After 118 years, the isolation of two common radical anion reductants as simple, stable solids†
Thomas A.
Scott
a,
Betty A.
Ooro
a,
David J.
Collins
a,
Michael
Shatruk
b,
Andrey
Yakovenko
c,
Kim R.
Dunbar
c and
Hong-Cai
Zhou
*ac
aDepartment of Chemistry and Biochemistry, Miami University, Oxford, OH 45056, USA. E-mail: zhouh@muohio.edu; Fax: +01 513 5290452; Tel: +01 513 5298091
bDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306, USA
cDepartment of Chemistry, Texas A&M University, College Station, TX 77842, USA
First published on 20th November 2008
Abstract
Two common radical anion reductants, potassium benzophenone ketyl (K(Ph2CO)) and potassium naphthalenide (K2(C10H8)2(THF)), have been isolated and characterized for the first time in solvent-free form or with low solvent content, allowing their use as pure solid reactants in preparative redox chemistry in accurate stoichiometric amounts.
The reactions of aromatic hydrocarbons with alkali metals have long been recognized as forming organic radical species,1–3 the first reported being benzophenone ketyl in 1891.4 These radical species are synthetically useful and have subsequently been used in a variety of organic5 and organometallic reactions6 as strong reductants.7 They are formed by the one-electron reduction of ketones or aldehydes using reducing metals, such as sodium and potassium. EPR solution studies of these anionic radicals suggest that they exist as monomeric and dimeric species.8 Despite the long existence and common usage of these radical species, structurally-characterized examples are very few due to their high reactivity, making them difficult to isolate.
Several naphthalenide compounds have been previously isolated with Li+ and Na+,8,9 while benzophenone ketyl compounds have been reported containing Sm3+,10Ca2+,11 and Na+.12 In these cases, however, there are large amounts of solvent ligands that serve to stabilize the metal centers by completing the coordination sphere. To date, no solvent-free alkali–metal organic radical compound has been isolated. In this communication we report, for the first time, the isolation and characterization in the solid state of solvent-free potassium benzophenone ketyl, K(Ph2CO), and of potassium naphthalenide, K2(C10H8)2(THF), with low solvent content.
Results obtained from single-crystal X-ray diffraction show that K2(C10H8)2(THF) crystallizes in the monoclinic space groupC2/c.‡ The asymmetric unit consists of one K+ cation, two symmetrically independent naphthalenide anions, which are located on inversion centers, and a bridging THF molecule located on a two-fold axis. The structure can be represented by a dinuclear unit. As shown in Fig. 1, the naphthalenide anions occupy two unique bonding environments. One is a [η12-C10H8]− anion that bridges two K+ centers from distinct dimer units through cation–π interactions. The other is a [η16-C10H8]− anion, which bridges four K+ centers forming two dimetal units through cation–π interactions. These multiple cation–π interactions between the radical anions and K+ cations lead to the formation of an extended 3D network and fulfill the coordination sphere of K+ ions.
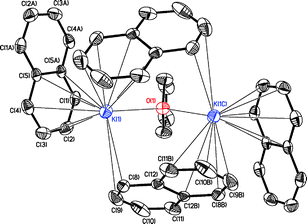 | ||
| Fig. 1 A dinuclear unit of K2(C10H8)2(THF); A −x + 0.5, −y + 0.5, −z + 1; B −x + 1, −y, −z + 1; C −x + 1, y, −z + 1.5. Thermal ellipsoids are shown at 50% probability level; hydrogen atoms are omitted for clarity. | ||
The K+ cations are arranged asymmetrically with respect to the ring centers of the naphthalene radicals. Distances between K+ cations and [η12-C10H8]− anions vary in the range of 3.088(3)–3.447(2) Å, the shortest distance being K(1)–C(2). The K–C distances between the [η16-C10H8]− anions and K+ anions fall in the range of 2.995(3)–3.523(2) Å. In the [η16-C10H8]− anion the peripheral carbon atoms C(8) and C(9) also form extra K+–π interactions (Fig. 1) at distances of 3.095(3) and 3.533(4) Å, respectively. These results are consistent with the observation made by Stucky.8
Single-crystal X-ray diffraction studies reveal that K(Ph2CO) crystallizes in the non-centrosymmetric tetragonal space groupI![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) .‡ Four K+ cations and four μ3-O atoms from the benzophenone radicals form a K4O4cubane-type structure, which is centered on a four-fold rotoinversion axis, as illustrated in Fig. 2(a). In the K4O4 fragment, each K atom form two long (2.735(3) and 2.766(4) Å) and one short (2.658(4) Å) K–O bonds leading to a D2d distortion of the K4O4-fragment from ideal Td symmetry.
.‡ Four K+ cations and four μ3-O atoms from the benzophenone radicals form a K4O4cubane-type structure, which is centered on a four-fold rotoinversion axis, as illustrated in Fig. 2(a). In the K4O4 fragment, each K atom form two long (2.735(3) and 2.766(4) Å) and one short (2.658(4) Å) K–O bonds leading to a D2d distortion of the K4O4-fragment from ideal Td symmetry.
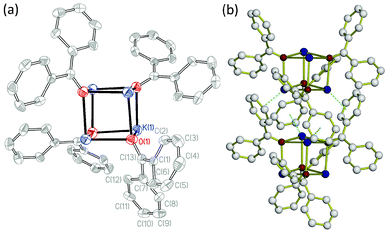 | ||
| Fig. 2 (a) K(Ph2CO) cubane-type structure; C = grey, K = blue, O = red. Thermal ellipsoids are shown at 50% probability level; hydrogen atoms are omitted for clarity. (b) Formation of a 1D chain through cation–π interactions between adjacent K(Ph2CO) cubanes. | ||
A comparison of benzophenone13 and K(Ph2CO) reveals that there is an elongation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond from 1.222 to 1.311(5) Å and a shortening of the C–CPh bond from 1.495 to 1.460(6) Å and from 1.497 to 1.460(7) Å. There are no significant changes in the average phenyl ring distances, indicating the additional electron is localized in the trigonal-planar C3O moiety.
O bond from 1.222 to 1.311(5) Å and a shortening of the C–CPh bond from 1.495 to 1.460(6) Å and from 1.497 to 1.460(7) Å. There are no significant changes in the average phenyl ring distances, indicating the additional electron is localized in the trigonal-planar C3O moiety.
Cubane formation can be viewed as the condensation of two dimers in the solid state, since ketyls are thought to exist as dimers in solution14 and have been isolated as dimeric species.15 The cubane structure is similar to that obtained by Hou for potassium fluorenone ketyl ([K(C13H8O)(HMPA)]4(μ-HMPA)),12 in which the K+ cations were stabilized by hexamethylphosphoric triamide (HMPA) coordination. In this case, however, there is an additional HMPA molecule bridging two K+ cations above one face of the cubane causing distortion from ideal Td symmetry. Hou also described the structurally similar benzophenone ketyl species (Na3(Ph2CO)3(HMPA)4),12 in which the ketyl O atoms bridge Na+ cations; one [Ph2CO]− anion contains a μ3-O atom, and the other two contain μ2-O atoms. The three Na+ cations are each stabilized by HMPA coordination with an additional bridging HMPA preventing cubane formation, resulting in a cuboidal-type structure. This example clearly illustrates that coordinating solvents affect the structure of these compounds.
In the crystal of K(Ph2CO), the K(Ph2CO) cubanes form chains along the c-axis via four K+–π interactions between phenyl rings and the nearest K+ centers located in the adjacent K(Ph2CO) cubane (Fig. 2(b)). The average K–C distance is 3.231(6) Å, with a range of 3.118(5)–3.351(5) Å. The cubanes are oriented 90° to each other to allow these interactions, which stabilize the structure by fulfilling the K+ coordination sphere.
The magnetic properties of the compounds were also investigated. The χT value at 300 K for K2(C10H8)2(THF) is 0.71 emu mol−1K, which is slightly lower than the value of 0.75 emu mol−1K expected for two non-interacting S = 1/2 organic spins. The χT curve decreases gradually with decreasing temperature and then abruptly increases below 30 K to a maximum value of 1.69 at 27 K; below this temperature χT abruptly decreases to zero as shown in Fig. 3(a). Similar behavior is observed for the temperature dependence of magnetic susceptibility, which remains nearly constant from 300 to 30 K, then abruptly increases and decreases again in a narrow temperature range below 30 K, as illustrated in Fig. 3(b).
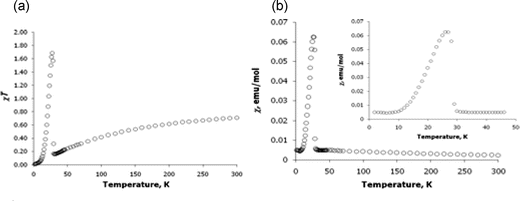 | ||
| Fig. 3 Magnetic properties of K2(C10H8)2(THF). (a) Temperature dependence of χT; (b) temperature dependence of magnetic susceptibility (χ). Inset: the expanded 0–50 K region of the χ vs. T dependence. | ||
Such magnetic behavior indicates that K2(C10H8)2(THF) undergoes long-range antiferromagnetic ordering below 30 K. To test this hypothesis, AC magnetic susceptibility and zero-field-cooled vs. field-cooled (ZFC/FC) magnetization studies were performed on samples of K2(C10H8)2(THF). A sharp out-of-phase signal was observed for the imaginary part (χ″) of the AC susceptibility (Fig. S1, ESI†). The signal maximum occurs at 28.6 K, and its position is independent of the frequency of the applied AC magnetic field. The ZFC and FC curves are essentially superimposable below 30 K (Fig. S2, ESI†), which, along with the AC susceptibility measurements, supports the conclusion that long-range antiferromagnetic ordering occurs in K2(C10H8)2(THF), with a Neel temperature of TN = 28.6 K.
The room-temperature χT value for K(Ph2CO) is 0.33 emu mol−1K, which is close to the theoretically expected value of 0.375 emu mol−1 K for an isolated S = 1/2 organic radical. As the temperature decreases, the value of χT decreases slowly (Fig. 4), and then more abruptly below 100 K, essentially reaching zero below 10 K. Such behavior is indicative of antiferromagnetic interactions between benzophenone ketyl radicals. An examination of the AC magnetic susceptibility data revealed no signature of magnetic ordering.
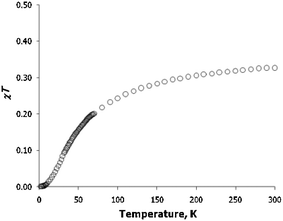 | ||
| Fig. 4 Temperature dependence of χT for K(Ph2CO). | ||
The differences in the magnetic behavior of K2(C10H8)2(THF) and K(Ph2CO) is attributed to their crystal structures. In the former case, the [C10H8]−radical anions exhibit interactions with K+ cations in all three dimensions. It is reasonable to suggest that magnetic coupling is mediated by these cation–π interactions which serve to bring the spin centers closer and hence increase dipolar interactions; the result is magnetic ordering for K2(C10H8)2(THF) at TN = 28.6 K.
In contrast, the packing motif in the crystal structure of K(Ph2CO) is such that the cation–π interactions produce a one-dimensional architecture. As previously mentioned, the K+ cations form cubane-type units with O atoms of the [Ph2CO]− anions, and these units are further connected into infinite chains. The [Ph2CO]− anions project outward from the K4O4 cubane units, illustrated in Fig. 5. Such an arrangement prevents close cation–π contacts in the other two dimensions hence magnetic coupling between the [Ph2CO]− radicals that belong to different chains is negligible and long-range magnetic ordering does not occur in K(Ph2CO).
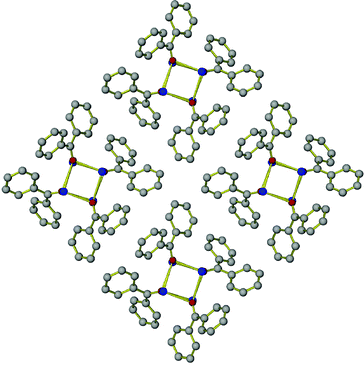 | ||
| Fig. 5 Packing of K(Ph2CO) chains, viewed in the c direction. Hydrogen atoms are omitted for clarity. | ||
The redox potentials of these isolated complexes agree with previously published values.15 The UV-Vis absorption spectrum of K(C10H8) exhibits bands at 364 and 434 nm, and a broad feature centered at 800 nm, in agreement with previously published values.16 The UV-Vis absorption spectrum of K(Ph2CO) exhibits bands at 326, 376 and 656 nm, giving this radical its intense blue color. These bands have been assigned to a D0→ D1 transition.17,18
In conclusion, the compounds K2(C10H8)2(THF) and neat K(Ph2CO) have been isolated and characterized in the solid state for the first time. Naphthalenide and benzophenone ketyl radical species are useful as strong reductants in preparative chemistry, but they have a limited shelf life in solution; moreover titration must be employed to determine exact concentrations. The compounds reported herein are stable as solids when stored under an inert atmosphere, thereby allowing for precise delivery in accurate stoichiometric amounts.
