Organic electrosynthesis using toluates as simple and versatile radical precursors†
Kevin
Lam
and
István E.
Markó
*
Département de Chimie, Bâtiment Lavoisier, Université catholique de Louvain, Place Louis Pasteur 1, B-1348, Louvain-la-Neuve, Belgium. E-mail: istvan.marko@uclouvain.be
First published on 17th November 2008
Abstract
The electrolysis of toluateesters leads smoothly to the formation of the radical of the alkyl fragment. This property has been used to develop a new electrochemical deoxygenation reaction.
The transformation of an alcohol into the corresponding alkane by removal of the OH function represents an important and often delicate conversion in organic chemistry. Apart from classical multistep sequences,1 the Barton–McCombie reaction and its variants have been considered as the acme of such transformations.2 Unfortunately, this procedure suffers from a number of shortcomings, including the preparation of sensitive xanthates and the use of toxic reagents, such as tin derivatives. Growing ecological concerns have provided chemists with renewed impetus to offer environmentally friendlier alternatives such as the use of hypophosphite3 or the trialkylborane/water system.4 Electrochemistry is a particularly economical and environmentally benign technology that possesses enormous, though still under exploited, potential for the development of eco-friendly transformations. For example, only a few preparative electrochemical deoxygenation reactions have been recounted in the literature. Usually, they require either a rather negative potential,5 and hence, the use of a lead or mercury cathode with high overvoltage, or they are limited to primary and secondary alcohols.6 Recently, we have described a method for generating radical species, directly from simple toluateesters, using samarium(II) iodide.7 In this Communication, we wish to report the successful application of electrochemistry to the efficient deoxygenation of a large variety of toluateesters. In contrast to our previous approach, no metals and no toxic co-solvents (e.g.HMPA) are required.
It has already been proposed that, under electrochemical conditions, aromatic esters were initially reduced to the corresponding radical anion 3, which partitions into the carboxylate 4 and the radical 5 (See Scheme 1).8 The rate of this decomposition can be directly correlated with the stability of the produced radical 5: the more stable is 5, the faster will be the breakdown of 3.
 | ||
| Scheme 1 Decomposition pathway of the radical anion. | ||
By recording cyclic voltammograms of aromatic esters such as 1 and by using DigitalSimulation,9 we have been able to measure the kinetics of decomposition of various radical anions (See ESI† ). The modulation of the aromatic substitution revealed that the toluate moiety displayed enhanced reactivity.10 It was thus selected for the deoxygenation studies.
Our initial attempts to deoxygenate 9-fluorenyl toluate6 were performed in an undivided electrolysis cell (Table 1). Unfortunately, using different sacrificial anodes or various solvents led to none of the desired product 7. However, when the reaction was conducted in a divided H-type cell, in N-methylpyrrolidone (NMP) at 130 °C using tetrabutylammonium tetrafluoroborate as supporting electrolyte,11 we were able to isolate the deoxygenated product 7 in a yield of 50%.12 Surprisingly, when the electrolysis was carried out in acetonitrile, only degradation occurred, probably due to side reactions involving the solvent. The yields in DMF and in NMP are comparable, but NMP is more convenient to use for long electrolysis since it doesn’t suffer degradation when heated. However, NMP is difficult to remove (high boiling point) and we have tried to replace it by THF. Although the electrochemical window of THF is too small to allow the reduction of the toluate (−2.6 V vs. Ag/AgCl in EtOH sat. LiCl), it is possible to use a mixture of THF/NMP (9/1 in volume) if the electrochemical cell is modified to be compatible with refluxing THF.
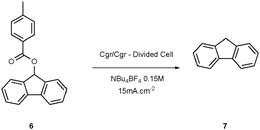
The temperature of the electrolysis also plays a key role (Table 2). The optimal temperature range lies between 70 °C and 100 °C. When the electrolysis was performed below 70 °C, only traces of the reduced compound were found among numerous degradation products.
