DOI:
10.1039/B809985E
(Paper)
CrystEngComm, 2009,
11, 143-150
“Click” synthesis of isomeric compounds for assessing the efficiency of the bifurcated Br⋯NO2 synthon†
Received
12th June 2008
, Accepted 20th August 2008
First published on 14th October 2008
Abstract
We report herein the extention of the azide–alkyne “click reaction” to crystal engineering and synthesize a collection of isomeric compounds with modular positioning of Br and NO2 on a tricyclic template and crystal structural analyses of the derived isomers. It is quite remarkable to notice that none of the isomers displayed the bifurcated three-center NO2⋯Br supramolecular synthon in their crystal structures
Introduction
The fundamental objective of crystal engineering is to design organic solids that can find applications in material chemistry.1 This needs to integrate our understanding about the non-covalent interactions of various types and strengths that glue the molecules in the crystal structures. Amongst various non-covalent interactions that govern molecular arrangement in crystal structures, those involving halogens have been a matter of debate for many years.2 After the disclosure of bifurcated three-centered NO2⋯X (X = Cl, Br, I) supramolecular synthon by Desiraju and co-workers,3 the occurrence of these soft and weak three-center interactions was analyzed using Cambridge Crystal Structure Database searches. Allen et al. used a combination of systemic database analysis and high-level ab initio molecular orbital calculations to provide a significant insight into the geometry of NO2⋯X interactions.4 They found that the C–X⋯O angle tends to linearity as the X⋯O distance shortens and the angle of approach of X to the C–NO2 plane is preferentially less than 45° for Cl, with an increasing tendency to in-plane X approach in the order C1 < Br < I. They classified NO2⋯X interactions according to the close approach of the X atom to NO2group into three different motifs; (I) symmetric bifurcated motif, (II) asymmetric bifurcated motif and (III) the motif in which halogen (X) forms mono-coordinate interactions with one nitro O atom. In the first two categories, the X atom approaches both nitro O atoms in a bifurcated manner (X⋯Otrans to C–N); the tendency to form such bifurcated motifs increases in the order C1 < Br < I whereas in the last category X⋯O is in a cis relationship to the nitro C substituent about the N–O bond. The results obtained from the ab initio calculations using the intermolecular perturbation theory (IMPT at 6–31G* level) was in agreement with crystal structure geometries.4,5
Though the NO2⋯X symmetric bifurcated motif has been regarded as a ‘discriminator synthon’,6 its predictability, when examined over a range of isomeric compounds revealed certain limitations. Glidewell and co-workers have examined the occurrence of I⋯NO2 synthon employing diaryl compounds connected through a variety of polar functional groups and varying the I and NO2groups positions systematically.7 It led to the conclusion that this three-center iodo⋯nitro interaction appears to behave predictably for certain isomers and in the remaining isomers it is not seen. The aggregation pattern for any one compound is not readily predictable from the knowledge of the patterns in the other isomers. Nevertheless, in the majority of the cases, the isomeric structural motifs subjected to analyses do contain influential strong non-covalent interactions too.
Apart from this, there are very few investigations that address the predictability of weak synthons in isomeric compounds, especially in absence of any strong non-covalent interactions. Partly, this is due to the complexity involved in synthesizing the systems with architectural control and flexibility in the incorporation of functional groups and an inherent suspicion about the crystallinity of all isomers.8 In this context, developing simple and efficient protocols for generation of isomeric compound libraries (devoid of polar functional units) with sufficient flexibility in placing complimentary functional groups will be instrumental for crystal engineers to understand these weak non-covalent interactions, the topological constraints for intermolecular connectivity and to examine the predictability of corresponding weak synthons.
Results and discussion
Focussing on the Br⋯NO2 synthon, we describe in this paper the synthesis and structural analysis of a collection of isomeric compounds with modular positioning of Br and NO2 on a flexible tricyclic template (Fig. 1). The Cu mediated Huisgen's azide-[3 + 2] cycloaddition [Fokin–Finn–Sharpless's “click reaction”]9,10 has served as a flexible molecular construct in our synthesis. Having no motifs that form the conventional hydrogen bond, this selected scaffold offer an opportunity for understanding molecular association either via Br⋯NO2 or Br⋯Br interacting synthons3 and for studying the interplay of other weak intermolecular interactions in molecular aggregation (Table 1).
