Control of polymorphism in NaNbO3 by hydrothermal synthesis†
Deena R.
Modeshia
a,
Richard J.
Darton
a,
Sharon E.
Ashbrook
b and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: r.i.walton@warwick.ac.uk; Fax: +44 2476524112; Tel: +44 2476523241
bSchool of Chemistry and EaStCHEM, University of St Andrews, St Andrews, UK KY16 9ST
First published on 12th November 2008
Abstract
The metastable ilmenite polymorph of NaNbO3, instead of the expected perovskite polymorph, may be prepared directly in one step under mild hydrothermal conditions by lowering pH and using close to stoichiometric amounts of metal precursors; in situ energy-dispersive X-ray diffraction shows that crystallisation occurs rapidly via a sequence of intermediate crystalline phases.
Control of crystal polymorphism in dense oxide phases that contain two or more metals is difficult by conventional synthetic methods since extreme temperatures and pressures are typically applied, resulting in the formation of only the most thermodynamically stable phases under the conditions used. Although chimie douce methods, such as sol–gel or co-precipitation, have been widely investigated for the preparation of complex oxides,1 these generally are multi-step procedures that involve the low-temperature formation of a precursor containing the desired combination of chemical elements and a final annealing or firing step to induce crystallinity, often at temperatures approaching those used in conventional synthesis. Hydrothermal techniques offer a useful alternative approach to low-temperature synthesis of dense oxides and in the past few years the preparation of a variety of complex phases has been described;2 this includes a number of perovskites such as titanates,3 manganites4 and ferrites.5 As well as providing rapid, one-step crystallisation, the advantage in these mild routes (below 250 °C) for such materials has so far largely been developed in offering control over crystal form, important for optimising properties of the materials for applications. For example, in the case of lead–zirconium titanate (PZT), control of crystal morphology from cuboid to fibrous and of size from the nanometre to the millimetre may be achieved by hydrothermal synthesis.6
NaNbO3 is well-known to exhibit a rich polymorphism based on the perovskite structure, with a number of displacive transitions occurring over a range of temperatures,7 which may also be sensitive to both pressure and crystallite size.8 Doped forms of the material are currently the focus of much attention because of their piezoelectric properties.9 The hydrothermal synthesis of perovskite NaNbO3 has been described by a number of groups directly from reaction between Nb2O5 and aqueous NaOH above 120 °C.10 Herein we show how by adjusting synthesis conditions another polymorph of the material can be isolated in phase-pure form in a single-step reaction. This demonstrates a further level of control offered by the hydrothermal synthesis of mixed-metal oxides: control of polymorphism.
The ilmenite polymorph of NaNbO3 is isolated from the hydrothermal reaction of Nb2O5 with 1 M NaOH solution after heating at 240 °C for 3 hours with a close to stoichiometric Na : Nb ratio.‡Powder neutron diffraction was used to verify the initial laboratory X-ray assignment of structure and allowed a refinement of the structure, using the isostructural NaBiO311 as a starting model, Fig. 1.‡EDX analysis, performed in the electron microscope, gave an average Na : Nb ratio of 1 : 0.9 (±0.1).
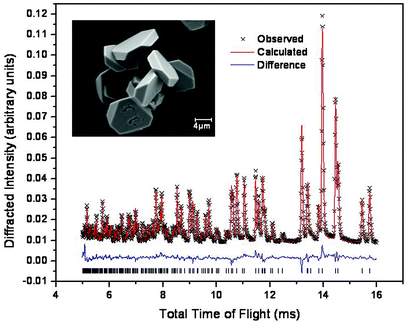 | ||
Fig. 1 Final Rietveld plot (GEM bank 6) for ilmenite NaNbO3 (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , a = 5.3430(9) Å, c = 15.6493(27) Å), and inset an SEM image of the specimen. , a = 5.3430(9) Å, c = 15.6493(27) Å), and inset an SEM image of the specimen. | ||
Structure and phase purity was further verified using MAS solid-state 23Na NMR, Fig. 2a, which shows a single Na site in the material with an isotropic chemical shift of 4.1 ppm and quadrupolar parameters CQ of 290 kHz and η of 0.2. A general trend of decreasing chemical shift with increasing coordination (and hence average Na–O distance) is established in 23Na NMR of oxide materials, although this can depend somewhat on the nature of the next nearest neighbour;12 however, the value we observe of 4.1 ppm can be compared directly with the values of −0.5 and −4.5 ppm reported for the two 12-coordinate Na sites in the perovskite NaNbO3,13 and is thus consistent with the lower coordination number of the distorted octahedral geometry of the single sodium site in the ilmenite polymorph. Importantly the 23Na spectrum shows no trace of the perovskite polymorph, to which the NMR is expected to be more sensitive than diffraction. The 93Nb spectrum, Fig. 2b, shows the presence of a single Nb site, consistent with the crystal structure, and the chemical shift of −1031.1 ppm (CQ of 36.8 MHz) is rather similar to values reported for the perovskite polymorph and for other materials that contain octahedral niobium(V).14