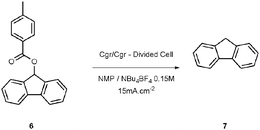
Finally, we have also investigated the influence of the cathode’s composition on the yield of the final product. As can be seen from Table 3, carbon graphite provides the best yield (entry 1). Surprisingly, with copper and lead electrodes, fluorenone was formed in 10% and 40% yields, respectively.13

In order to demonstrate the formation of a radical, akin to 5, during the reduction process, it was decided to capture it by a suitably positioned triple bond (See Scheme 2). Precursor 8 was thus assembled and subjected to the electrolytic conditions. Delightfully, the expected tetrahydrofuran 9 could be isolated in up to 60% yield.
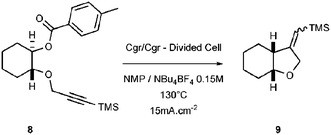 | ||
| Scheme 2 Radical cyclization. | ||
With this information in hand, the scope and limitations of this electrochemically-mediated deoxygenation methodology14 were investigated, with a special emphasis on functional group compatibility. Some results are collected in Table 4.












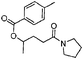



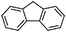
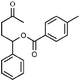
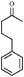
As can be seen from Table 4, a wide variety of functional groups are compatible with the deoxygenation reaction. The toluateester could be selectively deoxygenated in the presence of another ester (entry 4), an amide (entry 7) or a silyl ether (entry 5). Even ketones or unprotected alcohols are tolerated (entries 6 and 10). Only primary toluates tend to give moderate yields.
The inertness of the ketone function under these reductive conditions was rather puzzling and the deoxygenation of a few carbonyl-containing derivatives was next attempted (See Table 5).

As can be seen from Table 5, the substitution of the carbon atom bearing the toluate function by simple alkyl groups does not influence the fate of the reduction, since deoxygenation of primary, secondary and tertiary toluates gives only the reduced, non cyclic product 33, though the yields increase in the order 1°<2°<3° (entries 1, 2 and 3). On the other hand, if there is an anion stabilizing group α to the toluate, the radical could be rapidly reduced to the corresponding anion. This is illustrated by the electrolysis of substrate 32d, bearing a benzylic toluate (entry 4), which leads to the cyclic product 34d. In this case, the benzylic radical is rapidly reduced into the corresponding benzylic anion.
In summary, we have developed a novel, efficient and economical methodology for the deoxygenation of alcohols which tolerates a wide variety of functionalities. In contrast to previous methods, noxious or unstable xanthates and derivatives, expensive or sensitive metals and toxic co-solvents are no longer required. In addition, this methodology can also be useful for the electrogeneration of radicals or anions directly from the corresponding toluates.
Financial support of this work by the F.R.I.A. (Fond pour la formation à la Recherche dans l’Industrie et l’Agriculture, studentship to K.L.), the Université catholique de Louvain and Merck Sharp and Dohme (Merck Academic Development Program Award to I.E.M.) is gratefully acknowledged.
Notes and references
- K. Kaikiuchi, M. Ue, M. Takeda, T. Tadaki, Y. Kato, T. Nagashima, Y. Tobe, H. Koike, N. Ida and Y. Odaira, Chem. Pharm. Bull., 1987, 35, 617; T. H. Haskell, P. W. K. Woo and D. R. Watson, J. Org. Chem., 1977, 42, 1302 CrossRef CAS; E. L. O. Sauer and L. Barriault, Org. Lett., 2004, 6, 3329 CrossRef CAS.
- D. H. R. Barton and S. W. J. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574 RSC; W. Hartwig, Tetrahedron, 1983, 39, 2609 CrossRef CAS.
- D. H. R. Barton, D. O. Jang and J. Cs. Jaszberenyi, Tetrahedron Lett., 1992, 33, 5709 CrossRef CAS; S. R. Graham, J. A. Murphy and D. Coates, Tetrahedron Lett., 1999, 40, 2415 CrossRef CAS.