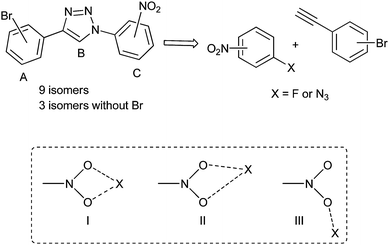 |
| | Fig. 1 Designed isomers and possible Br⋯NO2-synthon geometries. | |
Table 1 Geometrical parameters of intermolecular interactions
| |
D–H⋯A |
D–H/Å |
H⋯A/Å |
D⋯A/Å |
D–H⋯A/° |
Symmetry codes |
|
8
|
C1–H1⋯O1 |
0.94(3) |
2.47(3) |
3.270(3) |
143(2) |
1.5 − x, −1/2 + y, 1/2 − z |
| |
C11–H11⋯N2 |
0.97(3) |
2.71(3) |
3.642(4) |
162(2) |
x, −1 + y, z |
| |
C13–H13⋯O2 |
0.97(3) |
2.59(3) |
3.334(4) |
133(2) |
1 − x, −y, 1 − z |
|
9
|
C1–H1⋯O1 |
0.93 |
2.65 |
3.451(6) |
141 |
1.5 − x, 1/2 + y, 1.5 − z |
| |
C13–H13⋯O2 |
0.93 |
2.68 |
3.482(6) |
145 |
1 − x, 2 − y, 2 − z |
| |
C5–H5⋯Br1 |
0.93 |
2.92 |
3.690(6) |
145 |
x, −1 + y, z |
|
10
|
C1–H1⋯O1 |
0.93 |
2.32 |
3.153(4) |
150 |
1/2 − x, −1/2 + y, 1/2 − z |
| |
C11–H11⋯N2 |
0.93 |
2.54 |
3.360(4) |
147 |
x, −1 + y, z |
| |
C7–Br1⋯Br1 |
1.893(3) |
3.696(1) |
|
140.5(3) |
−1 − x, 1 − y, −z |
|
11
|
C1–H1⋯N2 |
0.86(3) |
2.66(3) |
3.447(4) |
152(2) |
−1 + x, y, z |
| |
C6–Br1⋯Br1 |
1.903(3) |
3.473(1) |
|
155.5(2) |
−x, 1 − y, 2 − z |
|
12
|
C1–H1⋯O1 |
0.93 |
2.53 |
3.409(3) |
158 |
−x, 2 − y, 1 − z |
| |
C14–H14⋯O1 |
0.93 |
2.48 |
3.408(3) |
176 |
−x, 2 − y, 1 − z |
| |
C8–H8⋯O2 |
0.93 |
2.70 |
3.593(3) |
160 |
−x, 2 − y, 1 − z |
| |
C6–H6⋯O2 |
0.93 |
2.57 |
3.372(3) |
144 |
x, 1.5 − y, 1/2 + z |
| |
C10–H10⋯N2 |
0.93 |
2.50 |
3.358(3) |
154 |
1 − x, −y, 1 − z |
|
13
|
C10–H10⋯O2 |
0.93 |
2.67 |
3.57(2) |
165 |
1/2 + x, 2.5 − y, 1/2 + z |
| |
C7–H7⋯N3 |
0.93 |
2.71 |
3.61(2) |
161 |
1/2 + x, 1.5 − y, 1/2 + z |
| |
C8–Br1⋯O2 |
1.913(2) |
3.317(17) |
|
144.28(3) |
1/2 + x, 2.5 − y, 1/2 + z |
|
14
|
C1–H1⋯O1′ |
0.93 |
2.47 |
3.395(3) |
173 |
x, −1 + y, z |
| |
C4–H4⋯O2′ |
0.93 |
2.67 |
3.478(4) |
146 |
x, −1 + y, z |
| |
C8–H8⋯Br1′ |
0.93 |
3.04 |
3.926(3) |
161 |
1 + x, y, 1 + z |
| |
C1′–H1′⋯O1 |
0.93 |
2.44 |
3.351(4) |
167 |
x, 1 + y, z |
| |
C4′–H4′⋯O2 |
0.93 |
2.55 |
3.391(4) |
151 |
x, 1 + y, z |
| |
C14′–H14′⋯O1 |
0.93 |
2.66 |
3.584(4) |
173 |
x, 1 + y, z |
| |
C8′–H8′⋯Br1 |
0.93 |
2.98 |
3.908(3) |
172 |
−1 + x, y, −1 + z |
|
15
|
C1–H1⋯O1 |
0.89(3) |
2.56(3) |
3.413(5) |
161(3) |
1 − x, −y, 1 − z |
| |
C4–H4⋯O2 |
0.86(4) |
2.70(4) |
3.482(5) |
153(3) |
1 − x, −y, 1 − z |
| |
C14–H14⋯O1 |
0.92(3) |
2.49(3) |
3.404(4) |
177(3) |
1 − x, −y, 1 − z |
| |
C10–H10⋯N2 |
0.93(4) |
2.56(4) |
3.342(5) |
142(3) |
2 − x, 2 − y, 1 − z |
| |
C6–Br1⋯O2 |
1.896(4) |
3.169(3) |
|
158.1(4) |
−1/2 + x, 1/2 − y, −1/2 + z |
|
16
|
C6–H6⋯O1 |
0.956(17) |
2.575(17) |
3.342(2) |
137.3(13) |
x, 1 + y, 1 + z |
| |
C6–H6⋯O2 |
0.956(17) |
2.660(17) |
3.333(2) |
127.7(12) |
1 + x, 1 + y, 1 + z |
|
17
|
C6–H6⋯O1 |
0.93 |
2.62 |
3.321(5) |
132 |
1/2 + x, 1/2 − y, 1/2 + z |
| |
C11–H11⋯N2 |
0.93 |
2.56 |
3.231(4) |
130 |
x, 1 − y, −1/2 + z |
| |
C8–Br1⋯O2 |
1.914(4) |
3.268(4) |
|
145.2(3) |
1/2 − x, 1/2 − y, −z |
|
18
|
C1–H1⋯O2 |
0.93 |
2.33 |
3.247(3) |
169 |
2 − x, −1/2 + y, 1.5 − z |
| |
C8–H8⋯O2 |
0.93 |
2.66 |
3.556(4) |
161 |
2 − x, −1/2 + y, 1.5 − z |
| |
C10–H10⋯O1 |
0.93 |
2.56 |
3.387(4) |
148 |
2 − x, −1/2 + y, 1.5 − z |
| |
C5–H5⋯N2 |
0.93 |
2.52 |
3.387(4) |
155 |
1 − x, −1/2 + y, 1/2 − z |
| |
C7–Br1⋯O1 |
1.897(3) |
3.292(3) |
|
160.1(3) |
x, −1 + y, z |
|
19
|
C1–H1⋯O2′ |
0.93 |
2.36 |
3.279(5) |
171 |
x, y, z |
| |
C10–H10⋯O2′ |
0.93 |
2.60 |
3.523(5) |
174 |
x, y, z |
| |
C1′–H1′⋯O2 |
0.93 |
2.31 |
3.230(5) |
168 |
−1 + x, y, 1 + z |
| |
C10′–H10′⋯O2 |
0.93 |
2.71 |
3.620(5) |
165 |
−1 + x, y, 1 + z |
| |
C4–H4⋯N3 |
0.93 |
2.65 |
3.401(5) |
139 |
−x, −y, −z |
| |
C4′–H4′⋯N3′ |
0.93 |
2.60 |
3.334(5) |
137 |
1 − x, 1 − y, 2 − z |
Considering the competitive formation of the regiomeric products on the one hand, and the known conversion of α-azido nitrobenzene to benzofuroxan11 under the thermal Hüisgen [3 + 2]-azidoalkyne cycloadditions conditions on the other hand, we opted for a SNAr reaction of fluoronitrobenzenes with sodium azide as a potential alternative in this regard.12 The feasibility of SNAr click reaction was examined with simple phenyl acetylene (4) employing three regiomeric fluoronitrobenzenes, adopting the conditions developed in general for copper mediated azide-alkyne cycloadditions. One reason to employ simple phenyl acetylene in this regard is to compare the packing of expected products with designed series where Br was placed at all three possible isomeric positions. In general, the reactions with 2-fluoronitrobenzene (1) are facile, whereas with 4-fluronitrobenzene (3), prolonged heating is required. As the reactions with 3- fluoronitrobenzene resulted only in the cycloaddition product of NaN3 with phenyl acetylene, 3-azidonitrobenzene (2) was employed under similar conditions to prepare the 3-NO2 isomer. These approaches have been extended with the three isomeric bromophenylacetylenes 5–7. As indicated in Scheme 1, the resulting compounds 8–19 were classified into three groups, according to the position of the nitro group.