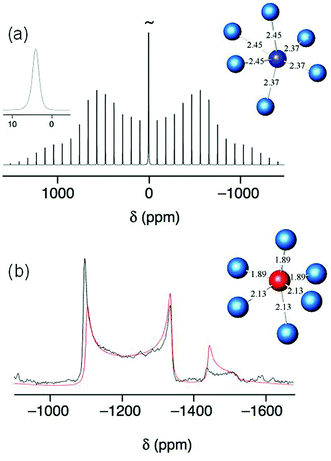 | ||
| Fig. 2 (a) 23Na (105.8 MHz) and (b) 93Nb (97.9 MHz) MAS NMR spectra of ilmenite NaNbO3. In (a), the inset spectrum is an expansion of the central-transition region. The local atomic arrangement of each atom type from the neutron structure determination are also shown (with interatomic distances in Å). | ||
The ilmenite polymorph of NaNbO3 has been reported previously by Kinomura and co-workers but a two-step synthesis method was used, involving first the preparation of Na8Nb6O19·13H2O followed by hydrothermal reaction with NaOH in a silver-lined vessel.15 The precise reaction conditions are very important in our synthesis route: if the reactions are performed for periods longer than 5 hours at 240 °C, then perovskite NaNbO3 is found along with the ilmenite polymorph, and if the concentration of NaOH is increased to 1.5 M then the perovskite is the sole product after 3 hours. This suggests that the ilmenite is metastable with respect to the perovskite polymorph under hydrothermal conditions. The same is true on heating the isolated ilmenite NaNbO3 in air: in situ high temperature powder X-ray diffraction experiments in the laboratory (ESI†) show an irreversible collapse into the orthorhombic perovskite polymorph above 650 °C.
In order to learn more about the hydrothermal crystallisation mechanism of NaNbO3 we have used time-resolved energy-dispersive X-ray diffraction (EDXRD) to follow the synthesis in a laboratory-sized autoclave using white-beam synchrotron X-rays.‡ This method has been widely used to study the crystallisation of zeolites and other open-framework materials,16 but rarely for the formation of mixed-metal oxide phases.17 Despite difficulties in dense oxide products settling out of the beam, by rapid stirring we have successfully measured time-resolved data that show the course of crystallisation of NaNbO3 in real time and under conditions representative of those used in the laboratory. Fig. 3 shows contour plots of EDXRD measured from reactions performed at two different NaOH concentrations. If a 1.5 M solution of NaOH is used (8 ml) and 1 g of Nb2O5, the product of the crystallisation at 240 °C is solely perovskite NaNbO3 after less than 1 hour (see ESI† for full assignment of in situ EDXRD spectra), and, as well as observing the disappearance of the crystalline Nb2O5 starting material, crystallisation of the perovskite is preceded by the formation of two other crystalline phases. Using the information from two of the detector elements these can be assigned as Na7(H3O)Nb6O19·14H2O, followed by Na2Nb2O6·nH2O (n ∼ 1). The former contains Nb6O198− Lindqvist ions,18 and the latter is the parent member of the Sandia octahedral molecular sieve (SOMS) materials.19 Similar phases have previously been reported on quenching the hydrothermal synthesis of NaNbO3 perovskite in its early stages,10 but our results show that intermediate crystalline materials are present under hydrothermal conditions, following the complete dissolution of Nb2O5 in less than 15 minutes. Upon lowering the sodium hydroxide concentration to 1 M, the ilmenite polymorph of NaNbO3 is the sole product after 3 hours, Fig. 3b. Here, the crystallisation initially follows the same pathway as for the perovskite, with the disappearance of Nb2O5 followed by the crystallisation of Na7(H3O)Nb6O19·14H2O then Na2Nb2O6·nH2O. At this point a third intermediate phase is observed to crystallise, coinciding with the disappearance of Na2Nb2O6·nH2O. This phase has so far proved difficult to identify unambiguously: none of the strong Bragg peaks matches any other known sodium niobate phase, although the pattern of Bragg peak positions differs little from those of Na2Nb2O6·nH2O, only their relative intensity changes, suggesting the unknown phase might be a structural variant of the SOMS material. Quenching experiments in the laboratory never yielded the unknown phase, suggesting that it might only be stable under hydrothermal conditions. These experiments also usually showed the presence of Nb2O5, even after it had been observed to dissolve completely in situ, indicating that quenching can result in recrystallisation, and thus does not offer a useful means of identifying phases present under hydrothermal conditions.