- D. A. Spiegel, K. Wiberg, L. Schacherer, M. Medeiros and J. L. Wood, J. Am. Chem. Soc., 2005, 127, 12513 CrossRef CAS.
- T. Shono, Y. Matsumura, K. Tsubata and Y. Sugihara, Tetrahedron Lett., 1979, 20, 2157 CrossRef.
- H. Ohmori, H. Maeda, M. Kikuoaka, T. Maki and M. Masui, Tetrahedron, 1991, 47, 767 CrossRef CAS; H. Maeda, T. Maki and H. Ohmori, Tetrahedron Lett., 1992, 33, 1347 CrossRef CAS; H. Maeda and H. Ohmori, Acc. Chem. Res., 1999, 32, 72 CrossRef CAS.
- K. Lam and I. E. Markó, Org. Lett., 2008, 10, 2773 CrossRef CAS.
- J. H. Wagenknecht, R. Goodin, P. Kinlen and F. E. Woodard, J. Electrochem. Soc., 1984, 131, 1559 CAS; R. D. Webster, A. M. Bond and R. G. Compton, J. Phys. Chem., 1996, 100, 10288 CrossRef CAS; R. D. Webster and A. M. Bond, J. Org. Chem., 1997, 62, 1779 CrossRef CAS.
- DigitalSimulations were performed by using DigiElch Pro software.
- The decomposition of the aromatic radical-anion is faster when the aromatic nucleus bears an inductive electron substituent, such as a methyl group. The methoxy-substituted benzoates are slightly more difficult to reduce but the rate of decomposition of the corresponding anion-radical is similar to that derived from the toluate. Finally, in the case of the simple benzoate, competing addition of the in situ generated radical to the para position of the aromatic ring has been observed.
- We suspect the tetrabutylammonium cation to be the hydrogen atom donor since only degradation occurred when lithium perchlorate was used as the supporting electrolyte.
- Standard electrolysis procedure: An H-type cell, with two compartments of 100 ml, separated by a sintered glass with a porosity of 40 μm, was dried during one night at 200 °C. Then, each cell was equipped with a graphite electrode of 6 cm2 and a magnetic stir bar. Both compartments were then flushed with argon during 10 min. After filling them with 5 g of NBu4BF4 and with 100 ml of NMP, freshly distilled under argon, 600 mg (0.6 mmol) of 9-fluorenyl toluate, dissolved in a little NMP, were added to the cathodic compartment and the solution was stirred and heated up to 130 °C. Then, the intensity of the current was fixed at 90 mA and the mixture was electrolysed until completion of the reaction, as shown by TLC or by GC. The cell was then cooled down to room temperature and the catholyte was carefully diluted with 100 ml of 4 N HCl. The resulting solution was extracted 4 times with 30 ml of ether. The organic phases were pooled, dried over sodium sulfate and the solvent was removed under reduced pressure. Finally, the crude product was purified by chromatography on silica gel, using pentane as eluent (Rf = 0.7), affording the title compound as a white powder in 50% yield. This material proved to be identical to an authentic sample of fluorene.
- A plausible explanation could be a reaction between the fluorenyl radical and the oxide layer of the electrode. Indeed, copper and lead electrodes are usually covered by a reactive oxide layer. In this regard, we have obtained a similar result when using Cgraphite electrodes in non-degassed NMP.
- No Kolbe-like dimers have been detected in this reaction. This could originate from the rapid capture of the radical by the large excess of tetra-alkylammonium salts or, as in the case of the benzylic substrates, by the fast reduction of this radical into the corresponding anion. (See Table 5, entry 4).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of new compounds, references to known compounds, an example of a cyclic voltammogram and kinetic rates determined by DigitalSimulation. See DOI: 10.1039/b813545b |
| This journal is © The Royal Society of Chemistry 2009 |