2-NO2 series
The crystal structures of the four compounds 8–11 contain one molecule per asymmetric unit. In all four structures, none of the aromatic ring is coplanar with the triazole ring. The crystal structures of 8–11 in general are dominated by the C–H⋯O and/or C–H⋯N, C–H⋯Br and Br⋯Br interactions and do not pack in their crystals via a Br⋯NO2 synthon. The molecules in compounds 8, 9 and 10 form isostructural helical assemblies along the crystallographic 21-screw axis linked via C–H⋯O interaction involving C1–H1 of the triazole ring and oxygen O1 of the nitro group [Fig. 2(a), (b) and (c)]. Along the helical assembly the unit translated molecules are weakly associated via C–H⋯N interactions with an additional C–H··Br contact in 9. These neighbouring helices are bridged via centrosymmetric weak C–H⋯O interactions engaging the nitro group phenyl ring in 8 and 9 whereas in 10 they are linked via a short centrosymmetric Br⋯Br contact. In 11, the molecules are linked via a rather weak C–H⋯N interaction involving C1–H1 of the triazole ring and N2 atom of the unit translated triazole ring forming a 2D layer structure along the a-axis [Fig. 2(d)]. The neighboring layers along the c-axis are linked in an anti-parallel fashion via short Br⋯Br contacts.
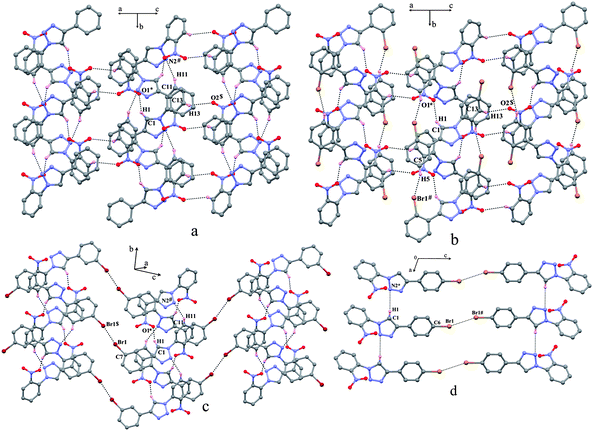 |
| | Fig. 2 Molecular organization in (a) 8, (b) 9, (c) 10 and (d) 11. The atoms marked with an asterisk (*), a hash (#) and a dollar sign ($) are at the following symmetry positions, respectively: For (a) (1.5 − x, −1/2 + y, 1/2 − z), (x, −1 + y, z), and (1 − x, −y, 1 − z); for (b) (1.5 − x, 1/2 + y, 1.5 − z), (x, −1 + y, z), and (1 − x, 2 − y, 2 − z); for (c) (1/2 − x, −1/2 + y, 1/2 − z), (x, −1 + y, z), and (−1 − x, 1 − y, −z); for (d) (−1 + x, y, z), and (−x, 1 − y, 2 − z). | |
3-NO2 series
The crystal structures of 3-nitro series are solely dominated by C–H⋯O and halogen bonding (Br⋯O) interactions. In all four structures, both aromatic rings are nearly coplanar with the triazole ring. The molecules in 12, 14 and 15 form a zero-dimensional face-to-face centrosymmetric dimeric assemblyvia C–H⋯O interactions [Fig. 3(a), (c) and (d)] except in 13 where the presence of the bromine atom at o-position prevented the face-to-face alliance. The molecular packing in 12 and 15 is almost isostructural [Fig. 3(a) and 3(d)].13 These face-to-face dimers are connected via centrosymmetric C–H⋯N interactions forming a planar sheet diagonal to the ab plane. These successive sheets along the c-axis are perpendicularly stitched through C–H⋯O interactions in 12 and Br⋯NO2 contact in 15 forming a square grid type network. In 14, the face-to-face dimers are formed between the two symmetry independent molecules in the asymmetric unit via C–H⋯O interactions. These dimers are extended in two dimensions forming a sheet pattern through the centrosymmetric weak C–H⋯Br and the short C–H⋯H–C (H⋯H = 2.240 Å) contacts [Fig. 3(c)]. Surprisingly, there are no significant interactions zipping the dimers. Therefore, the cohesion of dimers along the b-axis seems to be only via short H⋯H contacts.14 Along the third dimension these sheets are stacked one over the other with an interplanar spacing of ca. 3.5 Å. In 13, the nitro group carrying the phenyl ring has adapted almost an opposite orientation towards the respective phenyl ring in 12, 14 and 15. Due to this change in orientation, the formation of a face-to-face dimeric assembly is prevented. Instead, the molecules assembled as ‘zig-zag’ chains bridged via weak C–H⋯O and Br⋯NO2 contacts that resemble flat helical ribbons [Fig. 3(b)]. These flat ribbons are weakly associated via C–H⋯N interactions forming a planar sheet. Here, also the flat ribbons have ∼3.5 Å spacing along the third dimension.
