H. Chan and R. G. Xiong, Inorg. Chem., 2005, 55, 5278 CrossRef; (b) A. Rodríguez, R. Kivekäs and E. ; Colacio, Chem. Commun., 2005, 5228 RSC.
- (a) J. X. Guo, F. Fu, D. S. Li, J. Li, L. Tang and G. C. Qi, Z. Kristallogr., 2006, 221, 439 CAS; (b) P. Lin, W. Clegg, R. W. Harrington and R. A. Henderson, Dalton Trans., 2005, 2388 RSC.
- R. G. Xiong, X. Xue, H. Zhao, X. Z. You, B. F. Abrahams and Z. L. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800 CrossRef CAS.
- (a) L. Z. Wang, Z. R. Qu, H. Zhao, X. S. Wang, R. G. Xiong and Z. L. Xue, Inorg. Chem., 2003, 42, 3969 CrossRef CAS; (b) S. Stagni, A. Palazzi, S. Zacchini, B. Ballarin, C. Bruno, M. Marcaccio, F. Paolucci, M. Monari, M. Carano and A. J. Bard, Inorg. Chem., 2006, 45, 696; (c) Z. R. Qu, H. Zhao, X. S. Wang, Y. H. Li, Y. M. Song, Y. J. Liu, Q. Ye, R. G. Xiong, B. F. Abrahams, Z. L. Xue and X. Z. You, Inorg. Chem., 2003, 42, 7710 CrossRef CAS; (d) A. Palazzi, S. Stagni, S. Selva and M. Monari, J. Organomet. Chem., 2003, 669, 135 CrossRef CAS.
- (a) C. Jiang, Z. P. Yu, C. Jiao, S. J. Wang, J. M. Li, Z. Y. Wang and Y. , Cui, Eur. J. Inorg. Chem., 2004, 3662 CAS; (b) C. Jiang, Z. P. Yu, C. Jiao, S. J. Wang, J. M. Li, Z. Y. Wang and Y. Cui, Eur. J. Inorg. Chem., 2004, 4669 CrossRef CAS.
- (a) R. Bronisz, Inorg. Chim. Acta, 2002, 340, 215 CrossRef CAS; (b) Z. X. Yu, X. P. Wang, Y. Y. Feng and X. H. Zhong, Inorg. Chem. Commun., 2004, 7, 492 CrossRef CAS.
- X. Xue, B. F. Abrahams, R. G. Xiong and X. Z. You, Aust. J. Chem., 2002, 55, 495 CrossRef CAS.
- (a) H. Singh, A. S. Chawla, V. K. Kapoor, D. Paul and R. K. Malhotra, Prog. Med. Chem., 1980, 17, 151 Search PubMed; (b) R. J. Herr, Bioorg. Medicinial Chem., 2002, 10, 3379 Search PubMed.
- (a) H. Xue, Y. Gao, B. Twamley and J. M. Shreeve, Inorg. Chem., 2005, 44, 5068 CrossRef CAS; (b) C. F. Ye, J. C. Xiao, B. Twamley and J. M. Shreeve, Chem. Commun., 2005, 2750 RSC.
- (a) Z. P. Demko and K. B. Sharpless, Org. Lett., 2001, 3, 4091 CrossRef CAS; (b) F. Himo, Z. P. Demko and L. Noodleman, J. Org. Chem., 2003, 68, 9076 CrossRef CAS; (c) A. W. Bridge, R. H. Jone, H. Kabir, A. A. Kee, D. J. Lythgoe, M. Nakach, C. Pemberton and J. Wrightman, Org. Process Res. Dev., 2001, 5, 9 Search PubMed; (d) F. Ek, S. Manner, L. G. Wistrand and T. Frejd, J. Org. Chem., 2004, 69, 1346 CrossRef CAS.
- (a) Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2110 CrossRef CAS; (b) Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS; (c) Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2113 CrossRef CAS.
