A stable crystalline N-heterocyclic carbene with a 1,1′-ferrocenediyl backbone†‡
Ulrich
Siemeling
*,
Christian
Färber
and
Clemens
Bruhn
Institute of Chemistry, University of Kassel, Heinrich-Plett-Str. 40, D-34132, Kassel, Germany. E-mail: siemeling@uni-kassel.de; Fax: +49 561 804 4777; Tel: +49 561 804 4576
First published on 11th November 2008
Abstract
A stable six-membered N-heterocyclic carbene with a redox-active ferrocene-based backbone was prepared, characterised by X-ray crystallography and utilised as a ligand for the preparation of expected and unexpected metal complexes.
The chemistry of N-heterocyclic carbenes (NHCs)1 has developed rapidly since Arduengo et al. described the first stable crystalline NHC, 1,3-di-1-adamantylimidazol-2-ylidene (IAd),2 in 1991. Based on pioneering work by Herrmann and coworkers,3 they are widely applied as ligands in transition metal-catalysed reactions.1b,c Their steric and electronic properties can be modified by the exocyclic substituents at the nitrogen atoms as well as by the backbone which connects the two nitrogen atoms. While 5-membered NHCs based on imidazole and imidazoline have received most attention so far, there is great current interest in ‘non-standard’ NHCs, i.e. those with ring sizes other than 5 and/or with heteroatoms in the backbone.4 With a view to redox-tunable catalysts,5 we have focused our efforts on [3]ferrocenophane-type NHCs, which formally contain a 6-membered ring with a d-block metal atom (Scheme 1). The ferrocene-based backbone is expected to allow the redox-tuning of the electronic properties of such ligands much more efficiently than ferrocenyl substituents attached to the nitrogen atoms.6 Independently from us, Bielawski and coworkers have also been addressing this system. They recently communicated the first two examples of such NHCs, generated in situ by deprotonation of the corresponding formamidinium tetrafluoroborates 1 (Scheme 1). 2a (R = iBu) and 2b (R = Ph) proved to be too unstable for isolation.7 However, both could be trapped by coordination at [RhCl(L)2] fragments. Electrochemical data proved to be in support of an interaction between the two metal centres, and through-space π-backbonding between the Fe atom and the carbene C atom was invoked to be responsible for this. Unfortunately, only 2a was sufficiently stable for NMR spectroscopic characterisation in solution. The use of bulkier substituents (R = Mes, CH2tBu, CHPh2) was not possible, since the corresponding formamidinium salts could not be obtained. We can confirm the observations described by Bielawski and coworkers for 2b and we, too, have not succeeded so far in the preparation of formamidinium salts 1 with bulky aryl substituents. Since we have been more successful with bulky alkyl substituents, their report prompts us to disclose here preliminary results concerning the preparation, isolation and structural characterisation of 2c (R = 2-adamantyl) and its complexes [RhCl(2c)(cod)] (cod = 1,5-cyclooctadiene) and [Mo(2c)(CO)4].
 | ||
| Scheme 1 Structures of the ferrocene-based N-heterocyclic carbenes 2 and their formamidinium precursors 1. | ||
Carbene 2c was synthesised by deprotonation of 1c (prepared in three steps from 1,1′-diaminoferrocene) with lithium diisopropylamide in THF and isolated in 40% yield after standard work-up. It can be stored at room temperature in an inert atmosphere for weeks without noticeable decomposition. The 13C NMR signal of the divalent carbon atom is observed at 260.7 ppm, which compares well with the value of 259.6 ppm reported for 2a.7 The structures of the carbene 2c and its precursor 1c were determined by X-ray crystallography (Fig. 1 and Fig. 2).§ Owing to the very small size of the single crystal of 2c, the quality of the crystal structure determination was affected by a poor data/parameter ratio. It was further compromised by a disorder of the toluene molecule and of one of the adamantyl substituents in one of the two species present in the asymmetric unit. Due to the low resolution of the data for this structure, a discussion is meaningful only for the fundamental features and will be restricted to the non-disordered species shown in Fig. 2.