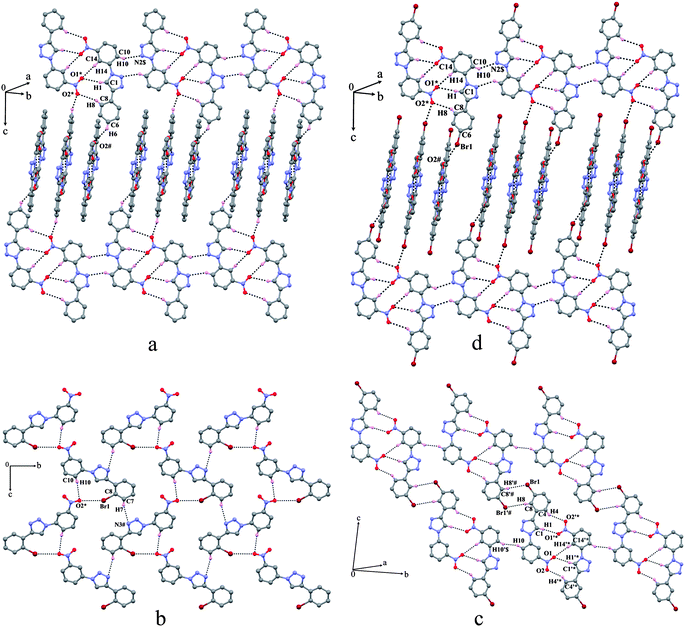 |
| | Fig. 3 Association of molecules in the structures of (a) 12, (b) 13, (c) 14 and (d) 15. The atoms marked with an asterisk (*), a hash (#) and a dollar sign ($) are at the following symmetry positions respectively: For (a) (−x, 2 − y, 1 − z), (x, 1.5 − y, 1/2 + z) and (1 − x, −y, 1 − z); for (b) (1/2 + x, 2.5 − y, 1/2 + z) and (1/2 + x, 1.5 − y, 1/2 + z); for (c) (x, −1 + y, z) and (1 + x, y, 1 + z); for (d) (1 − x, −y, 1 − z), (−1/2 + x, 1/2 − y, −1/2 + z) and (2 − x, 2 − y, 1 − z). | |
4-NO2 series
In this series of four structures both aromatic rings are nearly coplanar with the central triazole ring. In 19, there are two independent molecules in the asymmetric unit that differ by small dihedral angle difference between the triazole and bromophenyl ring. The crystal structures of compounds 16, 18, and 19 form a 2D-sheet structure [Fig. 4(a), (c) and (d)] and that of compound 17 displayed a corrugated strand structure [Fig. 4(b) and (e)]. In 16, the molecules form a 2D sheet pattern linked via bifurcated weak C–H⋯O interactions [Fig. 4(a)]. The spacing between the parallel sheets is about 3.9 Å. In compound 17, the molecules form centrosymmetric dimers viahalogen bonding (Br⋯NO2) contacts. These neighbouring halogen bonded dimeric units are linked via centrosymmetric weak C–H⋯O interactions along the a-axis whereas along the c-axis they are associated via moderate C–H⋯N interactions having c-glide symmetry resulting in the formation of corrugated network [Fig. 4(e)]. The structure of 18 revealed a flat helical self-assembly around the crystallographic two-fold screw axis through C–H⋯O and Br⋯NO2 contacts. These flat helices are bridged along the c-axis via C–H⋯N interactions forming a 2D sheet structure [Fig. (4(c)]. In compound 19 also the molecules form a 2D sheet network through weak C–H⋯N and C–H⋯O contacts [Fig. 4(d)]. Both symmetry independent molecules form centrosymmetric dimers via C–H⋯N contacts with their respective pair. These neighboring dimers are bridged through C–H⋯O contacts forming a flat sheet network. It is interesting to note here also that the molecules do not associate viaBr⋯Br contacts.