- (a) M. Biagini-Cingi, A. M. Manotti-Lanfredi, F. Ugozzoli and J. G. Haasnoot, Inorg. Chim. Acta, 1994, 227, 181 CrossRef CAS; (b) D. Hagrman and J. Zubieta, Chem. Commun., 1998, 2005 RSC; (c) J. Krober, I. Bkouche-Waksman, C. Pascard, M. Thomann and O. Kahn, Inorg. Chim. Acta, 1995, 230, 159 CrossRef; (d) M. G. B. Drew, P. C. Yates, J. Trocha-Grimshaw, K. P. McKillop and S. M. Nelson, J. Chem. Soc., Chem. Commun., 1985, 262 RSC; (e) K. D. Demadis, E.-S. El Samanody, T. J. Meyer and P. S. White, Inorg. Chem., 1998, 37, 838 CrossRef CAS; (f) R. Guilard, I. Perrot, A. Tabard, P. Richard, C. Lecomte, Y. H. Liu and K. M. Kadish, Inorg. Chem., 1991, 30, 27 CrossRef CAS; (g) R. Das, P. Paul, K. Nag and K. Venkatsubramanian, Inorg. Chim. Acta, 1991, 185, 221 CrossRef CAS; (h) S. F. Palopoli, S. J. Geib, A. L. Rheingold and T. B. Brill, Inorg. Chem., 1988, 27, 2963 CrossRef CAS; (i) M. Hill, M. F. Mahon and K. C. Molloy, J. Chem. Soc., Dalton Trans., 1996, 1857 RSC; (j) E. O. John, R. D. Willett, B. Scott, R. L. Kirchmeier and J. M. Schreeve, Inorg. Chem., 1989, 28, 893 CrossRef CAS; (k) L. A. Oro, M. T. Pinillos, C. Tejel, M. C. Apreda, C. Foces-Foces and F. H. Cano, J. Chem. Soc., Dalton Trans., 1988, 1927 RSC; (l) X. G. Zhou, Z. E. Huang, R. F. Cai, L. X. Zhang, X. F. Hou, X. J. Feng and X. Y. Huang, J. Organomet. Chem., 1998, 563, 101 CrossRef CAS; (m) H. P. H. Arp, A. Decken, J. Passmore and D. J. Wood, Inorg. Chem., 2000, 39, 1840 CrossRef CAS; (n) C. Yélamos, K. R. Gust, A. G. Baboul, M. J. Heeg, H. B. Schlegel and C. H. Winter, Inorg. Chem., 2001, 40, 6451 CrossRef CAS.
- Q. Ye, Y. M. Song, G. X. Wang, K. Chen, D. W. Fu, P. W. H. Chan, J. Z. Zhu, S. D. Huang and R. G. Xiong, J. Am. Chem. Soc., 2006, 128, 6554 CrossRef CAS.
- Q. Ye, Y. H. Li, Y. M. Song, X. F. Huang, R. G. Xiong and Z. L. Xue, Inorg. Chem., 2005, 44, 3618 CrossRef CAS.
- (a) P. J. Steel, Coord. Chem. Rev., 1990, 106, 227 CrossRef CAS; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (c) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef; (d) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (e) M. Fujita, Y. J. Kwon, O. Sasaki, K. Yamaguti and K. Ogura, J. Am. Chem. Soc., 1995, 117, 7278.
- (a) M. Eddaoudi, B. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS; (b) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS; (c) S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- (a) O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096 CrossRef CAS; (b) T. J. Prior and M. J. Rosseinsky, Chem. Commun., 2001, 495 RSC; (c) S. O. H. Gutschke, D. J. Price, A. K. Powell and P. T. Wood, Angew. Chem., Int. Ed., 2001, 40, 1920 CrossRef CAS.
- H. Kumagai, C. J. Kepert and M. Kurmoo, Inorg. Chem., 2002, 41, 3410 CrossRef CAS.
- S. S. Y. Chui, A. Siu, X. Feng, Y. Zhang, T. C. W. Mak and I. D. Williams, Inorg. Chem. Commun., 2001, 4, 467 CrossRef.
- (a) O. R. Evans and W. B. Lin, Acc. Chem. Res., 2002, 35, 511 CrossRef CAS; (b) M. Kondo, T. Okubo, A. Asami, S. Noro, T. Yoshitomi, S. Kitagawa, T. Ishii, H. Matsuzaka and K. Seki, Angew. Chem., Int. Ed., 1998, 38, 140.
- (a) A. L. Gavrilova and B. Bosnich, Chem. Rev., 2004, 104, 3553; (b) J. P. Zhang and X. M. Chen, Chem. Commun., 2006, 1689 RSC.
- (a) G. Nolze and W. Kraus, Powder Diffr., 1998, 13, 256; (b) http://www.ccdc.cam.ac.uk/products/mercury .
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105 CrossRef CAS.
- G. M. Sheldrick, University of Göttingen, Göttingen, Germany, 1996–2006.
- Bruker AXS, Madison, Wisconsin, USA, 2001 Search PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- Y. Song, J. Yu, Y. Li, G. Li and R. Xu, Angew. Chem., Int. Ed., 2004, 43, 2399 CrossRef.