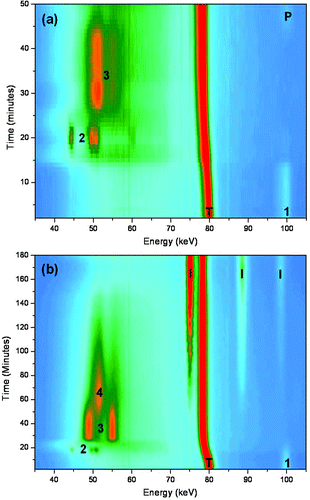 | ||
| Fig. 3 Contour plots of in situ EDXRD data from the crystallisation of NaNbO3 at 240 °C with NaOH concentration of (a) 1.5 M and (b) 1 M. The data shown are from a detector at 2θ = 1.81°. The labels on the Bragg peaks denote crystalline phases observed, 1: Nb2O5; 2: Na7(H3O)Nb6O10·14H2O; 3: Na2Nb2O6·nH2O; 4: unknown phase; P: perovskite NaNbO3; I: ilmenite NaNbO3; T: Teflon of the autoclave. | ||
Plots of the temporal evolution of the various phases present during the hydrothermal crystallisations, produced by integration of characteristic Bragg peaks, are shown in the ESI†. It is unlikely that the transient phases represent true intermediates in the building up of the dense products ultimately formed since they have very different structures, in particular in the mode of linking of constituent octahedral structural units (by various combinations of edge- and corner-sharing) differs between the materials. It is more reasonable to propose that each of these phases crystallises under certain conditions of pH and temperature before dissolving, providing the solution with further reactive chemical species for the crystallisation of the subsequent phase. The observation of the sequential hydrothermal crystallisation of several transient phases is unusual, although the process is similar to some results already seen in the synthesis of nanoporous phases, where the crystallisation of low-dimensional or lower density structures prior to the formation of three-dimensionally connected frameworks has been observed in situ for some zeotype phosphates.20
In summary, we have demonstrated the fine level of control of product offered by the hydrothermal synthesis of mixed-metal oxides. The in situ studies show the complexity of the crystallisations and how conditions must be carefully chosen for isolation of a desired phase. The extension of the synthetic work to more complex phases, such as doped analogues, containing three or more metals, could provide a fruitful approach to the discovery of new oxide materials not stable at high temperatures, something we are currently investigating.
We thank the EPSRC for provision of a DTA studentship for DRM and for funding under grant EP/F012721/1. The STFC is acknowledged for the award of beamtime at the Daresbury SRS and also access to ISIS via the Xpress service, and we thank Dr Ron Smith for measuring the data on GEM. We thank Professor Steve Wimperis, University of Glasgow, for access to his 400 MHz Bruker Avance NMR spectrometer.
Notes and references
- J. Gopalakrishnan, Chem. Mater., 1985, 7, 1265; J. Rouxel and M. Tournoux, Solid State Ionics, 1996, 84, 141 CrossRef CAS.
- G. Demazeau, J. Mater. Chem., 1999, 9, 15 RSC; R. I. Walton, Chem. Soc. Rev., 2002, 31, 230 RSC; R. E. Riman, W. L. Suchanek and M. M. Lencka, Anal. Chem., 2002, 27, 15 CAS.
- J. O. Eckert, C. C. Hung-Houston, B. L. Gersten, M. M. Lencka and R. E. Riman, J. Am. Ceram. Soc., 1996, 79, 2929.
- J. Spooren, A. Rumplecker, F. Millange and R. I. Walton, Chem. Mater., 2003, 15, 1401 CrossRef CAS; J. J. Urban, L. Ouyang, M. H. Jo, D. S. Wang and H. Park, Nano Lett., 2004, 4, 1547 CrossRef CAS.
- C. Chen, J. R. Cheng, S. W. Yu, L. J. Che and Z. Y. Meng, J. Cryst. Growth, 2006, 291, 135 CrossRef CAS.
- S.-B. Cho, M. Oledzka and R. E. Riman, J. Cryst. Growth, 2001, 226, 313 CrossRef CAS; X. Y. Liu, E. F. McCandlish, L. E. McCandlish, K. Mikulka-Bolen, R. Ramesh, F. Cosandey, G. A. Rossetti and R. E. Riman, Langmuir, 2005, 21, 3207 CrossRef CAS.