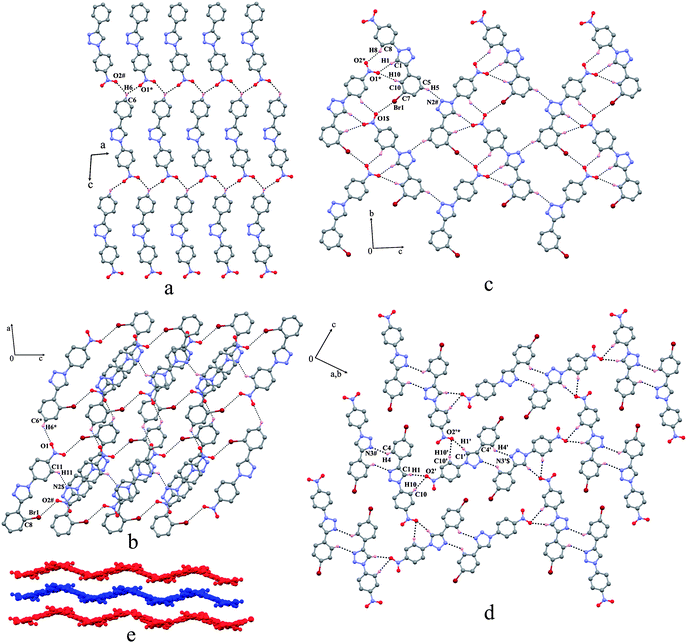 |
| | Fig. 4 Association of the molecules in the structures of (a) 16, (b) 17, (c) 18 and (d) 19; (e) shows corrugated strands in 17. The atoms marked with an asterisk (*), a hash (#) and a dollar sign ($) are at the following symmetry positions respectively: For (a) (x, 1 + y, 1 + z) and (1 + x, 1 + y, 1 + z); for (b) (1/2 + x, 1/2 + y, 1/2 − z), (1/2 − x, 1/2 − y, −z), and (x, 1 − y, −1/2 + z); for (c) (2 − x, −1/2 + y, 1.5 − z), (1 − x, −1/2 + y, 1/2 − z), and (x, −1 + y, z); for (d) (2 − x, 1 − y, −z), (x, −y, −z) and (1 − x, 1 − y, −z). | |
A comprehensive compilation of various weaker interactions observed in the crystal structures of compounds 8–19 revealed that out of nine compounds, the molecules in four compounds (13, 15, 17, and 18) have a Br⋯NO2 association (Type III geometry), though none of them exhibited an association with the bifurcated Br⋯NO2 synthon. In two compounds a Br⋯Br short contact (10, 11) and in one compound (9) C–H⋯Br electrostatic interaction were present. The similarity noticed in the supramolecular aggregation patterns of the crystal structures of 2-NO2 series and of 3-NO2 series groups due to weak C–H⋯O/N bonding demonstrate a control of weaker complimentary interaction patterns over the molecular packing in crystal structures.
Crystallographic details
The crystals of all compounds 8–19 were stable at room temperature and the diffraction data measurements were carried out at room temperature (297 K) on a Bruker SMART APEX CCD diffractometer with graphite-monochromatized (Mo Kα = 0.71073 Å) radiation. The X-ray generator was operated at 50 kV and 30 mA. Data were collected with an ω scan width of 0.3° at different settings of φ (0, 90, 180 and 270°) keeping the sample-to-detector distance fixed at 6.145 cm and the detector position (2θ) fixed at −28°. The X-ray data collection was monitored by SMART program (Bruker, 2003).15 All data were corrected for Lorentzian, polarization and absorption effects using SAINT and SADABS programs (Bruker, 2003). SHELX-97 was used for structure solution and full matrix least-squares refinement on F2.16 The hydrogen atoms for compounds 8, 11, 16 and 15 were located in a difference Fourier map and refined isotropically, whereas for the other compounds they were included in the refinement as per the riding model (Table 2).
| |
8
|
9
|
10
|
11
|
| Chemical formula |
C14H10N4O2 |
C14H9BrN4O2 |
C14H9BrN4O2 |
C14H9BrN4O2 |
|
M
r
|
266.26 |
345.16 |
345.16 |
345.16 |
| Temperature/K |
297(2) |
297(2) |
297(2) |
297(2) |
| Crystal size |
0.43 × 0.41 × 0.26 |
0.77 × 0.68 × 0.54 |
0.51 × 0.23 × 0.13 |
0.68 × 0.25 × 0.20 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
P21/n |
P21/c |
|
a/Å |
12.180(4) |
12.388(13) |
9.2835(11) |
5.4468(9) |
|
b/Å |
7.501(3) |
8.051(9) |
7.2943(9) |
20.778(3) |
|
c/Å |
14.277(5) |
14.256(15) |
20.794(2) |
12.482(2) |
|
α/° |
90 |
90 |
90 |
90 |
|
β/° |
100.286(7) |
103.305(18) |
97.444(2) |
102.427(3) |
|
γ/° |
90 |
90 |
90 |
90 |
|
V/Å3 |
1283.5(8) |
1384(2) |
1396.2(3) |
1379.6(4) |
|
Z, Dcalc/g cm−3 |
4, 1.378 |
2, 1.657 |
4, 1.642 |
4, 1.662 |
|
μ/mm−1, F(000) |
0.097, 552 |
2.981, 688 |
2.954, 688 |
2.989, 688 |
|
Ab. correction |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Transmission min / max |
0.9596 / 0.9753 |
0.2074 / 0.2959 |
0.3142 / 0.7001 |
0.2357 / 0.5962 |
| h, k, l (min, max) |
(−14, 15), (−9, 9), (−17, 17) |
(−12, 14), (−7, 9), (−16, 16) |
(−11, 10), (−8, 8), (−24, 24) |
(−6, 6), (−22, 24), (−9, 14) |
| Reflns collected |
9605 |
6344 |
8147 |
6843 |
| Unique reflns |
2513 |
2419 |
2448 |
2427 |
| Observed reflns |
1661 |
1685 |
1811 |
1896 |
|
R
int
|
0.0446 |
0.0517 |
0.0238 |
0.0221 |
|
No. of parameters |
221 |
190 |
190 |
226 |
| GoF |
1.098 |
0.960 |
1.030 |
1.045 |
|
R
1 [I > 2σ(I)] |
0.0619 |
0.0466 |
0.0602 |
0.0361 |
|
wR2 [I > 2σ(I)] |