- J. Fan, C. Slebodnick, R. Angel and B. E. Hanson, Inorg. Chem., 2005, 44, 552 CrossRef CAS.
- Y. Cui, M. He, G. Li, C. Chen, Z. Yi and W. Pang, Solid State Sci., 2006, 8, 1108 CrossRef CAS.
- Q. -X. Zeng, Y. Xiang, L. -W. Zhang, W. -H. Chen and J. -Z. Chen, Microporous Mesoporous Mater., 2006, 93, 270 CrossRef CAS.
- J. Fan and B. E. Hanson, Inorg. Chem., 2005, 44, 6998 CrossRef CAS.
- T. Y. Shvareva and T. E. Albrecht-Schmitt, Inorg. Chem., 2006, 45, 1900 CrossRef CAS.
- C. Chen, Y. Liu, S. Wang, G. Li, M. Bi, Z. Yi and W. Pang, Chem. Mater., 2006, 18, 2950 CrossRef CAS.
- (a) B. Q. Ma, H. L. Sun and S. Gao, Angew. Chem., Int. Ed., 2004, 43, 1374 CrossRef CAS; (b) A. J. Blake, M. T. Brett, N. R. Champness, A. N. Khlobystov, D. L. Long, C. Wilson and M. Schröder, Chem. Coummun., 2001, 2258 Search PubMed; (c) B. Q. Ma, H. L. Sun, S. Gao and G. X. Xu, Inorg. Chem., 2001, 40, 6247 CrossRef CAS; (d) R. J. Hill, D. L. Long, N. R. Champness, P. Hubberstey and M. Schröder, Acc. Chem. Res., 2005, 38, 335 CrossRef CAS.
- (a) R. Kefi, E. Jeanneau, F. Lefebvre and C. B. Nasr, Struct. Chem., 2007, 18, 923 CrossRef CAS; (b) A. Oueslati, A. Touati, C. B. Nasr and F. Lefebvre, Phosphorous Sulfur Silicon Relat. Mater., 2006, 181, 2117 Search PubMed; (c) S. Demir, V. T. Yilmaz and W. T. A. Harrison, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, O216 CrossRef; (d) A. Chtioui and A. Jouini, Mater. Res. Bull., 2006, 41, 569 CrossRef CAS; (e) A. Chtioui, L. Benhamada and A. Jouini, Mater. Res. Bull., 2005, 40, 2243 CrossRef CAS; (f) K. Kaabi, C. B. Nasr and M. Rzaigui, J. Solid State Chem., 2001, 161, 307 CrossRef CAS; (g) A. Criado, M. J. Dianez, S. Perez-Garrido, I. M. L. Fermandes, M. Belsley and E. D. Gorners, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 888 CrossRef.
- V. T. Yilmaz, S. Demir and W. T. A. Harrison, Z. Naturforsch., B: Chem. Sci., 2006, 61, 1067 CAS.
- (a) J. K. Gregory, D. C. Clary, K. Liu, M. G. Brown and R. J. Saykally, Science, 1997, 275, 814 CrossRef CAS; (b) S. Manikumari, V. Shivaiah and S. K. Das, Inorg. Chem., 2002, 41, 6953 CrossRef CAS.
- (a) I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, Cryst. Growth Des., 2008, 8, 519 CrossRef CAS; (b) Q. Fang, G. Zhu, M. Xue, Z. Wang, J. Sun and S. Qiu, Cryst. Growth Des., 2008, 8, 319 CrossRef CAS.
- (a) H. Chun, D. N. Nybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS; (b) B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390 CrossRef CAS; (c) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS; (d) B. Rather and M. J. Zaworotko, Chem. Commun., 2003, 830 RSC; (e) B. Chen, S. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H. C. Zhou, Inorg. Chem., 2007, 46, 1233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 3-D structure of [HOOC-pyH]+(NO3− (Fig. S1); structure of [(4,4′-bipyH2)(H2PO4)(H2O)2] (Fig. S2); PXRD data for [Zn(pytz)2(H2O)4]·2H2O (Fig. S3); 3D 6-connected contorted α-Po net in 2 with 412·63 topology (Fig. S4); 3D (4,5,10)-connected trinary net for 3 (Fig. S5); 3D (4,8)-connected binary (46)2(412·612·84) net for 4 (Fig. S6); The indexing of PXRD patterns of 4c (Fig. S7). CCDC reference numbers 688599–688605. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b808496c |
| This journal is © The Royal Society of Chemistry 2009 |