- C. N. W. Darlington and K. S. Knight, Acta Crystallogr., B: Struct. Sci., 1999, 55, 24 CrossRef.
- Y. Shiratori, A. Magrez, M. Kato, K. Kasezawa, C. Pithan and R. Wasert, J. Phys. Chem. C, 2008, 112, 9610 CrossRef CAS.
- M. D. Maeder, D. Damjanovic and N. Setter, J. Electroceram., 2004, 13, 385 CrossRef CAS.
- I. Santos, L. H. Loureiro, M. F. P. Silva and A. M. V. Cavaleiro, Polyhedron, 2002, 21, 2009 CrossRef CAS; G. K. L. Goh, F. L. Lange, S. M. Haille and C. G. Levi, J. Mater. Res., 2003, 18, 338 CrossRef CAS; H. Y. Zhu, Z. F. Zheng, X. P. Gao, Y. N. Huang, Z. M. Yan, J. Zou, H. M. Yin, Q. D. Zou, S. H. Kable, J. C. Zhao, Y. F. Xi, W. N. Martens and R. L. Frost, J. Am. Chem. Soc., 2006, 128, 2373 CrossRef CAS; A. J. Paula, M. A. Zaghete, E. Longo and J. A. Varela, Eur. J. Inorg. Chem., 2008, 1300 CrossRef CAS.
- N. Kumada, N. Kinomura and A. W. Sleight, Mater. Res. Bull., 2000, 35, 2397 CrossRef CAS.
- S. Antonijevic, S. E. Ashbrook, R. I. Walton and S. Wimperis, J. Mater. Chem., 2001, 12, 1469 Search PubMed.
- S. E. Ashbrook, L. Le Polles, R. Gautier, C. J. Pickard and R. I. Walton, Phys. Chem. Chem. Phys., 2006, 8, 3423 RSC.
- O. B. Lapina, D. F. Khabibulin, K. V. Romanenko, Z. H. Gan, M. G. Zuev, V. N. Krasil’nikov and V. E. Fedorov, Solid State Nucl. Magn. Reson., 2005, 28, 204 CrossRef CAS.
- N. Kinomura, N. Kumata and F. Muto, Mater. Res. Bull., 1984, 19, 299 CrossRef CAS; N. Kumada, N. Kinomura and F. Muto, J. Ceram. Soc. Jpn., 1990, 98, 384 CAS.
- G. Muncaster, A. T. Davies, G. Sankar, C. R. A. Catlow, J. M. Thomas, S. L. Colston, P. Barnes, R. I. Walton and D. O’Hare, Phys. Chem. Chem. Phys., 2000, 2, 3523 RSC; R. I. Walton and D. O’Hare, Chem. Commun., 2000, 2283 RSC.
- R. Kiebach, N. Pienack, W. Bensch, J. D. Grunwaldt, A. Michailovski, A. Baiker, T. Fox, Y. Zhou and G. R. Patzke, Chem. Mater., 2008, 20, 3022 CrossRef CAS.
- A. Goiffon, E. Philippot and M. Maurin, Rev. Chim. Miner., 1980, 17, 466 CAS.
- H. W. Xu, M. Nyman, T. M. Nenoff and A. Navrotsky, Chem. Mater., 2004, 16, 2034 CrossRef CAS.
- R. I. Walton, F. Millange, A. Le Bail, T. Loiseau, C. Serre, D. O’Hare and G. Ferey, Chem. Commun., 2000, 203 RSC; R. I. Walton, A. J. Norquist, S. Neeraj, S. Natarajan, C. N. R. Rao and D. O’Hare, Chem. Commun., 2001, 1990 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of NMR data collection, powder neutron structure refinement, results of variable temperature powder XRD, full analysis of EDXRD data, TGA data, IR data and H NMR spectra. See DOI: 10.1039/b814601b |
| ‡ Ilmenite NaNbO3 was synthesised by stirring 1 g of Nb2O5 into 8 ml of 1 M NaOH solution, followed by sealing in a Teflon-lined 20 ml stainless-steel autoclave and heating to 240 °C at 1 °C min−1 and holding for 3 hours. After cooling, the white solid product was recovered by filtration, washed with deionised water and dried. Time-of-flight powder neutron diffraction was measured using the GEM diffractometer at ISIS, the UK spallation neutron source, and the program GSAS used to refine the atomic structure (see ESI) Time-resolved EDXRD was performed on Station 16.4 of the Daresbury SRS with a 25 mL Teflon-lined stainless-steel autoclave with walls thinned to 0.25 mm and data measured in 60 s intervals using a three-element germanium detector. |
| This journal is © The Royal Society of Chemistry 2009 |