0.1304 |
0.1245 |
0.1213 |
0.0823 |
|
R
1 [all data] |
0.0997 |
0.0699 |
0.0954 |
0.0503 |
|
wR
2 [all data] |
0.1475 |
0.1376 |
0.1359 |
0.0889 |
| Δρmax, Δρmin/e Å−3 |
0.162, −0.146 |
0.923, −0.280 |
0.41, −0.27 |
0.657, −0.506 |
| CCDC deposition no. |
CCDC 679556 |
CCDC 679557 |
CCDC 679558 |
CCDC 679559 |
| |
12
|
13
|
14
|
15
|
| Chemical formula |
C14H10N4O2 |
C14H9BrN4O2 |
C14H9BrN4O2 |
C14H9BrN4O2 |
|
M
r
|
266.26 |
345.16 |
345.16 |
345.16 |
| Temperature/K |
297(2) |
297(2) |
297(2) |
297(2) |
| Crystal size/mm |
0.31 × 0.22 × 0.20 |
0.76 × 0.07 × 0.06 |
0.33 × 0.26 × 0.09 |
0.37 × 0.32 × 0.03 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P21/c |
P21/n |
P-1
|
P21/n |
|
a/Å |
8.502(3) |
7.228(7) |
7.4402(6) |
8.667(3) |
|
b/Å |
5.3559(18) |
13.308(13) |
12.4535(10) |
5.2323(18) |
|
c/Å |
27.363(9) |
14.521(15) |
14.6016(12) |
30.304(11) |
|
α/° |
90 |
90 |
91.2720(10) |
90 |
|
β/° |
95.564(6) |
93.675(19) |
98.9940(10) |
96.248(6) |
|
γ/° |
90 |
90 |
96.3090(10) |
90 |
|
V/Å3 |
1240.1(7) |
1394(2) |
1327.14(19) |
1366.0(8) |
|
Z
|
4 |
4 |
4 |
4 |
|
F(000)
|
552 |
688 |
688 |
688 |
|
D
calc/g cm−3 |
1.426 |
1.645 |
1.727 |
1.678 |
|
μ/mm−1 |
0.100 |
2.959 |
3.107 |
3.019 |
| Absorp. correction |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Tmin |
0.9696 |
0.2120 |
0.4270 |
0.4013 |
| Tmax |
0.9802 |
0.8518 |
0.7673 |
0.9149 |
| Reflns collected |
5402 |
9670 |
12926 |
6524 |
| Unique reflns |
2170 |
2455 |
4654 |
2403 |
| Observed reflns |
1779 |
1522 |
3565 |
1718 |
| h, k, l (min, max) |
(−10, 10), (−6, 6), (−32, 25) |
(−8, 8), (−15, 13), (−17, 17) |
(−8, 8), (−14, 14), (−17, 17) |
(−9, 10), (−6, 5), (−36, 27) |
|
R
int
|
0.0364 |
0.0571 |
0.0269 |
0.0289 |
|
No. of parameters |
181 |
190 |
379 |
226 |
|
R
1 [I > 2σ(I)] |
0.0525 |
0.1357 |
0.0342 |
0.0459 |
|
wR2 [I > 2σ(I)] |
0.1260 |
0.3958 |
0.0785 |
0.1064 |
|
R
1 [all data] |
0.0655 |
0.1755 |
0.0492 |
0.0675 |
|
wR
2 [all data] |
0.1328 |
0.4175 |
0.0847 |
0.1186 |
| GoF |
1.038 |
1.141 |
1.023 |
1.034 |
| Δρmax, Δρmin/e Å−3 |
0.171, −0.147 |
2.445, −0.631 |
0.436, −0.291 |
0.645, −0.540 |
| CCDC deposition no. |
CCDC 679560 |
CCDC 679561 |
CCDC 679562 |
CCDC 679563 |
| |
16
|
17
|
18
|
19
|
| Chemical formula |
C14H10N4O2 |
C14H9BrN4O2 |
C14H9BrN4O2 |
C14H9BrN4O2 |
|
M
r
|
266.26 |
345.16 |
345.16 |
345.16 |
| Temperature/K |
297(2) |
297(2) |
297(2) |
297(2) |
| Crystal size/mm |
0.69 × 0.52 × 0.08 |
0.68 × 0.25 × 0.20 |
0.88 × 0.13 × 0.08 |
0.82 × 0.43 × 0.12 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P-1
|
C2/c |
P21/c |
P-1
|
|
a/Å |
5.757(2) |
29.52(2) |
7.5222(8) |
9.6141(15) |
|
b/Å |
7.198(3) |
7.057(5) |
14.1697(15) |
12.0915(19) |
|
c/Å |
14.862(6) |
13.022(9) |
13.0479(14) |
12.900(2) |
|
α/° |
101.081(6) |
90 |
90 |
87.535(2) |
|
β/° |
99.217(6) |
97.405(12) |
90.391(2) |
71.407(2) |
|
γ/° |
90.859(6) |
90 |
90 |
76.183(2) |
|
V/Å3 |
595.8(4) |
2690(3) |
1390.7(3) |
1379.3(4) |
|
Z
|
2 |
8 |
4 |
4 |
|
F(000)
|
276 |
1376 |
688 |
688 |
|
D
calc/g cm−3 |
1.484 |
1.705 |
1.649 |
1.662 |
|
μ/mm−1 |
0.104 |
3.066 |
2.965 |
2.990 |
| Absorp. correction |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Tmin |
0.9315 |
0.2296 |
0.1800 |
0.1930 |
| Tmax |
0.9917 |
0.5791 |
0.7888 |
0.7155 |
| Reflns collected |
4652 |
6468 |
7835 |
13109 |
| Unique reflns |
2296 |
2377 |
2434 |
4834 |
| Observed reflns |
2006 |
1764 |
1859 |
3401 |
| h, k, l (min, max) |
(−7, 7), (−8, 8), (−16, 18) |
(−34, 28), (−5, 8), (−15, 15) |
(−8, 8), (−16, 14), (−15, 15) |
(−11, 11), (−14, 14), (−15, 15) |
|
R
int
|
0.0181 |
0.0378 |
0.0258 |
0.0356 |
|
No. of parameters |
221 |
199 |
190 |
379 |
|
R
1 [I > 2σ(I)] |
0.0363 |
0.0385 |
0.0387 |
0.0429 |
|
wR2 [I > 2σ(I)] |
0.0996 |
0.0953 |
0.0958 |
0.1159 |
|
R
1 [all data] |
0.0414 |
0.0556 |
0.0547 |
0.0674 |
|
wR
2 [all data] |
0.1042 |
0.1037 |
0.1033 |
0.1284 |