 | ||
| Fig. 1 Molecular structure of the cation of 1c in the crystal. Thermal ellipsoids are drawn at the 30% probability level. | ||
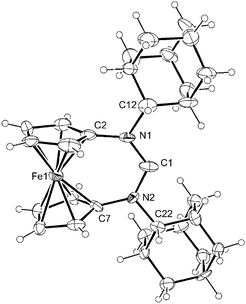 | ||
| Fig. 2 Molecular structure of 2c (non-disordered species) in the crystal. Thermal ellipsoids are drawn at the 30% probability level. | ||
A notable decrease of the N–C–N angle, accompanied by a slight increase of the average C–N bond length in this unit, is typically observed upon deprotonation of the formamidinium precursor. The limitations caused by the low resolution of the data for the structure of 2c notwithstanding, these general trends come out clearly in a comparison of 1c and 2c. The N–C–N angle of 1c is 131.1(6)°, which is indistinguishable within experimental error from the value of 129.6(3)° reported for 1b.7 This angle is considerably smaller in the case of the carbene 2c, viz. 119.2(7)°. In the case of 6-membered perimidine-based NHCs, which are structurally closely related to 2, a similar decrease of the N–C–N bond angle of ca. 10° has been observed upon deprotonation of the respective formamidinium precursor.8 Such a decrease of the N–C–N angle necessarily moves this C atom further away from the Fe atom. This iron–carbon distance is ca. 3.31 Å in the case of 1c, which is close to the corresponding value of 3.28 Å reported for 1b.7 The Fe–Ccarbene distance of ca. 3.44 Å observed for 2c is substantially longer than that and very similar to the corresponding distances found for the complexes [RhCl(2)(cod)] (vide infra). The average bond length in the N–C–N unit increases slightly, but significantly, upon deprotonation from 1.331(5) Å in 1c to ca. 1.39(1) Å in 2c, which is again in agreement with observations reported for the 6-membered perimidine-based NHCs and is consistent with a comparatively lower degree of π-delocalisation in the carbene.8 The cyclopentadienyl ring tilt angles are ca. 16° for all of these diaza-[3]ferrocenophane derivatives, which is larger than the values reported for [3]ferrocenophane (7.6°) and 7-oxa-[3]ferrocenophane (11.9°).9
We have started to investigate the coordination behaviour of the carbene 2c. Its reaction with [{Rh(μ-Cl)(cod)}2] afforded the expected [RhCl(2c)(cod)] in good yield. The product was structurally characterised by single-crystal X-ray diffraction (Fig. 3). The carbene C atom is in a trigonal planar environment (sum of angles ca. 360°). Its bond distance to the Rh atom is 2.088(10) Å, which compares well with the corresponding values of 2.044(4) and 2.059(4) Å reported for [RhCl(2a)(cod)] and [RhCl(2b)(cod)],7 respectively. The Fe–Ccarbene distance is ca. 3.43 Å in all three cases.
![Molecular structure of [RhCl(2c)(cod)] in the crystal. Thermal ellipsoids are drawn at the 30% probability level.](/image/article/2009/CC/b813809e/b813809e-f3.gif) | ||
| Fig. 3 Molecular structure of [RhCl(2c)(cod)] in the crystal. Thermal ellipsoids are drawn at the 30% probability level. | ||
The reaction of 2c with [Mo(CO)6] in THF under thermal conditions unexpectedly afforded the 16 valence electron complex [Mo(2c)(CO)4] in low yield. We ascribe the fact that the expected 18 valence electron analogue [Mo(2c)(CO)5] was not obtained to the steric shielding of the divalent C atom by the bulky 2-adamantyl substituents. A similar result was recently reported by Nolan and coworkers for the reaction of the bulky 5-membered NHC IAd with [Ni(CO)4], which afforded the 16 valence electron product [Ni(IAd)(CO)2].10 In the same vein, Bertrand and coworkers have been able to obtain coordinatively unsaturated complexes with a bulky cyclic (alkyl)(amino)carbene (CAAC) ligand.11 Very likely, 2c reacted with [Mo(CO)4(THF)2], which can form in small amounts by disproportionation of [Mo(CO)5(THF)] in THF.12 The complex can be prepared more rationally from 2c and [Mo(CO)4(nbd)] (nbd = 2,5-norbornadiene). [Mo(2c)(CO)4] was structurally characterised by X-ray crystallography (Fig. 4). The small number of independent reflections (due to problems detailed in the ESI†) caused some regrettable consequences, especially isotropic refinement of the equatorial carbonyl C atoms, which limits the discussion of geometric parameters. Again, the carbene C atom is in a trigonal planar environment (sum of angles ca. 360°). The coordination of the Mo atom is best described as distorted trigonal bipyramidal. The distortion is indicative of steric strain caused by the bulky carbene ligand, which occupies one of the equatorial positions and pushes the two equatorial carbonyl ligands together. The CeqMo–Ceq bond angle is only ca. 85°, while the CeqMo–Ccarbene angles are ca. 137°. The Cax–Mo–Cax unit is essentially linear. This coordination axis is canted with respect to the equatorial plane, as is reflected by the pronounced deviation of the Cax–Mo–Ccarbene angles (81° and 100°) from the ideal value of 90°. The bond distance between the carbene C atom and the Mo atom is ca. 2.26 Å, which agrees well with respective values found for other molybdenum carbonyl complexes containing 5-13 and 6-membered14 NHC ligands. Not surprisingly, the distances between the Mo atom and the C atoms of the carbonyl ligands, which are much stronger π-acceptors than any NHC, are considerably shorter and lie in the region typical for molybdenum CO complexes (av. Mo–COaxca. 