Notes and references
- W. Schlenk, J. Appenrodt, A. Michael and A. Thal, Ber. Dtsch. Chem. Ges., 1914, 47, 473–490 CAS.
- W. Schlenk and T. Weickel, Ber. Dtsch. Chem. Ges., 1911, 44, 1182–1189 CrossRef CAS.
- N. D. Scott, J. F. Walker and V. L. Hansley, J. Am. Chem. Soc., 1936, 58, 2442–2444 CrossRef CAS.
- F. Bechman and T. Paul, Justus Liebigs Ann. Chem., 1891, 266, 1–28 CrossRef.
- G. A. Molander, Acc. Chem. Res., 1998, 31, 603–609 CrossRef CAS.
- M. I. Bruce, D. C. Kehoe, J. G. Matisons, B. K. Nicholson, P. H. Rieger and M. L. Williams, J. Chem. Soc., Chem. Commun., 1982, 442–444 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- J. J. Brooks, W. Rhine and G. D. Stucky, J. Am. Chem. Soc., 1972, 94, 7346–7351 CrossRef CAS.
- H. Bock, C. Arad, C. Naether and Z. Havlas, J. Chem. Soc., Chem. Commun., 1995, 2393–2394 RSC.
- Z. Hou, T. Miyano, H. Yamazaki and Y. Wakatsuki, Kidorui, 1995, 26, 314–315 Search PubMed.
- Z. Hou, X. Jia, M. Hoshino and Y. Wakatsuki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1292–1294 CAS.
- Z. Hou, X. Jia, A. Fujita, H. Tezuka, H. Yamazaki and Y. Wakatsuki, Chem. Eur. J., 2000, 6, 2994–3005 CrossRef CAS.
- The geometric parameters for Ph2CO presented in the 2007 release of the Cambridge Structural Database [ConQuest, V. 1.10; Cambridge Crystallographic Data Centre (CCDC), 2007]. The Reference Code for Ph2CO is BphenO12.
- N. Hirota, J. Am. Chem. Soc., 1967, 89, 32–41 CrossRef CAS.
- K. R. Walczyk, G. S. Popkirov and R. N. Schindler, Ber. Bunsenges. Phys. Chem., 1995, 99, 1028–1036 CAS.
- T. Shida and S. Iwata, J. Am. Chem. Soc., 1973, 95, 3473–3483 CrossRef CAS.
- A. Henseler, M. von Raumer and P. Suppan, J. Chem. Soc., Faraday Trans., 1996, 92, 391–393 RSC.
- E. Wagner-Czauderna and M. K. Kalinowski, Collect. Czech. Chem. Commun., 2000, 65, 1573–1579 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, cyclic voltammetry, magnetic measurements, and UV-visible spectra. CCDC 689226 and 689227. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b815272a |
| ‡ Data reduction, structure solution and refinement calculations were performed using the Bruker SAINT+ and SHELXTL NT program packages. Crystal data for K(Ph2CO), C13H10KO, Mr = 221.31, tetragonal, space groupI, a = 16.927(4), c = 7.271(2) Å, V = 2083.3(9) Å3, T = 213(2) K, Z = 8, λ = 0.71073 Å, μ = 0.475 mm−1. 5597 reflections, 1767 unique (Rint = 0.088). Final R values: R1 = 0.0664 [I > 2σ(I)], wR2 = 0.0996 (all data). Flack parameter: 0.05(8). Crystal data for K2(C10H8)2(THF), C24H24K2O, Mr = 406.63, monoclinic, space groupC2/c, a = 17.865(3), b = 10.2540(15), c = 11.5701(16) Å, β = 97.655(4)°, V = 2100.6(6) Å3, T = 213(2) K, Z = 4, λ = 0.71073 Å, μ = 0.461 mm−1. 5599 reflections, 1861 unique (Rint = 0.027). Final R values: R1 = 0.0417 [I > 2σ(I)], wR2 = 0.0910 (all data). |
| This journal is © The Royal Society of Chemistry 2009 |