| GoF |
1.060 |
1.010 |
1.041 |
1.019 |
| Δρmax, Δρmin/e Å−3 |
0.195, −0.171 |
0.314, −0.234 |
0.489, −0.444 |
0.856, −0.469 |
| CCDC deposition no. |
CCDC 679564 |
CCDC 679565 |
CCDC 679566 |
CCDC 679567 |
Conclusions
In conclusion, the potential of Cu(I) catalysed azide-alkyne “click reaction” as a simple synthetic tool to build a collection of crystalline isomeric compounds with modular positioning of the Br and NO2 functional groups and evaluation of the occurrence of bifurcated Br⋯NO2 synthon was demonstrated. Considering the operational simplicity, architectural control and flexibility in the incorporation of functional groups, the present “click” synthesis of isomeric compounds could be a potential approach in crystal engineering to provide model systems for understanding directional preferences and topological constraints for intermolecular connectivites. The helical assembly of molecules through C–H⋯O interactions in 2-NO2 series, and the self-complimentary patterns displayed in the crystal structures of 3-NO2 series shows that one can design materials that require a reversible alternative for covalent bond by employing exclusively weak hydrogen bonding interactions like C–H⋯O/N to form either 2D sheets or 3D-helical networks. Efforts to extend the chemistry described herein to evaluate the efficiency of other weak supramolecular synthons are currently underway in our research group.
Acknowledgements
S.C. and K.A.D. thanks CSIR (New Delhi) for financial assistance in the form of a research fellowship.
References
-
(a) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS;
(b) J.-M. Lehn, Pure Appl. Chem., 1978, 50, 871–892 CrossRef CAS;
(c)
G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier: Amsterdam, 1989 Search PubMed;
(d)
J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed;
(e) C. B. Aakeröy, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 569–586 CrossRef;
(f) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS;
(g) G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS;
(h)
G. R. Desiraju, Crystal Design: Structure and Function, Wiley: New York, 2003 Search PubMed;
(i) G. R. Desiraju, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS;
(j) J. A. Bis and M. J. Zaworotko, Cryst. Growth Des., 2005, 5, 1169–1179 CrossRef CAS;
(k) D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 RSC;
(l) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS.
-
(a) For selected reviews see: J. M. Dumas, L. Gomel, M. Guerin, Halogen bonding: The Chemistry of Functional Groups Supplement D, ed. Wiley, New York, 1983 Search PubMed;
(b) A. C. Legon, Chem.–Eur. J., 1998, 4, 1890–1897 CrossRef CAS;
(c) A.C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef;
(d) P. Metrangolo and G. Resnati, Chem.–Eur. J., 2001, 7, 2511–2519 CrossRef;
(e) R. G. Gonnade, M. S. Shashidhar and M. M. Bhadbhade, J. Ind. Inst. Sci, 2007, 87, 149–165 Search PubMed;
(f) P. Metrangolo, G. Resnati, T. Pilati and S. Biella, Struct. Bonding, 2008, 126, 105–136 CAS.
-
(a) G. R. Desiraju and V. R. Pedireddi, J. Chem. Soc., Chem. Commun., 1989, 1112–1113 RSC;
(b) V. R. Pedireddi, J. A. R. P. Sarma and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1992, 311–320 RSC;
(c) G. R. Desiraju, V. R. Pedireddi, J. A. R. P. Sarma and D. E. Zacharias, Acta Chim. Hung., 1993, 451–465 CAS.
-
(a) F. H. Allen, J. P. M. Lommerse, V. J. Hoy, J. A. K. Howard and G. R. Desiraju, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 1006–1016 CrossRef;
(b) F. H. Allen, C. A. Baalham, J. P. M. Lommerse, P. R. Raithby and E. Sparr, in Acta ystallogr. Sect. B, 1997, 53, 1017–1024 Search PubMed.
-
(a) N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS;
(b) G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725–8726 CrossRef CAS;
(c) Y.-X. Lu, J.-W. Zou, Y.-H. Wang and Q.-S. , Yu, THEOCHEM , 2006, 767, 139–142 CrossRef CAS.