2.01 Å, av. Mo–COeqca. 1.87 Å). In the coordination chemistry of phosphanes hexacoordinate complexes [Mo(PR3)(CO)5] are formed even with the bulkiest ligands (R = tBu,15Cy16) (Cy = cyclohexyl). Photolytically generated pentacoordinate [Mo(PCy3)(CO)4] is unstable above ca. 115 K in solution.17 Related isolable 16 valence electron carbonyl complexes have only been obtained with at least two bulky phosphane ligands.18
![Molecular structure of [Mo(2c)(CO)4]. Thermal ellipsoids are drawn at the 30% probability level.](/image/article/2009/CC/b813809e/b813809e-f4.gif) | ||
| Fig. 4 Molecular structure of [Mo(2c)(CO)4]. Thermal ellipsoids are drawn at the 30% probability level. | ||
In summary, we have obtained a stable, crystalline NHC with a redox-active ferrocene-based backbone. Our preliminary results, in conjunction with those reported recently by Bielawski and coworkers for less stable, non-isolable analogues, augur well for the development of a rich coordination chemistry with such ligands. The redox-tunability of electronic properties will be a focus of our interest. The results of quantum-chemical calculations, spectroelectrochemical investigations and studies concerning applications in catalysis will be reported in due course.
We are grateful to the DFG for generous funding.
Notes and references
- For recent reviews, see: (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS; (b) N-Heterocyclic Carbenes in Transition Metal Catalysis (Top. Organomet. Chem., 2007, 21), ed. F. Glorius, Springer, Berlin, 2007 Search PubMed; (c) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006 Search PubMed; (d) R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schütz, Angew. Chem., Int. Ed., 2004, 43, 5896 CrossRef CAS; (e) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS; (f) K. J. Cavell and D. S. McGuinness, Coord. Chem. Rev., 2004, 248, 671 CrossRef CAS; (g) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (h) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951 CrossRef CAS; (i) E. Peris and R. H. Crabtree, C. R. Chim., 2003, 6, 33 CrossRef CAS; (j) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371 CrossRef CAS.
- See, for example: (a) M. Iglesias, D. J. Beetstra, J. C. Knight, L.-L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279 CrossRef CAS; (b) A. Kausamo, H. M. Tuononen, K. E. Krahulic and R. Roesler, Inorg. Chem., 2008, 47, 1145 CrossRef CAS; (c) T. D. Forster, K. E. Krahulic, H. M. Tuononen, R. McDonald, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2006, 45, 6356 CrossRef CAS; (d) C. Präsang, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2005, 127, 10182 CrossRef; (e) K. E. Krahulic, G. D. Enright, M. Parvez and R. Roesler, J. Am. Chem. Soc., 2005, 127, 4142 CrossRef CAS; (f) C. C. Scarborough, M. J. W. Grady, I. A. Guzei, B. A. Gandhi, E. E. Bunel and S. S. Stahl, Angew. Chem., Int. Ed., 2005, 44, 5269 CrossRef CAS; (g) E. Despagnet-Ayoub and R. H. Grubbs, J. Am. Chem. Soc., 2004, 126, 10198 CrossRef CAS; (h) P. Bazinet, G. P. A. Yap and D. S. Richeson, J. Am. Chem. Soc., 2003, 125, 13314 CrossRef CAS.
- See, for example: (a) C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410 CrossRef CAS; (b) I. M. Lorkovic, R. R. Duff, Jr and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617 CrossRef CAS.
- For examples of N-ferrocenylated NHCs, see: (a) B. Bildstein, M. Malaun, H. Kopacka, K. Wurst, M. Mitterböck, K.-H. Ongania, G. Opromolla and P. Zanello, Organometallics, 1999, 18, 4325 CrossRef CAS; (b) B. Bildstein, M. Malaun, H. Kopacka, K.-H. Ongania and K. Wurst, J. Organomet. Chem., 1999, 572, 177 CrossRef CAS.
- D. M. Khramov, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2008, 47, 2267 CrossRef CAS.
- P. Bazinet, T.-G. Ong, J. S. O’Brien, N. Lavoie, E. Bell, G. P. A. Yap, I. Korobkov and D. S. Richeson, Organometallics, 2007, 26, 2885 CrossRef CAS.
- M. Hillman and J. D. Austin, Organometallics, 1987, 6, 1737 CrossRef CAS.
- R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485 CrossRef CAS.
- V. Lavallo, I. Canac, A. DeHope, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 7236 CrossRef CAS.
- D. Sellmann, A. Brandl and R. Endell, J. Organomet. Chem., 1975, 97, 229 CrossRef CAS.
- See, for example: K. Öfele, W. A. Herrmann, D. Mihalios, M. Elison, E. Herdtweck, T. Priermeier and P. Kiprof, J. Organomet. Chem., 1995, 498, 1 Search PubMed , and references cited therein.
- C.-Y. Liu, D.-Y. Chen, G.-H. Lee, S.-M. Peng and S.-T. Liu, Organometallics, 1996, 15, 1055 CrossRef CAS.
- H. Schumann, O. Stelzer, J. Kuhlmey and U. Niederreuther, Chem. Ber., 1971, 104, 993 CrossRef CAS.
- H. Werner and R. Prinz, Chem. Ber., 1966, 99, 3582 CrossRef CAS.
- J. D. Black and P. S. Braterman, J. Organomet. Chem., 1973, 63, C19 CrossRef CAS.
- For [Mo(PR3)2(CO)3] (R = tBu, Cy), see: (a) S. P. Nolan and C. D. Hoff, J. Organomet. Chem., 1985, 290, 365 CrossRef CAS; for [Mo(PiPr3)2(CO)3], see: (b) G. J. Kubas, J. Chem. Soc., Chem. Commun., 1980, 61 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and analytical and spectroscopic data. CCDC 697881–697884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b813809e |
| ‡ Presented in part at the 38th International Conference on Coordination Chemistry, Jerusalem, July 2008. |
§ Crystal data for 1c: C32H41BCl2F4FeN2, M = 667.23, monoclinic, a = 7.9008(12), b = 18.7777(19), c = 10.3761(17) Å, β = 90.019(13)°, V = 1539.4(4) Å3, T = 223(2) K, space groupP 21/m (no. 11), Z = 2, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 075 reflections measured, 2819 unique (Rint = 0.1268) which were used in all calculations; final wR(F2) 0.098 (all data). For 2c: C32.75H40FeN2, M = 517.52, triclinic, a = 10.763(3), b = 16.153(5), c = 16.339(5) Å, α = 71.83(2), β = 70.42(2), γ = 81.36(2)°, V = 2539.7(12) Å3, T = 213(2) K, space groupP 075 reflections measured, 2819 unique (Rint = 0.1268) which were used in all calculations; final wR(F2) 0.098 (all data). For 2c: C32.75H40FeN2, M = 517.52, triclinic, a = 10.763(3), b = 16.153(5), c = 16.339(5) Å, α = 71.83(2), β = 70.42(2), γ = 81.36(2)°, V = 2539.7(12) Å3, T = 213(2) K, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 4, 11413 reflections measured, 6269 unique (Rint = 0.1921) which were used in all calculations; final wR(F2) 0.2023 (all data). For [RhCl(2c)(cod)]: C39H50ClFeN2Rh, M = 741.02, orthorhombic, a = 12.8330(12), b = 20.430(2), c = 12.3078(14) Å, V = 3226.8(6) Å3, T = 173(2) K, space group C cm21 (no. 36), Z = 4, 10266 reflections measured, 1546 unique (Rint = 0.0990) which were used in all calculations; final wR(F2) 0.0610 (all data). For [Mo(2c)(CO)4]: C35H38FeMoN2O4, M = 702.46, orthorhombic, a = 13.549(2), b = 20. 097(5), c = 11.2263(19) Å, V = 3056.7(10) Å3, T = 173(2) K, space group P nma (no. 62), Z = 4, 7347 reflections measured, 2583 unique (Rint = 0.1785) which were used in all calculations; final wR(F2) 0.1526 (all data). (no. 2), Z = 4, 11413 reflections measured, 6269 unique (Rint = 0.1921) which were used in all calculations; final wR(F2) 0.2023 (all data). For [RhCl(2c)(cod)]: C39H50ClFeN2Rh, M = 741.02, orthorhombic, a = 12.8330(12), b = 20.430(2), c = 12.3078(14) Å, V = 3226.8(6) Å3, T = 173(2) K, space group C cm21 (no. 36), Z = 4, 10266 reflections measured, 1546 unique (Rint = 0.0990) which were used in all calculations; final wR(F2) 0.0610 (all data). For [Mo(2c)(CO)4]: C35H38FeMoN2O4, M = 702.46, orthorhombic, a = 13.549(2), b = 20. 097(5), c = 11.2263(19) Å, V = 3056.7(10) Å3, T = 173(2) K, space group P nma (no. 62), Z = 4, 7347 reflections measured, 2583 unique (Rint = 0.1785) which were used in all calculations; final wR(F2) 0.1526 (all data). |
| This journal is © The Royal Society of Chemistry 2009 |