- For selected references that deal with X⋯NO2 3-centered symmetric synthon see
(a) J. M. A. Robinson, D. Philp, K. D. M. Harris and B. M. Kariuki, New J. Chem., 2000, 24, 799–806 RSC;
(b) P. K. Thallapally, G. R. Desiraju, M. Bagieu–Beucher, R. Masse, C. Bourgogne and J.-F. Nicoud, Chem. Commun., 2002, 1052–1053 RSC;
(c) P. K. Thallapally, S. Basavoju, G. R. Desiraju, M. Bagieu–Beucher, R. Masse and J.-F. Nicoud, Curr. Sci., 2003, 85, 995–1001 CAS;
(d) J. N. Moorthy, P. Venkatkrishnan, P. Mal, S. Dixit and P. Venugopalan, Cryst. Growth Des., 2003, 3, 581–585 CrossRef CAS;
(e) M. Kubicki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o341–o343 CrossRef;
(f) S. George, A. Nangia, C.-K. Lam, T. C. W. Mak and J.-F. Nicoud, Chem. Commun., 2004, 1202–1204 RSC;
(g) L. S. Reddy, S. K. Chandran, S. George, N. J. Babu and A. Nangia, Cryst. Growth Des., 2007, 7, 2675–2690 CrossRef CAS;
(h) I. Mossakowska and G. Wojcik, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o123–o125 CrossRef;
(i) R. Custelcean, Chem. Commun., 2008, 295–307 RSC.
-
(a) N. Maschiocchi, M. Bergamo and A. Sironi, Chem. Commun., 1998, 1347–1348 RSC;
(b) S. J. Garden, F. R. da Cunha, J. L. Wardell, J. M. S. Skakle, J. N. Low and C. Glidewell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o463–o466 CrossRef;
(c) S. J. Garden, S. P. Fontes, J. L. Wardell, J. M. S. Skakle, J. N. Low and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 701–709 CrossRef;
(d) C. J. Kelly, J. M. S. Skakle, J. L. Wardell, S. M. S. V. Wardell, J. N. Low and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 94–108 CrossRef;
(e) C. Glidewell, J. N. Low, J. M. S. Skakle, S. M. S. V. Wardell and J. L. Wardell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 472–480 CrossRef;
(f) C. Glidewell, J. N. Low, J. M. S. Skakle, S. M. S. V. Wardell and J. L. Wardell, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 227–237 CrossRef;
(g) J. L. Wardell, J. N. Low, J. M. S. Skakle and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 931–943 CrossRef.
- O. Navon, J. Bernstein and V. Khodorkovsky, Angew. Chem., Int. Ed. Engl., 1997, 36, 601–603 CrossRef CAS.
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS;
(b) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS;
(c) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS;
(d) M. V. Gil, M. J. Arévalo and Ó. Lopez, Synthesis, 2007, 1589–1620 CrossRef CAS;
(e) R. A. Evans, Aust. J. Chem., 2007, 60, 384–395 CrossRef CAS;
(f) O. S. Wolfbeis, Angew. Chem., Int. Ed., 2007, 46, 2980–2982 CrossRef;
(g) J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS.
- For a recent examples of triazole linker in non-covalent synthesis of porus metal organic frameworks, see
(a) D. Sun, S. Ma, Y. Ke, D. J. Collins and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef CAS;
(b) T. Devic, O. David, M. Valls, J. Marrot, F. Couty and G. Férey, J. Am. Chem. Soc., 2007, 129, 12614–12615 CrossRef CAS.
-
(a) A. R. Katritzky and M. F. Gordeev, Heterocycles, 1993, 35, 483–518 CrossRef CAS;
(b) T. Takayama, M. Kawano, H. Uekusa, Y. Ohashi and T. Sugawara, Helv. Chim. Acta, 2003, 86, 1352–1358 CrossRef CAS.
- When we are at an advanced stage, the SNAr of o-fluoronitrobenzene employing Cu(I) generated insitu by redox process was reported. More interestingly, it has been reported that when Cu(I) catalyst was employed directly, the reactions resulted with trace amounts of products B. Saha, S. Sharma, D. Sawant and B. Kundu, Synlett, 2007, 1591–1594 Search PubMed.
-
(a) L. Fábián, G. Argay, A. Kálmán and M. Báthori, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 710–720 CrossRef;
(b) L. Fábián and A. Kálmán, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 547–558 CrossRef;
(c) R. G. Gonnade, M. M. Bhadbhade and M. S. Shashidhar, CrystEngComm, 2008, 10, 288–296 RSC.
-
(a) R. H. Crabtree, P. E. M. Siegbahn, O. Eisenstein, A. L. Rheingold and T. F. Koetzle, Acc. Chem. Res., 1996, 29, 348–354 CrossRef CAS;
(b) R. H. Crabtree, Science, 1998, 282, 2000–2001 CrossRef CAS;
(c) R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS;
(d) P. Bombicz, M. Czugler, R. Tellgren and A. Kálmán, Angew. Chem., Int. Ed., 2003, 42, 1957–1960 CrossRef CAS;
(e) C.-C. Wang, T.-H. Tang, L.-C. Wu and Y. Wang, Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, 60, 488–493 CrossRef.
-
Bruker ( 2003). SADABS (Version 2.05), SMART (Version 5.631), SAINT (Version 6.45) and SHELXTL (Version 6.14)Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP drawings of 8–19, details of synthesis and spectral data for compounds 8–19. CCDC reference numbers 679556–679567. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b809985e |
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. ![Hüisgen [3 + 2] cycloaddition reaction between fluoronitrobenzenes 1, 3, and 3-azidonitrobenzene (2) and alkynes 4–7. Reagents and conditions: L-proline, Na2CO3, NaN3, sodium ascorbate, CuSO4·5H2O, DMSO–H2O (9 : 1), 70 °C, (a) 24 h; (b) 24 h; (c) 48 h.](/image/article/2009/CE/b809985e/b809985e-s1.gif)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: